1.本发明涉及生物技术领域,具体涉及一种个体差异表达蛋白质的识别方 法。
背景技术:
2.临床蛋白质组学肿瘤分析联盟(cptac)开发了关于人类肿瘤组织中基于 lc-ms的蛋白质组学测量的标准工作流程,并将该工作流程应用于来自癌症 基因组图谱的结直肠癌、卵巢癌和乳腺癌数据(tcga)。在这项初步研究之后, 还对其他13种癌症类型的蛋白质基因组学进行了全面表征。这些cptac研 究探索了蛋白质表达的反式作用基因组畸变,基于蛋白质组学重新对分子亚 型进行分类,并使用磷酸蛋白质组学确定通路。由于大多数cptac研究未对 正常组织进行分析,因此无法评估差异表达分析结果的准确性。样本异质性 进一步使蛋白质组学分析复杂化,并限制了分析个体改变的癌症蛋白质组的 能力。
3.目前,基于lc-ms的蛋白质组学已用于肝细胞癌、肺腺癌(luad)和胃 癌的配对肿瘤和邻近正常组织的研究。这些研究通过功能蛋白质组学提供了 癌症基因型与表型更全面的联系。通过使用t检验或wilcoxon符号秩检验来 鉴定癌症相关蛋白,然而,这些检验只能用于检测群体水平的差异表达基因, 导致其中缺乏个体水平的精确信息,掩盖了个体间的异质性。
4.到目前为止,研究者们已经针对mirna,incrna数据提出了个体差异 表达方法,如penda,rankcomp,quantile,peng method。但是由于蛋白质 数据与mirna,incrna数据的差异性。这些方法在蛋白质数据上表现仍有 待提升。
技术实现要素:
5.本发明提供了一种个体差异表达蛋白质的识别方法,该方法允许从精确 的蛋白质信息中识别患者特定的蛋白质缺陷,从而为研究癌症机制提供新的 见解。
6.为实现上述目的,本发明采用以下技术方案:
7.一种个体差异表达蛋白质的识别方法,该方法包括以下步骤:
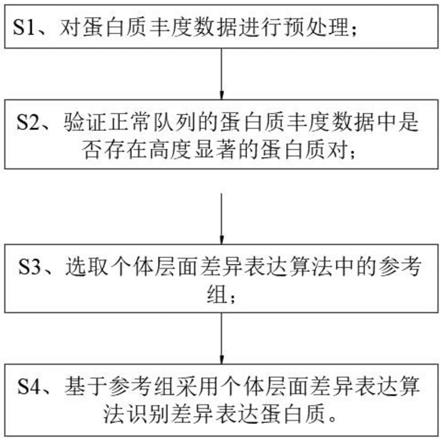
8.s1、对蛋白质丰度数据进行预处理;
9.s2、验证正常队列的蛋白质丰度数据中是否存在高度显著的蛋白质对;
10.s3、选取个体层面差异表达算法中的参考组;
11.s4、基于参考组采用个体层面差异表达算法识别差异表达蛋白质。
12.优选地,步骤s1的具体过程为:
13.s11、对蛋白质丰度数据中每个蛋白质在所有样本中缺失值的比例进行统 计,通过设置缺失值比例上限对蛋白质进行筛选,去除在蛋白质丰度数据中 缺失值比例超过阈值的蛋白质;
14.s12、通过计算蛋白质在同一队列的变异系数,判断蛋白质丰度在不同个 体间的差异是由于个体间的异质性还是定量误差导致的;
15.s13、对于输入的蛋白质丰度数据在样本上进行归一化,将蛋白质丰度数 据中还
存在的缺失值,基于贝叶斯的主成分分析方法对缺失值进行填充。
16.优选地,步骤s12中同一队列中的具有高变异系数的蛋白质被认定为定 量误差导致的,并对其进行筛除。
17.优选地,步骤s2的具体过程为:
18.s21、对于每一对蛋白质(pi,pj),ei与ej分别表示他们对应的丰度水平;
19.s22、通过二项分布检验计算偶然观察到该相对次序模式频率(k/n)的概 率为:
[0020][0021]
其中,p0为0.5,表示在正常样本中偶然观察到某种特定相对次序的概率; n表示样本的总数,k表示具有确定相对次序(ei》e
j or ei《ej)的样本数量;
[0022]
s23、采用benjamini andhochberg方法调整p值以控制fdr,再根据p 值判断该蛋白质对是否为高度稳定的蛋白质对。
[0023]
优选地,步骤s3的具体过程为:
[0024]
s31、确定正常样本与肿瘤样本中的稳定对;
[0025]
s32、对于同时存在于正常样本与肿瘤样本中的蛋白质(pi,pj),ei与ej分别表示他们的蛋白质丰度,若在正常样本队列与肿瘤样本队列中ei与ej关 系相同(ei》ej或ei《ej),则将蛋白质对(pi,pj)称为协同对;若在正常样 本与肿瘤样本中ei与ej关系相反,则将蛋白质对(pi,pj)称为逆转对,并将 所有与pi相关的逆转对中的蛋白质作为pi的参考组。
[0026]
优选地,步骤s4的具体过程为:
[0027]
s41、获取蛋白质pi的参考组后,比较正常队列中目标蛋白pi与所有参考 蛋白pj的表达量的相对关系,参考蛋白中pj《pi的数量记为a,pj》pi的数量记 为b;
[0028]
s42、比较肿瘤样本m中,目标蛋白pi与所有参考蛋白pj的表达量的相对 关系,将肿瘤样本m中pj《pi的数量记为c,pj》pi的数量记为d;
[0029]
s43、利用费雪精准检验得到蛋白质pi在肿瘤样本m中差异表达的显著 性,从而识别肿瘤样本m中的蛋白质pi是否差异表达。
[0030]
优选地,步骤s4还包括用于消除因参考组自身差异表达而引入误差的后 处理过程,所述后处理过程具体为:
[0031]
s44、对步骤s43中的差异表达结果进行迭代处理,将上一轮识别为差异 表达的蛋白质从参考组中去除;
[0032]
s45、重复步骤s41-s44,直到差异蛋白种类在前后两轮中的变化小于预 先设置的阈值,则迭代结束,否则继续重复步骤s41-s44。
[0033]
采用上述技术方案后,本发明与背景技术相比,具有如下优点:
[0034]
本发明提供一种个体差异表达蛋白质的识别方法(rank-prot方法),首 先基于蛋白质丰度数据的特征,对蛋白质丰度数据进行了预处理,去除了变 异系数高以及缺失值比例高的蛋白质,减少了蛋白质定量误差对个体差异蛋 白结果的影响;其次基于蛋白质稳定对选取参考组,并且利用迭代的方法, 不断优化参考组中存在差异表达的蛋白质,保证了参考组的稳定性。因此, 该方法可在个体水平上有效识别差异表达的蛋白质,识别精度高,应用前景 好。
附图说明
[0035]
图1为本发明的流程框图;
[0036]
图2为本发明的流程示意图;
[0037]
图3为本发明的rank-prot与其他算法在pn-score的比较结果图;
[0038]
图4为本发明的rank-prot方法与其他方法在预后蛋白的数量与显著性的 比较结果图。
具体实施方式
[0039]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及 实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实例 仅仅用以解释本发明,并不用于限定本发明。
[0040]
在本发明中需要说明的是,术语“上”“下”“左”“右”“竖直”“水 平”“内”“外”等均为基于附图所示的方位或位置关系,仅仅是为了便于 描述本发明和简化描述,而不是指示或暗示本发明的装置或元件必须具有特 定的方位,因此不能理解为对本发明的限制。
[0041]
实施例
[0042]
如图1至图4所示,本发明公开了一种个体差异表达蛋白质的识别方法, 该方法包括以下步骤:
[0043]
s1、对蛋白质丰度数据进行预处理;
[0044]
步骤s1的具体过程为:
[0045]
s11、对蛋白质丰度数据中每个蛋白质在所有样本中缺失值的比例进行统 计,通过设置缺失值比例上限对蛋白质进行筛选,去除在蛋白质丰度数据中 缺失值比例超过阈值的蛋白质;
[0046]
s12、通过计算蛋白质在同一队列的变异系数,判断蛋白质丰度在不同个 体间的差异是由于个体间的异质性还是定量误差导致的;
[0047]
步骤s12中同一队列中的具有高变异系数的蛋白质被认定为定量误差导 致的,并对其进行筛除;
[0048]
s13、对于输入的蛋白质丰度数据在样本上进行归一化,将蛋白质丰度数 据中还存在的缺失值,基于贝叶斯的主成分分析方法对缺失值进行填充;
[0049]
s2、验证正常队列的蛋白质丰度数据中是否存在高度显著的蛋白质对;
[0050]
步骤s2的具体过程为:
[0051]
s21、对于每一对蛋白质(pi,pj),ei与ej分别表示他们对应的丰度水平;
[0052]
s22、通过二项分布检验计算偶然观察到该相对次序模式频率(k/n)的概 率为:
[0053][0054]
其中,p0为0.5,表示在正常样本中偶然观察到某种特定相对次序的概率; n表示样本的总数,k表示具有确定相对次序(ei》e
j or ei《ej)的样本数量;
[0055]
s23、采用benjamini andhochberg方法调整p值以控制fdr,再根据p 值判断该蛋白质对是否为高度稳定的蛋白质对;
[0056]
s3、选取个体层面差异表达算法中的参考组;
[0057]
步骤s3的具体过程为:
[0058]
s31、确定正常样本与肿瘤样本中的稳定对;
[0059]
s32、对于同时存在于正常样本与肿瘤样本中的蛋白质(pi,pj),ei与ej分别表示他们的蛋白质丰度,若在正常样本队列与肿瘤样本队列中ei与ej关 系相同(ei》ej或ei《ej),则将蛋白质对(pi,pj)称为协同对;若在正常样 本与肿瘤样本中ei与ej关系相反,则将蛋白质对(pi,pj)称为逆转对,并将 所有与pi相关的逆转对中的蛋白质作为pi的参考组;
[0060]
s4、基于参考组采用个体层面差异表达算法识别差异表达蛋白质;
[0061]
步骤s4的具体过程为:
[0062]
s41、获取蛋白质pi的参考组后,比较正常队列中目标蛋白pi与所有参考 蛋白pj的表达量的相对关系,参考蛋白中pj《pi的数量记为a,pj》pi的数量记 为b;
[0063]
s42、比较肿瘤样本m中,目标蛋白pi与所有参考蛋白pj的表达量的相对 关系,将肿瘤样本m中pj《pi的数量记为c,pj》pi的数量记为d;
[0064]
s43、利用费雪精准检验得到蛋白质pi在肿瘤样本m中差异表达的显著 性,从而识别肿瘤样本m中的蛋白质pi是否差异表达;
[0065]
步骤s4还包括用于消除因参考组自身差异表达而引入误差的后处理过 程,所述后处理过程具体为:
[0066]
s44、对步骤s43中的差异表达结果进行迭代处理,将上一轮识别为差异 表达的蛋白质从参考组中去除;
[0067]
s45、重复步骤s41-s44,直到差异蛋白种类在前后两轮中的变化小于预 先设置的阈值,则迭代结束,否则继续重复步骤s41-s44。
[0068]
识别精度检测:
[0069]
由于肿瘤组织的之前的正常状态对于真实的肿瘤样本是未知的,因此使 用配对的肿瘤组织和肿瘤附近的正常组织作为金标准来比较差异表达算法识 别的差异蛋白质的精度。精度计算方法为tp/(tp fp),其中true positive(tp) 表示差异表达方向与金标准一致的差异蛋白质的数量,false positive(fp)表 示差异表达方向与金标准不一致的差异蛋白质的数量。
[0070]
根据dep的精度和数量计算pn-score,如下所示:
[0071][0072]
其中,prec_dep和num_dep分别表示差异蛋白质的精度与数量,β表 示计算pn-score时,精度与数量的权重。本发明的识别方法rank-prot与其他 算法在pn-score的比较结果如图3所示,其中横坐标表示β。
[0073]
由图3可知,在不同的β值下,rank-prot的pn-score都高于其他参考方 法。这表明在不同的个体差异蛋白的精度与数量权重下,本发明方法rank-prot 识别的个体差异蛋白不仅仅精度高并且兼顾了个体差异蛋白的数量。这为蛋 白质的下游分析,提供了可靠并且丰富的数据来源。
[0074]
预后蛋白质的发现:
[0075]
由于个体间的异质性,来自同一癌症类型的不同个体的蛋白质丰度可能 非常不同,因此个体中差异表达蛋白质可能具有预后意义。按如下步骤比较 不同个体差异表达算
法识别预后蛋白质的能力。首先,使用个性差异表达算 法来获得差异表达蛋白。对于每种蛋白质,计算显示为差异表达的样本数与 样本总数的比率,表示为差异表达率。再选择差异表达率在10%到90%范围 内的蛋白质作为候选预后蛋白质组。对于所有蛋白质,依次根据某一具体蛋 白质在样品中是否差异表达,将所有样品分为两组。使用单变量cox比例风 险回归模型来确定该蛋白质是否与生存相关。最后,比较了不同个体差异表 达算法获得的生存相关蛋白的数量和显著性。rank-prot方法与其他方法在预 后蛋白的数量与显著性的比较结果如图4所示。其中,图4中柱状图的高度 预后蛋白的数量,圆的直径对应显著性。本发明方法rank-prot对应的柱状图 的高度最高,说明发现的预后蛋白质数量最多。并且在p value小于0.01范围 内,预后蛋白对应的圆的数量明显多于参考方法(quantile,penda,peng)。 说明对于同一份蛋白质丰度数据,本发明方法能够挖掘出更多的与预后相关 的蛋白质,并且显著性高。
[0076]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不 局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可 轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明 的保护范围应该以权利要求的保护范围为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。