一种光诱导s-烷基二硫代氨基甲酸酯类化合物的合成方法及应用
技术领域
1.本发明涉及二硫代氨基甲酸酯类化合物技术领域,特别涉及一种光诱导s-烷基二硫代氨基甲酸酯类化合物的合成方法及应用。
背景技术:
2.s-烷基二硫代氨基甲酸酯是一种重要的含硫有机化合物,具有优异的化学特性与较强的生物活性,不仅广泛应用于抗肿瘤、抗真菌领域,在dna拓扑异构酶ii抑制剂的研究中也有重要应用价值。此外,s-烷基二硫代氨基甲酸酯还应用于农用化学品中,可以作为除草剂、驱虫剂和杀菌剂,具有较大的研究空间。如图2所示,列举了a-f六种市售药物分子。目前,s-烷基二硫代氨基甲酸酯类化合物的应用开发及新型高效合成方法的研究引起人们的较大关注,成为近年来研究热点之一。
3.合成s-烷基二硫代氨基甲酸酯的传统方法是二硫化碳、胺与亲电试剂(如卤代烷、卤代芳烃,有机硼试剂等)的一锅法反应。尽管该类方法有一些优点,开发更有效、经济和实用的合成方法来构建s-烷基二硫代氨基甲酸酯仍然是一项具有挑战性但非常有吸引力的任务。在过去几年中,可见光促进光氧化还原催化已成为有机合成的通用工具,并为构建具有挑战性的结构骨架和化学键开辟了绿色和可持续的途径。最近,文献报道,通过katritzky盐(烷基取代吡啶盐)的脱氨基策略已被证明是一种产生烷基自由基的新方法。然而,据我们所知,基于可见光诱导脱氨基策略将脂肪族伯胺间接转化为s-烷基二硫代氨基甲酸酯的方法至今未被开发。因此,开发一种更实用且环境友好的脱氨基方法来合成有用的s-烷基二硫代氨基甲酸酯衍生物尤为必要。
技术实现要素:
4.针对现有技术存在的不足,本发明提供一种光诱导s-烷基二硫代氨基甲酸酯类化合物的合成方法及应用,不需要苛刻的合成条件,能够一步合成各种s-烷基二硫代氨基甲酸酯类化合物及其衍生物,并且对多种官能团具有较高的普适性。
5.本发明为实现上述目的采用的技术方案是:一种光诱导s-烷基二硫代氨基甲酸酯类化合物,以取代的katritzky盐(烷基取代吡啶盐)、有机胺及其衍生物、二硫化碳为底物,使用碳酸盐或者含氮的有机碱作为碱,有机溶剂或水作为溶剂,在惰性气体环境和常温条件下,采用近蓝光照射反应液,“一锅法”合成s-烷基二硫代氨基甲酸酯类化合物。
6.进一步的,
7.所述s-烷基二硫代氨基甲酸酯类化合物的通式如式ⅰ所示;所述取代的katritzky盐的通式如式ⅱ所示;所述有机胺及其衍生物的通式如式ⅲ所示;
[0008][0009]
式中:
[0010]
r1选自氢、酯基或苄基中的一种;
[0011]
r2选自氢、苄基、烷基或酯基中的一种;
[0012]
r3选自烷基、芳基或氢中的一种;
[0013]
r4选自烷基、芳基、苄基、天然产物及药物分子衍生物中的一种;
[0014]
x选自pf6或bf4中的一种。
[0015]
本发明还包括一种光诱导s-烷基二硫代氨基甲酸酯类化合物的合成方法,所述合成方法包括如下步骤:
[0016]
(1)室温下,向装有磁力搅拌子的充满惰性气体的反应管中加入取代的katritzky盐、将有机胺及其衍生物、二硫化碳、碱加入到溶剂中,形成混合溶液,在惰性气体条件下,使用注射器加入到反应管中,常温条件下采用近蓝光照射反应液,促使反应发生;
[0017]
(2)反应结束后,向反应液中加入适量的去离子水,震荡以保证混合均匀,每次以3ml乙酸乙酯为萃取剂进行液相分离萃取,从反应液中提取粗产物,合并提取液,并通过旋转蒸发器除去溶剂;残渣经硅胶柱色谱纯化,制得s-烷基二硫代氨基甲酸酯类化合物。
[0018]
进一步的,
[0019]
所述惰性气体为氮气或氩气;
[0020]
所述近蓝光的波长范围为420nm-470nm;
[0021]
所述近蓝光的光源优选为蓝色led灯。
[0022]
进一步的,
[0023]
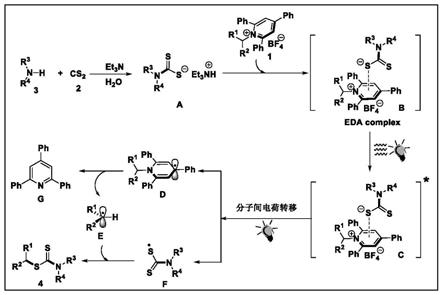
所述的有机胺及其衍生物物质的量为katritzky盐的1.0~2.5倍;
[0024]
所述二硫化碳的物质的量为katritzky盐的1.5~4.0倍;
[0025]
所述碱的物质的量为katritzky盐的1.0-3.0倍物质的量。
[0026]
进一步的,
[0027]
所述合成反应在标准大气压下进行,反应时间为12h~24h;
[0028]
所述碱为碳酸盐或含氮有机碱;
[0029]
所述的溶剂为有机溶剂和水中的一种。
[0030]
进一步的,
[0031]
所述碳酸盐为碳酸钾、碳酸钠或碳酸铯中的一种;
[0032]
所述含氮有机碱为三乙胺或吡啶中的一种;
[0033]
所述有机溶剂为dmso、ch3cn、dcm或etoh中的一种。
[0034]
进一步的,所述具体合成方法包括如下步骤:
[0035]
反应式为:
[0036][0037]
在室温下,将反应式中0.3mmol的取代katritzky盐加入到一25ml的schlenk管中,将0.8mmol的cs2,0.4mmol的有机胺,0.6mmol的et3n加入到2.0ml h2o中形成混合溶液,使用注射器将上述混合溶液在氮气环境中添加到含有取代katritzky盐的schlenk管中,schlenk管中充满氮气并配备有磁力搅拌器;并将反应管放置在距离10瓦蓝色led灯3cm处,蓝光照射并搅拌24小时;
[0038]
反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀,每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化,得到目标产物85mg,产率为82%。
[0039]
进一步的,所述合成的反应机理为:
[0040]
脂肪族伯胺(3)与二硫化碳(2)在碱的作用下形成硫醇盐(a),katritzky盐与硫醇阴离子通过阴离子交换过程产生eda复合物(b);经近蓝光照射激发产生激发态的eda复合物(c),再经近蓝光诱导从硫醇阴离子到katritzky盐(1)单电子转移,产生自由基中间体(d)与硫基自由基(f),自由基中间体(d)发生不可逆断裂产生烷基自由基(e)和三苯基吡啶(g);最后,烷基自由基(e)能够与硫基自由基(f)发生自由基交叉偶联反应,得到最终脱氨基产物s-烷基二硫代氨基甲酸酯类化合物(4)。
[0041]
本发明还包括一种光诱导s-烷基二硫代氨基甲酸酯类化合物的合成方法的应用,
[0042]
所述合成方法用于合成dna拓扑异构酶ii抑制剂;
[0043]
用于作为筛选抗肿瘤或抗真菌生物医药先导化合物;
[0044]
用于农用化学品中的除草剂、驱虫剂和杀菌剂。
[0045]
本发明光诱导s-烷基二硫代氨基甲酸酯类化合物的合成方法及应用的有益效果是:
[0046]
本发明不需要苛刻的合成条件,可一步完成反应,适用于合成各种s-烷基二硫代氨基甲酸酯类化合物,对芳环上的多种官能团具有较高的普适性,katritzky盐(烷基取代吡啶盐)的取代基的数量和类型没有特殊限制。此外,脂肪族伯胺的取代基的数量和类型不受特别限制。本发明的合成方法制备工艺和装置简单,以近蓝光为能量,在无过渡金属或其他光催化剂的条件下,采用有机溶剂或水作为溶剂,反应条件温和,绿色环保;由于合成方法只采用近蓝光作为能量激发诱导脂肪族伯胺间接转化,未采用金属或其他光催化剂,光无需复杂的预处理,具有无毒和友好环境的特点,符合绿色化学和原子经济的环保理念。
附图说明
[0047]
图1是本发明实施例合成方法的反应机理图;
[0048]
图2是本发明实施例含有s-烷基二硫代氨基甲酸酯骨架的药物活性分子。
具体实施方式
[0049]
下面结合附图与具体实施方式对本发明作进一步详细描述:
[0050]
实施例1:
[0051][0052]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),na2co3(0.4mmol)添加到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.3mmol),n-甲基苯胺(0.3mmol)溶解到h2o中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦455nm波长蓝色led灯3cm处,蓝光照射并搅拌24小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(20:1v/v))。得到目标产物85.0mg,产率为82%。
[0053]
所得产物核磁图谱数据为:
[0054]1h nmr(cdcl3,500mhz,ppm)δ7.44(d,j=7.4hz,3h),7.25-7.21(m,7h),4.97(t,j=7.0hz,1h),3.75(s,3h),3.57(s,3h),3.15(s,2h).
[0055]
13
c nmr(cdcl3,125mhz,ppm)δ196.7,171.5,144.4,137.6,129.9,129.3,128.4,127.0,126.9,56.0,52.5,46.5,38.4.
[0056]
高分辨质谱数据为:hrms calcd for c
18h19
no2s2na
[m na]
:368.0749;found 368.0752.
[0057]
实施例2:
[0058][0059]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),cs2co3(0.3mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.45mmol),4-甲氧基-n-甲基苯胺(0.4mmol)溶解到有机溶剂dmso中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦450nm波长蓝色led灯3cm处,蓝光照射并搅拌20小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(10:1v/v))。得到目标产物95.0mg,产率为85%。
[0060]
所得产物核磁图谱数据为:
[0061]1h nmr(cdcl3,500mhz,ppm)δ7.32-7.24(m,5h),7.17(d,j=8.4hz,2h),6.98(d,j=8.8hz,2h),4.99(t,j=15.0hz,1h),3.88(s,3h),3.76(s,3h),3.61(s,3h),3.18-3.12(m,2h).
[0062]
13
c nmr(cdcl3,125mhz,ppm)δ197.2,171.7,160.0,137.8,137.1,129.3,128.5,128.2,126.9,115.0,56.0,55.7,52.5,46.7,38.4.
[0063]
高分辨质谱数据为:hrms calcd for c
19h22
no3s
2
[m h]
:376.1036;found 376.1035.
[0064]
实施例3:
[0065][0066]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),k2co3(0.5mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.5mmol),二正丁胺(0.45mmol)溶解到有机溶剂dcm中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦420nm波长蓝色led灯3cm处,蓝光照射并搅拌22小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(20:1v/v))。得到目标产物79.4mg,产率为72%。
[0067]
所得产物核磁图谱数据为:
[0068]1h nmr(cdcl3,500mhz,ppm)δ7.31-7.26(m,4h),7.24-7.20(m,1h),5.08(t,j=15.3hz,1h),3.93-3.89(m,2h),3.69-3.59(m,5h),3.29-3.22(m,2h),1.72-1.64(m,4h),1.37-1.32(m,4h),0.95(dd,j=12.1,7.2hz,6h).
[0069]
13
c nmr(cdcl3,125mhz,ppm)δ193.4,171.9,137.7,129.4,128.5,127.0,55.5,52.8,52.6,38.5,29.5,28.5,20.3,20.2,13.9,13.8.
[0070]
高分辨质谱数据为:hrms calcd for c
19h30
no2s
2
[m h]
:368.1712;found 368.1708.
[0071]
实施例4:
[0072][0073]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),na2co3(0.6mmol)添加到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.7mmol),吗啉(0.6mmol)溶解到h2o中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦445nm波长蓝色led灯3cm处,蓝光照射并搅拌18小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(5:1v/v))。得到目标产物60.5mg,产率为67%。
[0074]
所得产物核磁图谱数据为:
[0075]1h nmr(cdcl3,500mhz,ppm)δ7.34-7.28(m,4h),7.27-7.24(m,1h),5.11(t,j=
7.5hz,1h),4.29(s,2h),3.97(s,2h),3.77(s,4h),3.69(s,3h),3.29(d,j=7.6hz,2h).
[0076]
13
c nmr(cdcl3,125mhz,ppm)δ195.0,171.5,137.3,129.3,128.6,127.1,66.3,55.3,52.7,51.0,38.8.
[0077]
高分辨质谱数据为:hrms calcd for c
15h20
no3s
2
[m h]
:326.0879;found 326.0884.
[0078]
实施例5:
[0079][0080]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.6mmol),吗啉(0.5mmol),吡啶(0.6mmol)溶解到etoh中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦470nm波长蓝色led灯3cm处,蓝光照射并搅拌12小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(8:1v/v))。得到目标产物70.3mg,产率为69%。
[0081]
所得产物核磁图谱数据为:
[0082]1h nmr(cdcl3,500mhz,ppm)δ7.25-7.22(m,2h),7.16(t,j=6.6hz,3h),4.82(t,j=7.0hz,1h),4.35-4.13(m,4h),3.97(s,2h),3.75-3.69(m,4h),2.79-2.69(m,2h),2.31-2.18(m,2h),1.26(t,j=7.1hz,3h)
[0083]
13
c nmr(cdcl3,125mhz,ppm)δ195.4,171.5,140.8,128.6,128.6,126.3,66.3,61.9,54.3,51.1,33.7,33.6,14.3.
[0084]
高分辨质谱数据为:hrms calcd for c
17h24
no3s
2
[m h]
:354.1192;found 354.1190.
[0085]
实施例6:
[0086][0087]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol)添加到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.4mmol),吡咯烷(0.6mmol),et3n(0.5mmol)溶解到dcm中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦420nm波长蓝色led灯3cm处,蓝光照射并搅拌20小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残
渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(15:1v/v))。得到目标产物62.8mg,产率为62%。
[0088]
所得产物核磁图谱数据为:
[0089]1h nmr(cdcl3,500mhz,ppm)δ7.29-7.26(m,2h),7.21-7.17(m,3h),4.90(t,j=7.0hz,1h),4.23-4.19(m,2h),3.91(t,j=6.9hz,2h),3.74-3.61(m,2h),2.83-2.72(m,2h),2.35-2.21(m,2h),2.11-2.05(m,2h),2,00-1.95(m,2h),1.30(t,j=7.1hz,3h).
[0090]
13
c nmr(cdcl3,125mhz,ppm)δ190.6,171.7,141.0,128.5,128.5,126.2,61.7,55.3,53.8,50.8,33.9,33.5,26.2,24.3,14.3.
[0091]
高分辨质谱数据为:hrms calcd for c
17h24
no2s
2
[m h]
:338.1243;found 338.1246.
[0092]
实施例7:
[0093][0094]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),na2co3(0.9mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.8mmol),n-甲基苯胺(0.5mmol)溶解到dmso中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦445nm波长蓝色led灯3cm处,蓝光照射并搅拌14小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(25:1v/v))。得到目标产物79.6mg,产率为71%。
[0095]
所得产物核磁图谱数据为:
[0096]1h nmr(cdcl3,500mhz,ppm)δ7.48-7.43(m,3h),7.26(t,j=7.4hz,4h),7.19-7.14(m,3h),4.73(t,j=7.1hz,1h),4.21-4.15(m,2h),3.74(d,j=15.1hz,3h),2.75-2.62(m,2h),2.25-2.08(m,2h),1.28(t,j=7.1hz,3h).
[0097]
13
c nmr(cdcl3,125mhz,ppm)δ197.2,171.6,140.9,130.0,129.3,128.5,127.0,126.2,61.6,55.0,46.4,33.6,33.6,14.3.
[0098]
高分辨质谱数据为:hrms calcd for c
20h24
no2s
2
[m h]
:374.1243;found 374.1242.
[0099]
实施例8:
[0100][0101]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),k2co3(0.7mmol)添加到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.6mmol),n-甲基苄胺(0.4mmol)溶解到dmso中,使用注射器将上述混合溶液在氮气环
境中添加到反应管中,并将反应管放置在距离10瓦460nm波长蓝色led灯3cm处,蓝光照射并搅拌18小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(7:1v/v))。得到目标产物80.1mg,产率为83%。
[0102]
所得产物核磁图谱数据为:
[0103]1h nmr(cdcl3,500mhz,ppm)δ7.33(t,j=7.2hz,2h),7.28(d,j=6.9hz,1h),7.24(d,j=5.1hz,4h),7.18(d,j=8.2hz,2h),5.04(s,2h),4.83(d,j=5.1hz,2h),3.16(s,3h).
[0104]
13
c nmr(cdcl3,125mhz,ppm)δ182.6,136.8,136.4,133.3,129.2,129.0,128.9,127.8,127.1,56.8,49.6,38.0.
[0105]
高分辨质谱数据为:hrms calcd for c
16h16
clns2na
[m na]
:344.0305;found 344.0301.
[0106]
实施例9:
[0107][0108]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.6mmol),二正丁胺(0.4mmol),et3n(0.8mmol)溶解到h2o中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦455nm波长蓝色led灯3cm处,蓝光照射并搅拌20小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(10:1v/v))。得到目标产物73.2mg,产率为74%。
[0109]
所得产物核磁图谱数据为:
[0110]1h nmr(cdcl
3,
500mhz,ppm)δ7.28(d,j=8.4hz,2h),7.24(d,j=8.4hz,2h),4.84(d,j=5.1hz,2h),3.62-3.50(m,4h),1.64-1.57(m,4h),1.36-1.28(m,4h),0.91(t,j=7.3hz,6h).
[0111]
13
c nmr(cdcl
3,
125mhz,ppm)δ181.1,137.1,133.3,129.2,128.9,51.2,49.3,29.7,20.3,13.9.高分辨质谱数据为:hrms calcd for c
16h25
clns
2
[m h]
:330.1111;found 330.1108.
[0112]
实施例10:
[0113][0114]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.7mmol),二苄
胺(0.6mmol),吡啶(0.6mmol)溶解到h2o中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦450nm波长蓝色led灯3cm处,蓝光照射并搅拌16小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(4:1v/v))。得到目标产物102.7mg,产率为86%。
[0115]
所得产物核磁图谱数据为:
[0116]1h nmr(cdcl3,500mhz,ppm)δ7.34-7.27(m,6h),7.23(d,j=7.0hz,4h),7.14(d,j=8.3hz,2h),6.91(d,j=8.3hz,2h),4.96(s,4h),4.75(d,j=5.1hz,2h).
[0117]
13
c nmr(cdcl3,125mhz,ppm)δ183.1,136.4,135.9,133.2,129.1,128.8,128.7,128.0,127.1,54.4,49.7.
[0118]
高分辨质谱数据为:hrms calcd for c
22h20
clns2na
[m na]
:420.0618;found 420.0612.
[0119]
实施例11:
[0120][0121]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),cs2co3(0.7mmol)添加到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.6mmol),n-乙基苯胺(0.4mmol)溶解到dmso中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦425nm波长蓝色led灯3cm处,蓝光照射并搅拌14小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(15:1v/v))。得到目标产物62.6mg,产率为57%。
[0122]
所得产物核磁图谱数据为:
[0123]1h nmr(cdcl3,500mhz,ppm)δ7.49(t,j=7.7hz,2h),7.40(t,j=7.4hz,1h),7.35-7.31(m,1h),7.29(s,1h),7.21(d,j=7.5hz,2h),7.15-7.09(m,2h),4.79(d,j=5.7hz,2h),4.28(q,j=7.1hz,2h),1.21(t,j=7.1hz,3h).
[0124]
13
c nmr(cdcl3,125mhz,ppm)δ181.9,140.8,140.8,130.8,130.5,130.3,130.2,129.1,128.3,125.9,122.7,50.2,48.8,13.1.
[0125]
高分辨质谱数据为:hrms calcd for c
16h17
brns
2
[m h]
:365.9980;found 365.9984.
[0126]
实施例12:
[0127][0128]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),na2co3(0.5mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将
cs2(0.8mmol),吗啉(0.6mmol)溶解到dcm中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦455nm波长蓝色led灯3cm处,蓝光照射并搅拌20小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(10:1v/v))。得到目标产物70.8mg,产率为71%。
[0129]
所得产物核磁图谱数据为:
[0130]1h nmr(cdcl3,500mhz,ppm)δ7.53(s,1h),7.39(d,j=7.9hz,1h),7.32(d,j=7.6hz,1h),7.17(t,j=7.8hz,1h),4.56(s,2h),4.33(s,4h),3.95(s,2h),3.76(s,4h).
[0131]
13
c nmr(cdcl3,125mhz,ppm)δ196.6,138.6,132.4,130.8,130.2,128.1,122.6,66.3,51.2,41.1.
[0132]
高分辨质谱数据为:hrms calcd for c
12h15
brnos
2
[m h]
:331.9773;found 331.9782.
[0133]
实施例13:
[0134][0135]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),k2co3(0.5mmol)添加到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.9mmol),二正丁胺(0.7mmol)溶解到h2o中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦440nm波长蓝色led灯3cm处,蓝光照射并搅拌24小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(5:1v/v))。得到目标产物60.3mg,产率为69%。
[0136]
所得产物核磁图谱数据为:
[0137]1h nmr(cdcl3,500mhz,ppm)δ4.80(q,j=7.4hz,1h),3.93-3.89(m,2h),3.75(s,3h),3.69-3.55(m,2h),1.74-1.65(m,4h),1.59(d,j=7.4hz,3h),1.39-1.32(m,4h),0.98-0.93(m,6h).
[0138]
13
c nmr(cdcl3,500mhz,ppm)δ193.8,173.1,55.4,52.8,49.2,29.5,28.5,20.3,17.5,14.0,13.8.
[0139]
高分辨质谱数据为:hrms calcd for c
13h26
no2s
2
[m h]
:292.1399;found 292.1400.
[0140]
实施例14:
[0141][0142]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol)添加
到一个25ml的schlenk管中,该管中充满氩气并配备有磁力搅拌器。将cs2(0.7mmol),n-甲基苄胺(0.6mmol),吡啶(0.8mmol)溶解到etoh中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦460nm波长蓝色led灯3cm处,蓝光照射并搅拌22小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(20:1v/v))。得到目标产物51.9mg,产率为61%。
[0143]
所得产物核磁图谱数据为:
[0144]1h nmr(cdcl3,500mhz,ppm)δ7.40-7.27(m,4h),7.22(d,j=6.4hz,1h),5.42-4.89(m,2h),4.85-4.78(m,1h),3.77(s,3h),3.34(d,j=85.4hz,3h),1.63(t,j=10.0hz,3h).
[0145]
13
c nmr(cdcl
3,
125mhz,ppm)δ196.7,172.8,135.5,128.9,127.9,127.3,59.7,58.0,52.9,49.8,17.5.
[0146]
高分辨质谱数据为:hrms calcd for c
13h18
no2s
2
[m h]
:284.0773;found 284.0776.
[0147]
实施例15:
[0148][0149]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol),cs2co3(0.8mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(0.8mmol),4-甲氧基-n-甲基苯胺(0.7mmol)溶解到ch3cn中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦420nm波长蓝色led灯3cm处,蓝光照射并搅拌24小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(20:1v/v))。得到目标产物82.1mg,产率为81%。
[0150]
所得产物核磁图谱数据为:
[0151]1h nmr(cdcl3,500mhz,ppm)δ7.22(d,j=8.4hz,2h),7.12(d,j=8.9hz,4h),6.94(d,j=8.8hz,2h),4.75(d,j=5.6hz,2h),3.79(s,3h),3.64(s,3h).
[0152]
13
c nmr(cdcl
3,
125mhz,ppm)δ182.6,159.5,136.9,135.0,133.0,128.7,128.7,128.2,115.9,55.6,48.9,43.8.
[0153]
高分辨质谱数据为:hrms calcd for c
16h17
clnos
2
[m h]
:338.0435;found 338.0429.
[0154]
实施例16:
[0155][0156]
在室温下,将上述反应式中取代的katritzky盐(烷基取代吡啶盐)(0.3mmol)添加到一个25ml的schlenk管中,该管中充满氮气并配备有磁力搅拌器。将cs2(1.0mmol),二苄
胺(0.5mmol),et3n(0.9mmol)溶解到dmso中,使用注射器将上述混合溶液在氮气环境中添加到反应管中,并将反应管放置在距离10瓦445nm波长蓝色led灯3cm处,蓝光照射并搅拌16小时。反应结束后,向反应液中加入2ml饱和食盐水,搅拌均匀。每次以3ml乙酸乙酯为萃取剂,通过液相分离萃取操作从反应液中萃取粗产物,合并萃取液,通过旋转蒸发器除去溶剂;残渣用硅胶柱纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯(15:1v/v))。得到目标产物77.7mg,产率为72%。
[0157]
所得产物核磁图谱数据为:
[0158]1h nmr(cdcl3,500mhz,ppm)δ7.38-7.33(m,6h),7.26-7.24(m,4h),5.35-5.22(m,2h),4.95-4.85(m,3h),3.78(s,3h),1.64(d,j=7.5hz,3h).
[0159]
13
c nmr(cdcl3,125mhz,ppm)δ197.6,172.7,135.4,134.5,129.1,128.9,128.2,128.1,127.96,127.4,56.2,54.3,52.9,50.0,17.3.
[0160]
高分辨质谱数据为:hrms calcd for c
19h22
no2s
2
[m h]
:360.1086;found 360.1090.
[0161]
在本发明的制备方法中,各种材料的加成顺序和具体反应步骤可由本领域技术人员进行调整,不仅适用于实验室的小规模制备,也适用于化工厂的工业化大规模生产。在工业批量生产中,具体反应参数可由本领域技术人员通过实验确定。
[0162]
若无特殊说明,下述实施例中所用的实验方法,均为常规方法。
[0163]
若无特殊说明,下述实施例中所用到的试剂、材料等,均可从商业途径获到或由商业途径所获原料合成。
[0164]
上述实施例只是为了说明本发明的技术构思及特点,其目的是在于让本领域内的普通技术人员能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡是根据本发明内容的实质所做出的等效的变化或修饰,都应涵盖在本发明的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
