钨亚氨基亚烷基o-bitet和o-binol配合物及其在烯烃复分解反应中的用途
技术领域
1.本发明涉及钨亚氨基亚烷基o-bitet配合物,其中本公开中使用的术语“o-bitet”是指衍生自5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚的配体,该配体通过从酚oh基团获取质子以其酚负离子的形式(olate-form)与钨结合。在另一个实施方案中,bitet配体以其芳族形式使用,即它衍生自1,1
’‑
联萘-2-酚,在本文中称为“o-binol”。该配合物可用于各种烯烃复分解反应,优选用于乙烯醇分解和交叉复分解,如不饱和脂肪酸酯的交叉复分解,以及用于闭环复分解反应。
背景技术:
2.过渡金属催化剂催化的烯烃复分解反应是有机合成化学中最重要的反应之一。一种有价值的已知催化剂类型是金属亚氨基亚烷基配合物类。其功效取决于金属、亚烷基和配体的类型。然而,直到现在,对这样的催化剂和待再复分解的底物之间各自的结构-活性关系的知识是有限的。因此,在特定复分解反应中催化剂的选择、合成和使用通常需要研究计划以找到最佳方案。
3.发明目的
4.本发明的目的是提供一组定制的且密切相关的金属亚氨基亚烷基化合物或多组密切相关的金属亚氨基亚烷基化合物,其被设计成在烯烃复分解反应中有效,并且优选在乙烯醇分解和交叉复分解如不饱和脂肪酸酯的交叉复分解以及在闭环复分解反应中有效。
技术实现要素:
5.该目的已通过所附独立权利要求中定义的特定钨亚氨基亚烷基o-bitet和o-binol配合物和使用该配合物的方法实现。优选实施方案在从属权利要求中限定。
6.钨亚烷基配合物的亚烷基部分被设计为基于
7.=ch-c(ch3)
2-c6h5[本文中表示为化学式(i)的化合物],或
[0008]
=ch-c(ch3)
2-苯基,其中苯环在邻位带有(或包含)选自o-(c
1-c6烷基)和-ch
2-o-(c
1-c6烷基)的基团[本文中表示为化学式(ii)的化合物],或
[0009]
=ch-苯基,其中苯环在邻位带有(或包含)选自o-(c
1-c6烷基)和-ch
2-o-(c
1-c6烷基)的基团[本文中表示为化学式(iii)的化合物],或
[0010]
=c(苯基)2,其中至少一个苯环在邻位带有(或包含)分别选自o-(c
1-c6烷基)和-ch
2-o-(c
1-c6烷基)的基团[本文中表示为化学式(iv)化合物],或
[0011]
=ch-ar,其中ar[本文中表示为化学式(vi)的化合物]选自苯基[本文中表示为化学式vi-a的化合物]、萘基[本文中表示为化学式vi-b的化合物]和蒽基(本文中表示为化学式vi-c的化合物)。优选地,当ar=苯基,即钨亚烷基部分是=ch-c6h5时,苯基残基未被取代或可以被取代,但在邻位不带有(或不包含)o-(c
1-c6烷基)基团。
[0012]
亚氨基残基优选为苯基亚氨基残基。
[0013]
优选地,所述苯基亚氨基残基被吸电子基团如卤素或三氟甲基取代,例如苯基残基是2,6-二氯苯基、五氟苯基或邻三氟甲基苯基。
[0014]
发明人发现,这样的配合物可以在各种烯烃复分解反应(例如不饱和脂肪酸酯的乙烯醇分解和交叉复分解以及闭环复分解反应)中提供优异的活性。
[0015]
不受理论的束缚,本发明人假设所选的金属(即钨)、含苯基的亚烷基部分、o-bitet配体或o-binol配体和亚胺基配体的组合在催化剂和待复分解的底物之间提供了有益的结构-活性关系。
具体实施方式
[0016]
包含=chc(ch3)2c6h5部分的化学式(i)的化合物
[0017]
根据第一方面,本发明涉及化学式(i)的化合物
[0018][0019]
其中
[0020]
m=w;
[0021]
r1选自被卤素或cf3中的一种或多种取代的苯基;
[0022]
r2选自吡咯-1-基或吲哚-1-基,分别任选地被取代;
[0023]
r3和r4中的一个是h,另一个是c(ch3)2c6h5;
[0024]
lo-是
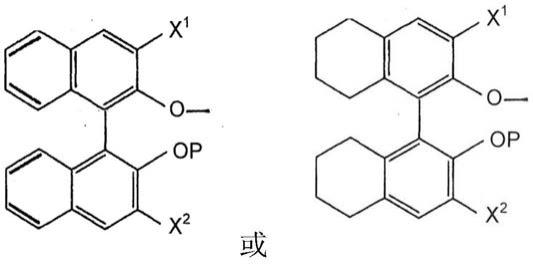
[0025][0026]
其中x1和x2独立地选自卤素、cf3和c6f6;或
[0027]
x1=x2=卤素、cf3或c6f5;
[0028]
p是c
1-c6烷基或甲硅烷基;且
[0029]
n是结合至m的中性配体,
[0030]
其中当lo-是o-bitet配体时,n是1或2,或
[0031]
其中当lo-是o-binol配体时,n是0、1或2。
[0032]
在一个优选的实施方案中,r1是2,6-二氯苯基、五氟苯基或邻cf
3-c6h4。
[0033]
如果没有另外说明,在本文定义的所有方面的整个公开内容中使用的术语“吡咯-1-基或吲哚-1-基,任选地被取代”是指各自的取代基可以选自c
1-4
烷基、c
1-4
烷氧基、卤素、腈和苯基中的一种或多种。
[0034]
在一个优选实施方案中,r2选自由吡咯-1-基、2,5-二甲基-吡咯-1-基、2,5-二乙基-吡咯-1-基、2,5-二苯基-吡咯-1-基和吲哚-1-基组成的组。
[0035]
在一个实施方案中,取代的吲哚-1-基是2-甲基-吲哚-1-基。
[0036]
已知loh可以以各种光学形式存在,即外消旋形式和对映异构体形式,即(r)和(s)形式。如果复分解反应产生的产物是手性的,则使用(r)或(s)对映异构体形成化学式(i)化合物中的o-bitet配体可以是有利的。然后,如果需要,可以形成复分解产物的光学活性形式。
[0037]
如果不希望形成光学活性形式,则优选使用外消旋形式的loh来形成式(i)化合物中的bitet配体。这在经济方面是有利的,因为外消旋loh通常比其分离的对映异构体更便宜。
[0038]
在一个实施方案中,lo-具有(r)构型。
[0039]
在另一个实施方案中,lo-具有(s)构型。
[0040]
在另一个实施方案中,lo-是外消旋的。
[0041]
与op部分中的p结合使用的术语“甲硅烷基”可以是在硅和氧之间形成共价键的任何甲硅烷基。
[0042]
已知的基团是例如叔丁基二甲基甲硅烷基(tbs,tbdms)、三甲基甲硅烷基(tms)、三乙基甲硅烷基(tes)、三异丙基甲硅烷基(tips)、叔丁基二苯基甲硅烷基(tbdps)和三苯基甲硅烷基。
[0043]
在一个优选的实施方案中,所述中性配体n是腈。
[0044]
优选地,所述腈是乙腈。
[0045]
腈通过n结合至m。
[0046]
在另一个优选的实施方案中,所述中性配体n是膦。
[0047]
优选地,所述膦选自由二甲基苯基膦、甲基二苯基膦和三(环己基)膦组成的组。
[0048]
膦通过p结合至m。
[0049]
在另一个优选的实施方案中,所述中性配体是吡啶。
[0050]
优选地,所述吡啶是吡啶本身,或2,2
’‑
联吡啶,或1,10-菲咯啉。
[0051]
所述吡啶可以被一个或多个独立地选自c
1-4
烷基、c
1-4
烷氧基、苯基、苯氧基和卤素的取代基取代。
[0052]
所述吡啶通过n结合至m,或者作为单齿配体如吡啶本身,或者作为双齿配体如2,2
’‑
联吡啶和1,10-菲咯啉。
[0053]
化学式(i)的示例性化合物是例如o-bitet配合物1、2、3和17、18和19:
[0054][0055]
[0056][0057]
化学式(i)的化合物可以由不带有中性配体的相应配合物通过分别用所述中性配体处理来制备,其根据已知方法在配体存在下制备。
[0058]
化合物4
[0059][0060]
已由ximo ag/hungary开发,并从wo 2014/139679中已知,其中它用于烯丙基苯(其表16中的化合物207)的自身复分解(homo metathesis)。在此,bitet配体lo-作为r-对映异构体提供。
[0061]
从wo 2017/087710(provivi公司)的权利要求26进一步已知该化合物。该参考文献公开了使用化合物4在两种内烯烃之间进行交叉复分解以产生信息素。
[0062]
通常,不带有中性配体的配合物如化合物4在合成后以非结晶形式或油状形式存在,或当用于复分解反应时甚至必须原位制备。将油状形式转化为固体形式的尝试通常导致严重的产率损失,这在经济和工业要求下是不可接受的。
[0063]
然而,与中性配体如腈(例如乙腈)配合的配合物可以以结晶形式提供。这是有利的,例如考虑到化合物在复分解反应中的处理、功效和商业方面。
[0064]
令人惊讶的是,还发现提供有作为外消旋体的lo-的化合物4结晶非常好,与用r-lo开发的化合物相反。
[0065]
在第一方面的另一个实施方案中,化学式(i)的示例性化合物是o-binol化合物5:
[0066][0067]
在关于化学式(i)的化合物的其他方面,r3也可以是c
1-5
烷基,其中其他残基具有如上关于所述化学式(i)的化合物所定义的含义。
[0068]
包含=chc(ch3)2苯基部分的化合物,其中苯基残基在邻位包含选自o-(c
1-6
烷基)和-ch
2-o-(c
1-6
烷基)的基团
[0069]
根据第二方面,本发明涉及化学式(ii)的化合物
[0070][0071]
其中
[0072]
m=w;
[0073]
r1选自芳基、烷基和环烷基,其各自任选地被取代;
[0074]
r2选自吡咯-1-基和吲哚-1-基,分别任选地被取代;
[0075]
r3和r4中的一个是h,另一个是碳(ch3)2苯基,其中c(ch3)2苯基部分的苯基另外在邻位被选自o-(c
1-c6烷基)和-ch
2-o-(c
1-c6烷基)的基团取代;
[0076]
lo-是
o-(c
1-6
烷基)的基团
[0103]
根据第三方面,本发明涉及化学式(iii)的化合物
[0104][0105]
其中
[0106]
m是w;
[0107]
r1选自芳基、烷基和环烷基,其各自任选地被取代;
[0108]
r2是吡咯-1-基或吲哚-1-基,分别任选地被取代;
[0109]
r3选自h;
[0110]
r4选自o-(c
1-c6烷基)和-ch
2-o-(c
1-c6烷基);
[0111]
r5是独立地选自h、c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素、no2、cn和nhc(o)-(c
1-c6烷基)的一个或多个残基;
[0112]
lo-是
[0113][0114]
其中
[0115]
x1和x2独立地选自卤素、cf3和c6f6;或
[0116]
x1=x2=卤素、cf3或c6f5;
[0117]
p是c
1-c6烷基或甲硅烷基;且
[0118]
n是结合至m的中性配体,其中n是0、1或2;
[0119]
条件是排除以下化学式的化合物
2017/087710(provivi公司)的权利要求27中已知。该参考文献公开了使用该被排除的化合物在两种内烯烃之间进行交叉复分解以产生信息素。
[0143]
结构(iii)的新化合物可以根据已知方法制备,例如通过wo2015/155593(ximo ag)中公开的亚烷基交换。在卡宾交换之前,可以通过根据已知方法使双吡咯与例如锂盐loli反应来将o-bitet配体引入配合物中。
[0144]
在一个优选的实施方案中,化学式(iii)的化合物选自一种化合物,其中
[0145]
m=w,r1=2,6-二氯苯基;r2=2,5-二甲基-吡咯-1-基;r3=h;r4=och3;r5=h;x1=x2=f;p=tbs(化合物7):
[0146][0147]
m=w,r1=2,6-二氯苯基;r2=2,5-二甲基-吡咯-1-基;r3=h;r4=och3;r5=h;x1=x2=cl;p=tbs(化合物8):
[0148][0149]
m=w,r1=2,6-二氯苯基;r2=2,5-二甲基-吡咯-1-基;r3=h;r4=och3;r5=r6=h;x1=x2=i;p=tbs(化合物9):
[0150][0151]
m=w,r1=2,6-二氯苯基;r2=2,5-二甲基-吡咯-1-基;r3=h;r4=och3;r5=h;x1=x2=cf3;p=tbs(化合物10),
[0152]
m=w,r1=2,6-二氯苯基;r2=2,5-二甲基-吡咯-1-基;r3=h;r4=och3;r5=h;x1=x2=c6f5;p=tbs(化合物11),和
[0153]
m=w,r1=2,6-二氯苯基;r2=2,5-二甲基-吡咯-1-基;r3=h;r4=och3;r5=h;x1=x2=br;p=tbs;n=1,10-菲咯啉;n=1(化合物12):
[0154][0155]
化合物12(其中lo-残基作为r-对映异构体提供)的特征在于改善的空气稳定性。其特征还在于,在溶液中配合物在释放菲咯啉时解离。剩余的亚烷基配合物在烯烃复分解中具有活性。鉴于在已知的亚烷基-菲咯啉配合物中中性菲咯啉配合物的去除需要加入路易斯酸如氯化锌,这是有利的。
[0156]
化合物12
[0157][0158]
也可以以其中lo-是外消旋体(或其中lo-是s-对映异构体)的形式提供。
[0159]
在关于化学式(iii)的化合物的另一方面,r3也可以是c
1-5
烷基,其中其他残基具有如以上关于所述化学式(iii)的化合物所定义的含义。
[0160]
包含=ch(苯基)2部分的化合物,其中苯基残基在邻位包含选自o-(c
1-6
烷基)和-ch
2-o-(c
1-6
烷基)的基团
[0161]
根据第四方面,本发明涉及化学式(iv)的化合物
[0162][0163]
其中
[0164]
m是w;
[0165]
r1选自芳基、烷基和环烷基,其各自任选地被取代;
[0166]
r2是吡咯-1-基或吲哚-1-基,分别任选地被取代;
[0167]
r3是
[0168][0169]
其中*表示r3和亚烷基碳之间的键;
[0170]
r4选自o-(c
1-c6烷基)和-ch
2-o-(c
1-c6烷基);
[0171]
r5是独立地选自h、c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素、no2、cn和nhc(o)-(c
1-c6烷基)的一个或多个残基;
[0172]
lo-是
[0173][0174]
其中
[0175]
x1和x2独立地选自卤素、cf3和c6f6;或
[0176]
x1=x2=卤素、cf3或c6f5;
[0177]
p是c
1-c6烷基或甲硅烷基;且
[0178]
n是结合至m的中性配体,其中n是0、1或2。
[0179]
在一个优选的实施方案中,r1选自由以下组成的组:被c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素和cf3中的一种或多种取代的苯基;叔丁基和1-金刚烷基。
[0180]
在一个实施方案中,r1选自被卤素或cf3中的一种或多种取代的苯基。
[0181]
在一个优选的实施方案中,r1是2,6-二氯苯基、五氟苯基或邻cf
3-c6h4。
[0182]
优选地,r2选自吡咯-1-基、2,5-二甲基-吡咯-1-基、2,5-二乙基-吡咯-1-基、2,5-二苯基-吡咯-1-基和吲哚-1-基。
[0183]
在一个实施方案中,lo-具有(r)构型。
[0184]
在另一个实施方案中,lo-具有(s)构型。
[0185]
在另一个实施方案中,lo-是外消旋的。
[0186]
外消旋lo-的使用在经济方面可以是有利的,因为外消旋loh通常比其对映异构体更便宜。
[0187]
与op部分中的p结合使用的术语“甲硅烷基”可以是在硅和氧之间形成共价键的任何甲硅烷基。
[0188]
已知的基团是例如叔丁基二甲基甲硅烷基(tbs,tbdms)、三甲基甲硅烷基(tms)、三乙基甲硅烷基(tes)、三异丙基甲硅烷基(tips)、叔丁基二苯基甲硅烷基(tbdps)和三苯基甲硅烷基。
[0189]
在一个优选的实施方案中,所述中性配体n是腈。
[0190]
优选地,所述腈是乙腈。
[0191]
腈通过n结合至m。
[0192]
在另一个优选的实施方案中,所述中性配体n是膦。
[0193]
优选地,所述膦选自由二甲基苯基膦、甲基二苯基膦和三(环己基)膦组成的组。
[0194]
膦通过p结合至m。
[0195]
在进一步优选的实施方案中,所述中性配体是吡啶。
[0196]
优选地,所述吡啶是吡啶本身,或2,2
’‑
联吡啶,或1,10-菲咯啉。
[0197]
所述吡啶可以被一个或多个独立地选自c
1-4
烷基、c
1-4
烷氧基、苯基、苯氧基和卤素的取代基取代。
[0198]
所述吡啶通过n结合至m,作为单齿配体或双齿配体。
[0199]
在化学式(i)、(ii)、(iii)或(iv)或(vi)的化合物存在下的复分解反应
[0200]
根据第五方面,本发明涉及进行复分解反应的方法,该方法包括:
[0201]
在如第一方面、第二方面、第三方面或第四方面或第八方面(定义如下)或其任何实施方案中定义的化学式(i)、(ii)、(iii)或(iv)或(vi)的化合物的存在下进行复分解反应。
[0202]
在一个优选的实施方案中,所述复分解反应选自内烯烃的乙烯醇分解、烯烃的交叉复分解、和闭环复分解反应。
[0203]
在一个优选的实施方案中,内烯烃的乙烯醇分解是乙烯与不饱和脂肪酸酯的反应。
[0204]
在另一个优选的实施方案中,交叉复分解反应是不饱和脂肪酸酯的自身复分解。
[0205]
在一个实施方案中,所述不饱和脂肪酸酯是天然油。
[0206]
术语“天然油”包括甘油三酯,例如植物油、藻油、鱼油和动物脂肪。
[0207]
在一个优选的实施方案中,不饱和脂肪酸酯是甲酯(fame),其中fame选自油酸甲酯、亚油酸甲酯和亚麻酸甲酯及其两种或三种的混合物。
[0208]
在一个特别优选的实施方案中,所述不饱和脂肪酸酯是油酸甲酯。
[0209]
乙烯醇分解反应允许由内烯烃通过与乙烯的交叉复分解反应形成末端烯烃。作为从生物质获得有用化学品的方法,包含内烯烃例如天然油或脂肪酸甲酯例如油酸甲酯的天然产物的有效乙烯醇分解是有吸引力的。
[0210]
在第五方面的另一个实施方案中,复分解反应是闭环复分解反应。
[0211]
在化学式(v)的化合物存在下使用不饱和脂肪酸酯的复分解反应
[0212]
根据第六方面,本发明涉及一种进行复分解反应的方法,其中所述复分解反应为不饱和脂肪酸酯的乙烯醇分解、不饱和脂肪酸酯的自身复分解,或闭环反应,所述方法包括:
[0213]
在化学式(v)的化合物的存在下进行复分解反应
[0214][0215]
其中
[0216]
m=w;
[0217]
r1选自被卤素或cf3中的一种或多种取代的苯基;
[0218]
r2选自吡咯-1-基或吲哚-1-基,分别任地选被取代;优选吡咯-1-基、2,5-二甲基-吡咯-1-基、2,5-二乙基-吡咯-1-基、2,5-二苯基-吡咯-1-基和吲哚-1-基;
[0219]
r3和r4中的一个是h,另一个是c(ch3)2c6h5;
[0220]
lo-是
[0221][0222]
其中x1和x2独立地选自卤素、cf3和c6f6;或
[0223]
x1=x2=卤素、cf3或c6f6;
[0224]
p是c
1-c6烷基或甲硅烷基;且
[0225]
n是结合至m的中性配体,其中n是0、1或2。
[0226]
在第六方面的一个实施方案中,lo-具有(r)或(s)构型;或lo-是外消旋的。
[0227]
p和n如第一方面所定义。
[0228]
在一个优选的实施方案中,在根据第六方面的方法中使用的化学式(v)的化合物中,r1是2,6-二氯苯基、五氟苯基或邻cf
3-c6h4。
[0229]
在一个实施方案中,在第六方面的方法中使用的化学式(v)的化合物中
[0230]
r1是2,6-二氯苯基,r2是2,5-二甲基吡咯-1-基,x1=x2=f;或
[0231]
r1是2,6-二氯苯基,r2是2,5-二甲基吡咯-1-基,x1=x2=cl;或
[0232]
r1是2,6-二氯苯基,r2是2,5-二甲基吡咯-1-基,x1=x2=br;或
[0233]
r1是2,6-二氯苯基,r2是2,5-二甲基吡咯-1-基,x1=x2=i。
[0234]
在一个实施方案中,化学式(v)的化合物选自由化合物13、14、15和16组成的组:
[0235][0236]
在第六方面的一个实施方案中,所述不饱和脂肪酸酯是天然油。
[0237]
在一个优选的实施方案中,所述不饱和脂肪酸酯是甲酯(fame)。
[0238]
在一个还更优选的实施方案中,所述甲酯是油酸甲酯或亚油酸甲酯或亚麻酸甲酯或其两种或三种的混合物。
[0239]
优选地,所述甲酯是油酸甲酯。
[0240]
在第六方面的另一个实施方案中,所述复分解反应是闭环复分解反应。
[0241]
可以根据本领域已知的方法在复分解之前纯化待进行复分解的化合物。例如,合适的方法描述于wo 2014/139679(ximo ag)中。
[0242]
具体的=chc(me)2c6h5配合物
[0243]
根据第七方面,本发明涉及化学式14、15、16或20的化合物:
[0244][0245]
化学式(vi)的包含=ch(芳基)部分(例如苯亚甲基)的化合物
[0246]
根据第八方面,本发明涉及化学式(vi)的化合物
[0247][0248]
其中
[0249]
m是w;
[0250]
ar选自苯基、萘基和蒽基,分别任选地被取代;
[0251]
r1选自芳基、烷基和环烷基,其各自任选地被取代;
[0252]
r2是吡咯-1-基或吲哚-1-基,任选地被取代;
[0253]
r3选自h;
[0254]
lo-是
[0255][0256]
其中
[0257]
x1和x2独立地选自卤素、cf3和c6f6;或
[0258]
x1=x2=卤素、cf3或c6f5;
[0259]
p是c
1-c6烷基或甲硅烷基;且
[0260]
n是结合至m的中性配体,其中n是0、1或2。
[0261]
如本文所用,术语“苯基、萘基和蒽基,分别任选地被取代”是指芳基残基可以独立地带有(或包含)c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素、no2、cn和nhc(o)-(c
1-c6烷基)中的一种或多种。
[0262]
在一个优选的实施方案中,r1选自由以下组成的组:被c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素和cf3中的一种或多种取代的苯基;叔丁基和1-金刚烷基。
[0263]
在一个实施方案中,r1选自被卤素或cf3中的一种或多种取代的苯基。
[0264]
在一个优选的实施方案中,r1是2,6-二氯苯基、五氟苯基或邻cf
3-c6h4。
[0265]
优选地,r2选自吡咯-1-基、2,5-二甲基-吡咯-1-基、2,5-二乙基-吡咯-1-基、2,5-二苯基-吡咯-1-基和吲哚-1-基。
[0266]
在一个实施方案中,lo-具有(r)构型。
[0267]
在另一个实施方案中,lo-具有(s)构型。
[0268]
在另一个实施方案中,lo-是外消旋的。
[0269]
外消旋lo-的使用在经济方面可以是有利的,因为外消旋loh通常比其对映异构体更便宜。
[0270]
与op部分中的p结合使用的术语“甲硅烷基”可以是在硅和氧之间形成共价键的任何甲硅烷基。
[0271]
已知的基团是例如叔丁基二甲基甲硅烷基(tbs,tbdms)、三甲基甲硅烷基(tms)、三乙基甲硅烷基(tes)、三异丙基甲硅烷基(tips)、叔丁基二苯基甲硅烷基(tbdps)和三苯基甲硅烷基。
[0272]
在一个优选的实施方案中,所述中性配体n是腈。
[0273]
优选地,所述腈是乙腈。
[0274]
腈通过n结合至m。
[0275]
在另一个优选的实施方案中,所述中性配体n是膦。
[0276]
优选地,所述膦选自由二甲基苯基膦、甲基二苯基膦和三(环己基)膦组成的组。
[0277]
膦通过p结合至m。
[0278]
在进一步优选的实施方案中,所述中性配体是吡啶。
[0279]
优选地,所述吡啶是吡啶本身,或2,2
’‑
联吡啶,或1,10-菲咯啉。
[0280]
所述吡啶可以被一个或多个独立地选自c
1-4
烷基、c
1-4
烷氧基、苯基、苯氧基和卤素的取代基取代。
[0281]
所述吡啶通过n结合至m,作为单齿配体或双齿配体。
[0282]
根据第八方面,本发明涉及化学式(vi-a)的化合物
[0283][0284]
其中
[0285]
m是w;
[0286]
r1选自芳基、烷基和环烷基,其各自任选地被取代;
[0287]
r2是吡咯-1-基或吲哚-1-基,任选地被取代;
[0288]
r3选自h;
[0289]
r4是r5;
[0290]
r5是独立地选自h、c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素、no2、cn和nhc(o)-(c
1-c6烷基)中的一个或多个;其中o-(c
1-c6烷基)不在邻位;
[0291]
lo-是
[0292][0293]
其中
[0294]
x1和x2独立地选自卤素、cf3和c6f6;或
[0295]
x1=x2=卤素、cf3或c6f5;
[0296]
p是c
1-c6烷基或甲硅烷基;且
[0297]
n是结合至m的中性配体,其中n是0、1或2。
[0298]
在一个优选的实施方案中,r1选自由以下组成的组:被c
1-c6烷基、o-(c
1-c6烷基)、苯基、卤素和cf3中的一种或多种取代的苯基;叔丁基和1-金刚烷基。
[0299]
在一个实施方案中,r1选自被卤素或cf3中的一种或多种取代的苯基。
[0300]
在一个优选的实施方案中,r1是2,6-二氯苯基、五氟苯基或o-cf
3-c6h4。
[0301]
优选地,r2选自吡咯-1-基、2,5-二甲基-吡咯-1-基、2,5-二苯基-吡咯-1-基和吲哚-1-基。
[0302]
在一个实施方案中,lo-具有(r)构型。
[0303]
在另一个实施方案中,lo-具有(s)构型。
[0304]
在另一个实施方案中,lo-是外消旋的。
[0305]
外消旋lo-的使用在经济方面可以是有利的,因为外消旋loh通常比其对映异构体更便宜。
[0306]
与op部分中的p结合使用的术语“甲硅烷基”可以是在硅和氧之间形成共价键的任何甲硅烷基。
[0307]
已知的基团是例如叔丁基二甲基甲硅烷基(tbs,tbdms)、三甲基甲硅烷基(tms)、三乙基甲硅烷基(tes)、三异丙基甲硅烷基(tips)、叔丁基二苯基甲硅烷基(tbdps)和三苯基甲硅烷基。
[0308]
在一个优选的实施方案中,所述中性配体n是腈。
[0309]
优选地,所述腈是乙腈。
[0310]
腈通过n结合至m。
[0311]
在另一个优选的实施方案中,所述中性配体n是膦。
[0312]
优选地,所述膦选自由二甲基苯基膦、甲基二苯基膦和三(环己基)膦组成的组。
[0313]
膦通过p结合至m。
[0314]
在进一步优选的实施方案中,所述中性配体是吡啶。
[0315]
优选地,所述吡啶是吡啶本身,或2,2
’‑
联吡啶,或1,10-菲咯啉。
[0316]
所述吡啶可以被一个或多个独立选自c
1-4
烷基、c
1-4
烷氧基、苯基、苯氧基和卤素的取代基取代。
[0317]
所述吡啶通过n结合至m,作为单齿配体或双齿配体。
[0318]
在另一个实施方案中,本发明涉及化学式(vi-b)的化合物,其中在化学式(vi)的化合物中,ar=萘基,任选地被取代。
[0319]
在一个实施方案中,化合物具有化学式(vi-ba),其中萘基是萘-1-基,任选地被取代。
[0320]
在另一个实施方案中,化合物具有化学式(vi-bb),其中萘基是萘-2-基,任选地被取代。
[0321]
在一个实施方案中,化合物具有化学式(vi-c),其中在化学式(vi)的化合物中,ar=蒽基,任选地被取代。
[0322]
在一个实施方案中,化合物具有化学式(vi-ca),其中蒽基是蒽-9-基,任选地被取代。
[0323]
在另一个实施方案中,化合物具有化学式(vi-cb),其中蒽基是蒽-1-基,任选地被
取代。
[0324]
在另一个实施方案中,化合物具有化学式(vi-cc),其中蒽基是蒽-2-基,任选地被取代。
[0325]
化学式(vi)的化合物也可用于第五方面所定义的复分解反应。
[0326]
在另一方面,本发明涉及一种组合物,其包含化学式(i)、(ii)、(iii)、(iv)、(v)或(vi)的化合物和待复分解的烯烃,其中所述待复分解的烯烃在复分解之前已经用三烷基铝化合物处理。
[0327]
在一个优选的实施方案中,在化学式(i)、(ii)、(iii)、(iv)、(v)或(vi)的化合物中,lo-是外消旋的。
[0328]
实施例
[0329]
3,3
’‑
二取代的1,1
’‑
联萘二酚(binol)衍生物和3,3
’‑
二取代的5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1-联萘二酚(bitet)衍生物根据已知方法合成,如e.s.sattely等人,j.am.chem.2009,131,943-953报道的。
[0330]
实施例1(比较例):
[0331]
wnar
cl
(me2pyrr)((r)-br-tbsobiteto)(ch(me)2ph)(化合物4)的合成:
[0332][0333]
由双吡咯前体(wnar
cl
(me2pyrr)2(chcme2ph))和(r)-3,3
’‑
取代-2
’‑
(叔丁基二甲基甲硅烷基氧基)-5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚制备原液(c=0.1m,以苯-d6为溶剂)。将100μl原液混合并在室温下搅拌过夜。然后加入500μl苯-d6并通过1h nmr 300mhz测量样品。该溶液用于催化反应,无需进一步转化。
[0334]
主要非对映异构体,1h-nmr(c6d6参比1h溶剂=7.16ppm):-0.06(s,3h),0.11(s,3h),0.93(s,3h),1.25-1.60(m br,8h),1.69(s,h),1.73(s,h),2.26(br,6h),2.00-2.60(m,4h),5.97(br,2h),6.23(t,1h,3jhh=8.1hz),6.85(d,2h,3jhh=8.1hz),6.93(m,1h),7.09(m,2h),7.16(s,1h),7.24(s,1h),7.42(m,2h),9.73(s,1h,1jch_syn=117.8hz,2jwh=16.0hz)ppm。
[0335]
实施例2
[0336]
wnar
cl
(chcme2ph)(me2pyr)((r)-br-tbsbitet-o))(mecn)(化合物1)的合成:
[0337][0338][0339]
反应在充满n2的手套箱中进行。圆底烧瓶配备有磁力搅拌棒。向烧瓶中加入起始的w(narcl)(chcme2ph)(2,5-me2pyr)2配合物(0.20g,0.30mmol),然后将其与甲苯(6ml)混合,得到棕黄色均匀溶液。然后在环境温度下将配体(r)-3,3
’‑
取代-2
’‑
(叔丁基二甲基甲硅烷基氧基)-5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚,0.17g,0.30mmol)以固体形式添加到该溶液中。将反应混合物搅拌过夜;通过nmr监测反应进程。在减压下去除溶剂。将残余物溶解在正戊烷(3ml)中,得到橙红色的均匀溶液。在室温向该溶液中加入mecn(18.5mg,24μl,0.45mmol)。加入mecn后,淡黄色沉淀从溶液中析出。将混合物放入手套箱的冰箱中一天。滤出黄色沉淀,用冷正戊烷(3ml)洗涤并减压干燥,得到黄色粉末状产物(m=217mg,62%)。
[0340]1h nmr(c6d6,300mhz):δ9.85ppm(s,1h,chcme2ph),7.41(d,2h,芳族),7.25(s,1h,芳族),7.16(s,1h,芳族),7.08(t,2h,芳族),6.92(t,1h,芳族),6.85(d,2h,芳族),6.22(t,1h,芳族),5.99(br s,2h,nc4h2),2.27(s,6h,me2nc4h2),2.15(m,8h,bitet),1.72(s,3h,phcme2),1.70(s,3h,phcme2),1.39(m,8h,bitet),0.93(s,9h,tbs),0.59(s,3h,mecn),0.14(s,3h,tbs),-0.10(s,3h,tbs)。
[0341]
实施例3
[0342]
w(nar
cl
)(chcme2ph)(me2pyr)((r)-br-tbsobitet-o)(py)(化合物3)的合成:
[0343][0344]
反应在充满n2的手套箱中进行。圆底烧瓶配备有磁力搅拌棒。向烧瓶中加入起始的w(nar
cl
)(chcme2ph)(me2pyr)2配合物(0.20g,0.30mmol),然后将其与甲苯(6ml)混合,得到棕黄色均匀溶液。然后在环境温度下将配体(r)3,3
’‑
取代-2
’‑
(叔丁基二甲基甲硅烷基氧基)-5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚,0.17g,0.30mmol)以固体形式添加到该溶液中。将反应混合物搅拌过夜;通过nmr监测反应进程。在减压下去除溶剂。将残余物溶
解在正戊烷(3ml)中,得到橙红色的均匀溶液。在室温向该溶液中加入几滴吡啶。加入吡啶后,淡黄色沉淀从溶液中析出。将混合物放入手套箱的冰箱中一天。滤出黄色沉淀,用冷正戊烷(3ml)洗涤并减压干燥,得到黄色粉末状产物(m=227mg,62%)。
[0345]1h nmr(甲苯-d8,300mhz,70℃):δ9.63ppm(s,1h,chcme2ph),8.45(d,2h,芳族),7.34(d,2h,芳族),7.15(s,2h,芳族),7.06-6.86(m,芳族),6.69(m,2h,芳族),6.32(t,1h,芳族),5.81(br s,2h,nc4h2),2.36(m,8h,bitet),2.16(s,6h,me2nc4h2),1.68(s,3h,phcme2),1.65(s,3h,phcme2),1.42(m,8h,bitet),0.84(s,9h,tbs),0.03(s,3h,tbs),-0.06(s,3h,tbs)。
[0346]
实施例4
[0347]
wnar
cl
(me2pyrr)(r)-tbsbinol)(chcme2ph)(化合物5)的合成
[0348][0349]
将双吡咯前体wnar
cl
(me2pyrr)2(chcme2ph)(0.035mmol,23.3mg)溶解在苯-d6(0.35ml)中。将((r)-3,3
’‑
二溴-2
’‑
(叔丁基二甲基甲硅烷基氧基)-1,1
’‑
联萘-2-酚(0.035mmol,19.5mg)溶解在苯-d6(0.35ml)中并在室温下加入到双吡咯前体中。将混合物室温下搅拌过夜。在1h nmr测量以确认结构后,使用催化剂溶液而无需进一步转化。
[0350]
实施例5
[0351]
wnar
cl
(me2pyrr)((r)-i-tbsobiteto)(ch(2-meo-c6h4))(化合物9)的合成:
[0352][0353]
将wnar
cl
(me2pyrr)((r)-i-bitet-o)(chcme2ph),(0.15mmol,191mg)溶解在甲苯(2ml)中并向其中加入2-meo-苯乙烯(0.165mmol,22.1mg)。将混合物在室温搅拌1天。添加额外量的2-meo-苯乙烯0.1mmol(14mg),并将混合物在室温下进一步搅拌。然后将混合物蒸发至干燥,用戊烷(4ml)研磨,然后用乙腈研磨并冷却至-40℃过夜。通过过滤分离固体并用
乙腈洗涤。分离后的产量:16mg,8.7%。
[0354]1h nmr(c6d6,δ
参比1h溶剂
=7.16ppm,25℃,300mhz):11.28ppm,特征性亚烷基信号。
[0355]
实施例6
[0356]
wnarcl(me2pyrr)((r)-f-bitet-o)(chcme2ph)(化合物7)的合成
[0357][0358]
将双吡咯前体wnar
cl
(me2pyrr)2(chcme2ph)(0.5mmol,332mg)溶解在甲苯(1ml)中。将(r)-3,3
’‑
氟-2
’‑
(叔丁基二甲基甲硅烷基氧基)-5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚(0.5mmol,222mg)溶解在甲苯(2ml)中并在室温下加入到双吡咯前体中。将混合物在室温下搅拌过夜,然后蒸发至干燥,用乙腈(3ml)研磨并冷却至-40℃过夜。滤出固体并用乙腈洗涤。然后溶于苯并蒸发至干燥以去除乙腈。分离后的产量:229mg,45%。
[0359]1h-nmr(c6d6,δ
参比1h溶剂
=7.16ppm,25℃,300mhz):9.53ppm;特征亚烷基信号。
[0360]
19
f-nmr(c6d6,δ
参比1h溶剂
=7.16ppm,25℃,282.4mhz):-134.8(s,1f),-133.2(s,1f)。
[0361]
实施例7wnar
cl
(me2pyrr)(chme2ph)(cl-(r)-tbsobitet-o)(化合物14)的合成
[0362][0363]
将双吡咯前体wnar
cl
(me2pyrr)2(chcme2ph)(0.5mmol,332mg)溶解在甲苯(1ml)中。将(r)-3,3
’‑
氯-2
’‑
(叔丁基二甲基甲硅烷基氧基)-5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚(0.5mmol,239mg)溶解在甲苯(2ml)中并在室温下加入到双吡咯前体中。将混合物在室温下搅拌过夜,然后蒸发至干燥,用乙腈(2ml)研磨并冷却至-40℃过夜。滤出固体并用乙腈洗涤。分离后的产量:287mg,54.8%。
[0364]1h-nmr(c6d6,δ
参比1h溶剂
=7.16ppm,25℃,300mhz)9.83ppm,特征亚烷基信号:
[0365]
实施例8
[0366]
在9-癸烯酸甲酯(9-dame)的自身复分解反应中测试根据本发明的化合物:
[0367][0368]
根据wo2014/139679(ximo)中已知的方法,通过吸附方法或三乙基铝(teal)处理来纯化底物。通过吸附法来纯化底物的反应在24小时后终止,通过三乙基铝来纯化底物的反应在4小时后终止(rt=室温)。
[0369]
通过色谱法测定转化率和形成的e-异构体和z-异构体。结果示于表1:
[0370]
表1
[0371][0372]
实施例9
[0373]
在闭环复分解反应中测试根据本发明的化合物:
[0374]
[0375]
在手套箱的气氛下,在烘箱干燥的4ml小瓶中,通过自动移液器添加底物并精确测量其重量。将其溶解在1ml甲苯中,然后将催化剂原料加入其中。用穿孔盖封闭小瓶并将反应混合物在室温下搅拌4小时。然后将1ml meoh添加到样品中以淬灭催化剂。用0.5ml二氧化硅层填充20ml塑料注射器,将1ml反应混合物通过其过滤并用20ml乙酸乙酯洗涤。通过gcms分析样品以确定转化率。通过手性hplc(agilent 1200plus hplc,带有256nm的二极管阵列检测器)测定产物的对映异构体比率。柱:kromasil 5-amycoat 4.6
×
150mm,使用h2o-meoh梯度洗脱)。
[0376]
结果示于表2:
[0377]
表2
[0378][0379]
实施例10
[0380]
在油酸甲酯的乙烯醇分解中测试了根据本发明的化合物。
[0381]
根据wo 2014/139679(ximo)已知的方法使用三乙基铝(teal)来纯化底物。将油酸甲酯与700ppmwt teal混合并将混合物在室温下搅拌4小时。
[0382]
在充满氮气的手套箱中,将脂肪酸甲酯量入30ml玻璃小瓶中,并与三乙基铝(在甲苯中23wt%)的原液混合。最佳三乙基铝量是先前确定的,为700ppm。将混合物在室温下搅拌1小时。将催化剂作为原液(在苯中0.01m)添加。将小瓶放入配备有alublock的不锈钢高压釜中,并在50℃在10atm乙烯气体过压下搅拌18小时。在具有公共气体空间的同一高压釜中进行五个反应。过量的乙烯被排出。从反应混合物中取出2.0μl并用正戊烷稀释至1.5ml,并通过gcms-fid分析,(shimadzu 2010plus,柱:zebron zb-35ht inferno,30m
×
0.25mm
×
0.25μm)。
[0383]
*与12-rac的反应以250ml的规模进行,因为催化剂由于其不溶性而以粉末形式分配到其中。通过gc对液相进行定量表明了下表3中给出的转化率:
[0384]
表3
[0385]
[0386]
1:化合物6,其中lo是外消旋的
[0387]
2:化合物6,其中2,5-二甲基-吡咯-1-基已经被2,5-二乙基-吡咯-1-基取代
[0388]
3:化合物12,其中lo是外消旋的
[0389]
在相当的条件下,使用化合物4的mo类似物进行的油酸甲酯的乙烯醇分解产生约30%的9-dame产率和约40%的总转化率。
[0390]
实施例11
[0391]
wnar
cl
(chcme2ph)(me2pyr)((s)-br-tbsbitet-o))(mecn)
[0392][0393]
在室温下,向溶解于甲苯(30ml)中的双吡咯w(nar-2,6-dicl)(char-o-ome)(2,5-me2pyr)2(1000mg,1.51mmol)中缓慢分批加入固体((s)-3,3
’‑
二溴-2
’‑
(叔丁基二甲基甲硅烷基氧基)-1,1
’‑
联萘-2-酚(853mg,1.51mmol)。将反应混合物室温下搅拌过夜。在处理前通过nmr分析确认完全转化为14-电子map配合物。在真空下蒸发溶剂。将残余物在ch3cn中研磨,得到标题化合物,为黄色粉末。产量:360mg(20%)。
[0394]1h nmr(c6d6,300mhz):δ9.89ppm(s,1h,chcme2ph)特征亚烷基信号。
[0395]
实施例12
[0396]
wnar
cl
(chcme2ph)(me2pyr)((rac)-br-tbsbitet-o))(mecn)
[0397][0398]
反应在充满n2的手套箱中进行。向100ml烧瓶中加入起始的w(nar-2,6-dicl)(chcme2ph)(2,5-me2pyr)2配合物(1.00g,1.51mmol),然后将其与甲苯(30ml)混合,得到棕黄色均匀溶液。然后在环境温度下将配体((rac)-3,3
’‑
二溴-2
’‑
(叔丁基二甲基甲硅烷氧基)-1,1
’‑
联萘-2-酚,0.853g,1.51mmol)以固体形式添加到该溶液中。将反应混合物搅拌过夜,通过nmr监测反应进程。在减压下去除溶剂。将残余物在乙腈中研磨,产生黄色沉淀。
滤出黄色沉淀,用冷正戊烷(10ml)洗涤并减压干燥,得到黄色粉末状产物(m=1102mg,62%)。
[0399]1h nmr(c6d6,300mhz):δ9.92ppm(s,1h,chcme2ph)特征亚烷基信号。
[0400]
实施例13
[0401]
wnar
cl
(me2pyrr)((r)-br-tbsobitet-o)(ch(2-meoc6h4))(化合物6)的合成
[0402][0403]
将2-甲氧基苯乙烯(1.19g,8.85mmol)溶于甲苯中,并加入到预先原位制备的w(nar
cl
)(me2pyrr)((r)-br-tbsobiteto)(ch(me)2ph)(7.63mmol,形式为在甲苯中的溶胶0.1mol/l)溶液中,在室温下搅拌混合物一周末。通过1h nmr监测反应的完成。在减压下蒸发反应混合物至干燥。将深红棕色残余物与戊烷(20ml)混合,红色沉淀从溶液中析出。滤出沉淀,用戊烷洗涤并干燥。分离后的产量:5624mg,65%。
[0404]1h-nmr(c6d6;δ
参比1h溶剂
=7.16ppm):-0.24(s,3h,ch
3 tbs),0.24(s,3h,ch
3 tbs),0.88(s,3h,c(ch3)3tbs),1.28-1.53(m br,8h,c6-h2,c6
’‑
h2,c7-h2,c7
’‑h2 bitet),1.76(br,3h,ch
3 dime-吡咯),1.86-2.40(m,4h,c8-h2,c8
’‑h2 bitet),2.06,2.11(m,2h,c5-h
2 bitet),2.47(br,2h,c5
’‑h2 bitet),3.16(br,3h,ch
3 dime-吡咯),3.67(s,3h,meo苯亚甲基ch3),6.15(br,2h,c
ar
h dime-吡咯),6.20(t,1h,3j
hh
=8.1,hz n-ar c
para-h),6.23(dd,1h,j=7.4,1.2hz,meo苯亚甲基c6-h),6.48(ddd,1h,j=8.1,7.4,1.2hz,meo苯亚甲基c4-h),6.64(d,1h,j=8.1hz,meo苯亚甲基c3-h),6.68(s,1h,c3-h bitet),6.84(td,1h,j=7.4,1hz,meo苯亚甲基c5-h),6.84(d,2h,3j
hh
=8.1hz,n-ar c
meta-h),7.28(s,1h,c3
’‑
h bitet),11.28(s,1h,w=ch,1j
ch_anti
=155hz,2j
wh
=7.2hz)ppm。
[0405]
wnar
cl
(me2pyrr)((s)-br-tbsobitet-o)(ch(2-meoc6h4))的合成
[0406]
[0407]
将双吡咯前体w(nar
cl
)(me2pyr)2(chcme2ph)(1mmol,664mg)溶解在苯(2ml)中。将(s)-3,3
’‑
二溴-2
’‑
(叔丁基二甲基甲硅烷基氧基)-5,5’,6,6’,7,7’,8,8
’‑
八氢-1,1
’‑
联萘-2-酚(1mmol,566mg)溶解在苯(2ml)中并在室温下加入到双吡咯前体中。通过1h nmr监测反应。将混合物室温下搅拌过夜然后蒸发至干燥。将残余物重新溶解在4ml苯中并加入2-meo-苯乙烯(1.5mmol,201mg),并将混合物在室温下搅拌过夜。然后将混合物蒸发至干燥,用正戊烷(5ml)研磨,然后冷却至-40℃过夜。通过过滤分离固体并用正戊烷(3ml)洗涤。获得红棕色固体(500mg,产率44%)。
[0408]1h nmr(c6d6,300mhz):δ11.28ppm(s,1h,chcme2ph)特征亚烷基信号。
[0409]
wnar
cl
(me2pyrr)((rac)-br-tbsobitet-o)(ch(2-meoc6h4))的合成
[0410][0411]
反应在充满n2的手套箱中进行。圆底烧瓶配备有磁力搅拌棒。向烧瓶中加入起始的w(nar-2,6-dicl)(chcme2ph)(2,5-me2pyr)2配合物(359mg,0.54mmol),然后将其与甲苯(10.5ml)混合,得到棕黄色均匀溶液。然后在环境温度下将配体((rac)-3,3
’‑
二溴-2
’‑
(叔丁基二甲基甲硅烷基氧基)-1,1
’‑
联萘-2-酚,0.296g,0.524mmol)以固体形式添加到该溶液中。将反应混合物搅拌过夜,通过nmr监测反应进程。在减压下去除溶剂。将残余物溶解在正戊烷(4ml)中,过滤去除固体,浓缩滤液至干燥。将残余物溶解在甲苯(6ml)中,加入2-甲氧基苯乙烯(0.594mmol,80mg)。将反应混合物搅拌过夜,减压蒸发溶剂至干燥。将深红色残余物溶于干燥的正戊烷(约5ml)中,通过过滤分离得到的红色结晶固体,用正戊烷洗涤并真空干燥。(m=299mg,49%)。
[0412]1h nmr(c6d6,300mhz):δ9.92ppm(s,1h,chcme2ph)特征亚烷基信号。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。