用一代测序评估样本dna中碱基损伤、错配和变异的方法
技术领域
1.本发明属于基因检测技术领域,尤其涉及一种用一代测序评估样本dna中碱基损伤、错配和变异的方法。
背景技术:
2.随着技术的发展,在dna检测领域,特别是癌症检测领域,人们越来越关注低比例的突变信息,比如0.1%的体突变信息是当前液体活检领域重点关注的指标之一,逐渐地,人们也会不再满足于0.1%这个指标,而要求更进一步,如果到了0.01%这个层面,就会面临如何区分突变与错配和碱基损伤的问题。
3.首先明确一下突变和错配这两个概念的含义。单拷贝细胞层面上,比如单个精子和卵子,它们是单倍体,突变这个概念在单倍体细胞比较难以适用,常规的突变更多是一种群体或者集体意义上的概念,比如人基因组hg19,chr1:2,000这个位置的碱基是c,那么如果1000个精子细胞中有1个精子细胞出现了c》t的突变,其他细胞保持野生型的c,那么我们说这个位置上出现了0.1%的c》t突变,而对于这个含有t的精子细胞中,chr1:2,000这个位置是正常t:a配对,并没有出现突变,而本专利所述的错配,是指在单个精子细胞中chr1:2,000并不是正常的c:g配对,而是t:g配对这种不符合碱基配对原则的情况,这种在双链中出现碱基配对错误,称之为碱基错配,这种碱基错配如果没有被修复系统修复,在某种情况下,被dna聚合酶复制过一次后,就变成了正常配对的t:a和c:g,并传给了子代,那么就形成了突变,因此突变在概念上,是有一个群体的语境的。
4.碱基损伤和碱基错配可以是先天形成的,也可以是后天形成的;先天形成的碱基错配是指,在生物体的细胞中,细胞在进行分裂增殖的过程中,由于体内dna复制体系的错误,为g错误的匹配上了非c的碱基,而这种错误并没有得到体内修复系统的修复,进而保留了下来;后天形成的碱基损伤是指,在我们提取dna的过程当中,由于技术、方法和条件的不恰当或局限而发生的损伤,比如胞嘧啶c在氧化条件下,发生氧化损伤,发生了脱氨反应,变成了脱氨基的胞嘧啶,然后在复制过程中,脱氨基的胞嘧啶被认为是尿嘧啶,进而与a发生匹配;又比如g在氧化条件下,容易形成8-oxog,然后在复制过程中,也容易oxo-g:a匹配;总体来说,一旦这些损伤的碱基和错配在生物体内稳定遗产下来,就会形成突变,在关键基因的关键位置发生的突变,并累积到一定程度,就有可能成为严重疾病的病因,比如癌症,也有可能成为耐药的原因。很明显能看出,如果是后天造成的碱基损伤,很容易给万分之一或者千分之一这个指标造成困扰,因此,评估样本中脱氧核糖核苷酸的损伤和错配是非常重要的,特别是对一些关键突变热点尤其显得重要,这些位置上碱基损伤导致的c》t、g》a会造成假阳性干扰。
5.由于这种损伤和错配发生的概率和比例是很低的,目前已知在万分之几左右,低于常规技术平台的灵敏度,比如二代测序平台的错误率在千分之几左右,因此,二代测序平台的检测灵敏度在1%左右;qpcr平台的某些技术最好的检测灵敏度在0.2%。因此在技术方面,如果要检测低比例的变异信息,是一定离不开分子标签标记技术的,但是分子标签技
术严重依赖于高深度测序,而且时间周期长,不利于检测项目的推广。
技术实现要素:
6.为解决上述技术问题,本发明公开一种评估dna样本中低比例碱基损伤或者碱基错配的方法,同时也可以评估生物体内天然存在的低比例碱基错配和碱基变化的比例,能够有利于优化样本dna提取技术和保存方法,帮助评估样本dna的质量。
7.本发明的第一个目的是提供一种用一代测序评估样本dna中碱基损伤、错配和变异的方法,包括如下步骤:
8.s1、加入能够抑制dna样本中非目标区域(非目标区域指的是对应发生碱基损伤、错配和变异的未发生碱基损伤、错配和变异的区域)的核酸组合物,以及带有可纠错的分子标签库的扩增引物,对dna样本进行扩增,采用一代测序技术对pcr扩增后的产物进行测序;
9.其中,抑制dna样本中非目标区域的核酸组合物根据dna样本中的采样区域进行设计;
10.s2、获得s1步骤pcr扩增产物的测序数据,采用基于富集放大作用的评估方法和基于分子标签种类数量的评估方法分别对产物的测序数据进行分析,获得评估样本dna中碱基损伤、错配和变异的比例值;
11.s3、在基于富集放大作用的评估方法和基于分子标签种类数量的评估方法结果同时具有可信结果时,采用基于分子标签种类数量的评估方法结果作为评估样本dna中碱基损伤、错配和变异的比例值。
12.其中,能够抑制dna样本中非目标区域的核酸组合物的设计方法参见申请号为2020115796048的中国专利。
13.可纠错的分子标签库的扩增引物的设计方法参见申请号为2020115404605的中国专利。
14.进一步地,所述的基于富集放大作用的评估方法,是通过如下步骤进行分析:
15.s01、用efold值代表每个采样区域的富集放大作用,计算公式为:
16.efold=(vrf/vaf)
×
[(1-vaf)/(1-vrf)],
[0017]
其中,vaf是样本中的变异信息的初始比例;vrf是检测结果中样本的变异信息比例;
[0018]
s02、通过对标准品测试,获得每个采样区域的efold值,并通过pcr扩增产物的测序数据中不同碱基的峰值比例计算vrf值,当vrf满足5%《=vrf《=95%时,通过如下公式计算vaf值:
[0019]
vaf=vrf/(efold-efold
×
vrf vrf),
[0020]
当vrf不满足5%《=vrf《=95%时,基于富集放大作用的评估方法结果不可信。
[0021]
例如,已知标准品样本中的变异信息的比例是0.1%,那么vaf=0.1%,经过富集放大之后,pcr产物经过测序分析,发现含有变异信息占比是50%,那么vrf=50%,此时:
[0022]
efold=(50%/0.1%)
×
[(1-0.1%)/(1-50%)]=999
[0023]
如果一个pcr反应没有富集放大作用,即vaf=0.1%,vrf也将是0.1%,那么,
[0024]
efold==(0.1%/0.1%)
×
((1-0.1%)/(1-0.1%))=1
[0025]
由此可见,efold=1的时候,整个反应体系对于变异信息来说是没有富集放大作
用的;efold对于一个具体的反应体系来说,体现了这个反应体系的内在特征,下表举例说明通过不同vaf和vrf计算出来的efold。
[0026] efoldvaf=0.1%,vrf=0.1%1vaf=0.1%,vrf=50%999vaf=0.1%,vrf=90%8991vaf=1%,vrf=50%99vaf=1%,vrf=90%891vaf=1%,vrf=99%9801vaf=5%,vrf=99%1881
[0027]
由上表可以看出,当vrf无限趋近于100%的时候,vaf值与efold无法继续成为正比的关系,比如,vaf=1%,vrf=99%和vaf=5%,vrf=99%的情况,表示某个反应在1%的时候,其放大富集作用就已经饱和,如果用vaf=5%时的efold值来表示某个反应的富集放大作用,反而是不准确的,因此,我们规定:某个具体反应的efold值,必须要在5%《=vrf《=95%的情况下获得。
[0028]
s02步骤具体如下表举例所示,当efold已知后,不同的vrf可以推算出待测目标样本中的vaf
[0029][0030][0031]
需要注意的是,如果在sanger信号中会呈现出变异信息的纯合峰,代表着信号可能已经饱和了,即vrf已经很接近100%了,vrf和vaf之间很可能无法成正比关系了,比如,vaf=5%的时候,sanger测序结果中的vrf=99%;vaf=10%的时候,sanger的vrf也还是99%,这样的话其实就无法区分vaf到底是5%还是10%,因此,当5%《=vrf《=95%的范围内时,vaf=vrf/(efold-efold
×
vrf vrf)才能在线性范围内合理成立,当vrf》95%或vrf《5%时,意味着待测目标样本的碱基损伤和/或碱基错配比例超出本专利公开方法的检测范围。
[0032]
进一步地,所述的基于分子标签种类数量的评估方法,是通过如下步骤进行分析:
[0033]
s001、基于pcr扩增产物的测序数据和已知分子标签序列的dna序列识别方法,所述dna序列识别方法输出一个分子标签序列的种类数量uminum;
[0034]
s002、当uminum《=10时,碱基损伤、错配和变异的比例pdm%计算公式如下:
[0035]
pdm%=uminum/(ng
×
1000
×
2/6.67)
×
100%,
[0036]
其中ng=反应中投入的dna的质量;
[0037]
当uminum》10时,基于分子标签种类数量的评估方法结果不可信。
[0038]
比如ng=10ng,uminum=5时,
[0039]
碱基损伤和错碱基错配的比例pdm%=5/2998.5
×
100%=0.17%。
[0040]
进一步地,在计算vrf值前或输出参数uminum之前,包括识别变异信息的步骤:
[0041]
s0001、获取sanger测序信号基线值noisec;方法在于:
[0042]
a)读取sanger ab1文件,得到文件中每个荧光通道每次信号采样的信号值fluor
cs
,以及每个碱基所在的信号采样数sk;
[0043]
fluor
ck
为荧光通道c在碱基k所在信号采样数sk±
i区域内的最大值,fluor
ck
的计算方法为:
[0044]
fluor
ck
=max{fluor
cs
:s=s
k-i..sk i}
[0045]
其中i可以是0~5之内的正整数;
[0046]
b)对每个荧光通道在所有n个碱基位置的最大值有从中去掉一代测序中碱基识别为荧光通道c对应的m个碱基(sanger ab1文件中有给出)的最大值,得到新的最大值集合:
[0047][0048]
c)计算的平均绝对偏差,去除与中位值之差超过平均绝对偏差n倍的值,n可以是取值在2~5,计算余下最大值的平均值noisec作为荧光通道c的背景噪音基线;
[0049]
d)对所有荧光通道信号采样的信号值减去对应荧光通道的背景噪音值,得到fluornn
cs
(no noise):
[0050]
fluornn
cs
=fluor
cs-noisec[0051]
s0002、根据各荧光通道的信号变化寻找区域信号峰值:
[0052]
遍历荧光通道的峰值,当一个碱基宽度wk的区域内只有任一通道存在峰值,则该区域有一个碱基,且该碱基的类型是有峰值的通道对应的碱基类型;当一个碱基宽度wk的区域内有多个通道存在峰值时,则该区域可能存在多个碱基,峰值最高的通道所对应的碱基类型是该区域的主要碱基,其他通道的峰值,以该峰值数据占主要碱基通道峰值的比例为依据,比例高于阈值时,则该通道对应的碱基类型是该区域的一个备选碱基类型,否则不存在备选碱基类型;得到由主要碱基和备选碱基组成的候选碱基序列a,并在存在备选碱基的位置标注备选的碱基类型;
[0053]
其中,所述一个碱基宽度wk的区域定义为:如果sanger ab1包含n个碱基,那么碱基k所处的信号采样数为sk,前一碱基所处信号采样数为s
k-1
,后一碱基所处信号采样数为s
k 1
,那么碱基k的碱基宽度区域起始位置wsk由如下公式得到:
[0054][0055]
碱基k的碱基宽度区域终止位置wek由如下公式得到:
[0056][0057]
其中,所述一个碱基宽度wk的区域内存在峰值的定义为:使用scipy的find_peaks算法对荧光通道c在s∈(wsk,wek)区域的去背景噪音后信号值fluornn
cs
计算区域峰值;若没有峰值存在,则荧光通道c在碱基宽度wk的区域内不存在峰值;若存在一个或多个峰值,则取信号值最大的峰值作为荧光通道c在碱基宽度wk的区域内的峰值;
[0058]
s0003、根据一代测序结果获得由iupac编码的候选碱基序列b:
[0059]
候选碱基序列b代表pcr产物的全长序列,包括候选碱基序列b1、候选碱基序列b2和候选碱基序列b3,其中,候选碱基序列b1为分子标签库位置的序列,候选碱基序列b2为样本dna采样区域的序列,候选碱基序列b3为除分子标签库位置的序列和样本dna采样区域的序列之外的其他序列;使用iupac(international union of pure and applied chemistry)推荐碱基编码规则合并所述候选碱基序列a中的主要碱基和备选碱基,得到由iupac编码的候选碱基序列b;如:
[0060][0061]
iupac编码表:
[0062][0063]
s0004、识别一代测序结果中的变异信息:
[0064]
1)用对位信息计算方法识别出候选碱基序列b与已知参考序列r(即参考序列基因组的序列,比如说,hg19)不同的信息;
[0065]
所述的对位信息计算方法是对所述的iupac编码的候选碱基序列b和已知参考序列r使用序列比对算法gotoh’s algorithm和nuc.4.4 iupac编码比对分数表进行比对;挑选比对分数最高的结果,作为候选碱基序列b和已知参考序列r的对齐结果,得到候选碱基序列b和已知参考序列r的对位信息;2)使用对位信息计算方法,得到候选碱基序列b2和b3和已知参考序列r的对位信息,并使两条序列对齐;扫描已对齐的候候选碱基序列b2和b3和已知参考序列r,得到iupac序列中与已知参考序列r不同的碱基信息,即为变异信息;
[0066]
其中,定义basek为某一碱基位置,参考碱基base
kr
是参考序列中的碱基信息,与已知参考序列r不同的碱基为base
km
;候选碱基序列b2和b3中某个具体位置的碱基basek由参考碱基base
kr
和代表损伤、错配或变异信息的base
km
组成。
[0067]
比如某一碱基位置basek在iupac序列中是“m”(对应着“a”或者“c”),而参考序列中是“a”,那么认为该位置存在一个碱基类型为“c”的变异信息,我们定义,参考碱基base
kr
是参考序列中的碱基信息,比如上述的“a”,与参考序列r不同的碱基称之base
km
,比如上述的“c”,可见,base
km
包含有碱基损伤、错配、变化或变异等信息,所述的base
km
是指代某一种具体的碱基类型,因此同一个位置可能具有多个base
km
。
[0068]
进一步地,vrf值通过如下公式计算得到:
[0069][0070]
其中peak(base
km
)是碱基base
km
的峰值荧光信号,是basek中碱基(包括主要碱基和备选碱基)的峰值荧光信号的总和。
[0071]
进一步地,分子标签序列的种类数量uminum通过如下方法得到:
[0072]
以b1相邻的扩增引物作为已知参考序列,对候选碱基序列b使用对位信息计算方法,得到候选碱基序列b和扩增引物的对位信息,并使两条序列对齐;通过b1序列已知长度信息,从对齐序列中得到候选碱基序列b1;
[0073]
提取候选碱基序列b1每个位置的n-信息作为特征值,所述的n-信息是指basek不包含的碱基类型,比如,候选碱基序列b1的1号位是w(a/t),那么1号位的n-信息是s(g/c),如果候选碱基序列b1的2号位是h(a/t/c),那么2号位的n-信息是g,将候选碱基序列b1的n-信息的集合定义为indexb,将标签序列库中的各个已知序列定义为index
l
,将index
l
的各个位置用indexb信息做排除,标签序列库index
l
中剩下的分子标签个数即为uminum。
[0074]
本发明的第二个目的是提供一种用一代测序评估样本dna中碱基损伤、错配和变异的分析装置,所述的分析装置包括:
[0075]
数据提取模块,用于获取一代测序ab1文件中碱基序列信息和荧光信号数据;
[0076]
预处理模块,用于荧光信号去背景噪音和候选碱基序列的生成;
[0077]
分析模块,用于分析获取一代测序结果中的变异信息;
[0078]
标签处理模块,用于分析计算pcr产物中分子标签种类数量uminum。
[0079]
本发明的第三个目的是提供一种服务器,包括一个或多个处理器和存储器,
[0080]
所述存储器,用于保存计算机程序;
[0081]
所述处理器,用于执行所述计算机程序,以实现所述的用一代测序评估样本dna中碱基损伤、错配和变异的方法。
[0082]
本发明的第四个目的是提供一种计算机可读存储介质,用于保存计算机程序,其中,所述计算机程序被处理器执行时实现所述的用一代测序评估样本dna中碱基损伤、错配和变异的方法。
[0083]
借由上述方案,本发明至少具有以下优点:
[0084]
本发明在pcr扩增过程中一方面采用分子标签来标记带有损伤或者错配的dna原始分子,另一方面对抽样区域进行富集放大扩增,将小于0.1%的损伤或者错配信息放大到10~99%,然后采用基于富集放大作用的评估方法和基于分子标签种类数量的评估方法分别评估样本dna中碱基损伤、错配和变异的比例值,根据两种方法的可信结果,判断样本dna中碱基损伤、错配和变异的比例值。本发明的方法能够采用经济快速的sanger测序方法准
确的确认损伤或者错配真实存在,能够有利于优化样本dna提取技术和保存方法,帮助评估样本dna的质量。
[0085]
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合详细说明如后。
附图说明
[0086]
图1为外周血dna样本碱基损伤sanger评估结果;
[0087]
图2为100种分子标签序列位次表;
[0088]
图3为获取n-信息示意图;
[0089]
图4为实施例2中利用碱基位次和n-信息在excel中排除不可能出现的分子标签示意图;
[0090]
图5为实施例2中利用n-信息进行排除并验证分子标签存在示意图;
[0091]
图6a和图6b为实施例3中100种分子标签序列位次表;
[0092]
图7为实施例3中的sagner测序结果;
[0093]
图8为实施例3中利用碱基位次和n-信息在excel中排除不可能出现的分子标签示意图;
[0094]
图9为实施例3中利用n-信息进行排除并验证分子标签存在示意图。
具体实施方式
[0095]
实施例1:用一代测序来评估样本dna中碱基损伤的程度
[0096]
1、在人类基因组的4个位置设置采样区域,并设计用于pcr的引物对组合,如下表:
[0097]
nameseq(5
’‑3’
)(seq id no.1~13)50mm,25℃,deltagdmde1-fpccctgacaacatagttggaatca-27.4dmde1-rpactccaggataatacacatcacagt-29.2dmde1-bltggaatcactcatgatatctcgagccat-34.0dmde2-fpagcagtctctgcctcgc-24.5dmde2-rpagaagattcggcagaactaagca-28.5dmde2-blcctcgccaagcggctcatgttaatatt-35.0dmde4-fpagaagatgtggaaaagtcccaatg-28.4dmde4-rpgtgcccaggtcagtggat-24.7dmde4-bltcccaatggaactatccggaacatcca-34.1dmde6-fptcctttaaccacataattagaatcattcttga-33.9dmde6-rpagttagttttcactctttacaagttaaaatga-33.5dmde6-blatcattcttgatgtctctggctagaccaaa-35.6unitagtgtaaaacgacggccagtaca [0098]
注意:表中的rp序列只是特异性序列部分,在制备过程中,加上unitaq序列构建5-tgtaaaacgacggccagtaca(n28)-rp结构,其中,n28是实例2中100种umi序列。
[0099]
2、根据hg19参考序列信息,定制合成阳性突变质粒模板。阳性突变模板中采样区域附近区域的序列如下:
[0100]
nameseq(5
’‑3’
)(seq id no.14~17)plasmid01tggaatcactcatgata
‑‑
tcgagccaplasmid02cctcgccaagc
‑‑
ctcatgttaplasmid04tcccaatggaactat
‑‑
ggaacatccplasmid06atcattcttgatgtctctg
‑‑
tagaccaaa
[0101]
其中,
“‑‑”
指2个碱基的缺失。
[0102]
3、制备0.1%的变异标准品。标准品的配置:先通过qubit定量,根据质粒模板的分子质计算理论的分子数,逐步配置成0.1%的变异标准品,然后通过ddpcr进行校正和调整,配置成相对误差较小的0.1%,后续继续通过二代测序的结果进行校正。
[0103]
4、通过ngs测序获得每个采样区域的efold值
[0104]
a)5
×
oligo mix with bl体系的配置
[0105]
组分引物浓度(μm)体积(μl)fp10020rp10020bl1001000.1
×
te 补齐至1000μltotal 1000μl
[0106]
b)5
×
oligo mix w/o bl体系的配置(供评估样本时作为对照使用,pcr体系使用量与with bl组相同)
[0107]
组分引物浓度(μm)体积(μl)fp10020rp100200.1
×
te 补齐至1000μltotal 1000μl
[0108]
c)pcr体系的配置
[0109]
试剂组成体积(μl)5
×
oligo mix with bl6μl2
×
dna聚合酶master mix15μl0.1%标准品300ngnuclease free water补齐至30μl
[0110]
d)umi-pcr扩增程序
[0111][0112]
[0113]
pcr结束后,每个反应中加入1个单位的核酸外切酶i,37℃孵育30分钟,80℃灭活30分钟。再加入2μl 10μm fp和2μl 10μm unitag,进行后续的pcr扩增程序。
[0114]
e)后续pcr过程
[0115][0116]
5、反应后的pcr产物用商业化的二代测序建库试剂盒进行建库,在illumina平台上进行测序,测序之后,分析含有2bp deletion变异信息的reads的分子标签种类数量,同时分析野生型信息的reads的分子标签种类数量,两者之比为校正后的vaf;分析含有变异信息的reads数和分析野生型信息的reads数,两者之比为vrf。计算每个采样位置的efold值。
[0117] ngs校正前vafngs校正后vafvrfefolddmde10.1%0.25%57.2%533.2dmde20.1%0.31%83.5%1627.4dmde40.1%0.15%48.4%624.4dmde60.1%0.23%61.0%678.5
[0118]
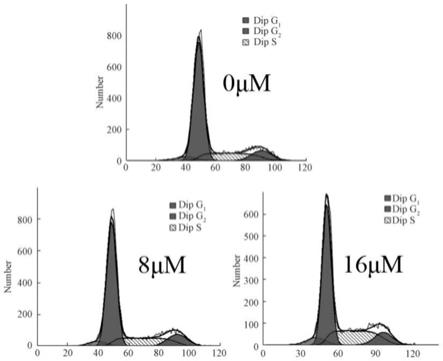
6、选择需要评估的外周血dna样本,dna input=30ng,然后with bl和w/o bl组同时做,确保没有污染,同时在两组的比较中可以看到富集和放大效果,部分结果如图1所示,可以看到w/o bl组显示的是野生型信息,意味着没有任何富集放大作用。
[0119]
7、根据ngs结果中获得的efold和sanger分析步骤获得的vrf,根据公式:vaf=vrf/(efold-efold
×
vrf vrf)计算原始样本中的vaf:
[0120]
采样区域名称碱基位置信息efoldvrfvafdmde19g》a533.273%0.50%dmde211c》t1627.49%0.01% 12c》t1627.411%0.01% 13g》a1627.447%0.05% 14c》t1627.429%0.03%dmde46t》c624.48%0.01% 10g》a624.431%0.07%dmde610g》a678.550%0.15% 12g》a678.535%0.08%
[0121]
由于采样区域中可能存在多个碱基位置可能发生损伤或者错配,因此,我们将最终损伤或者错配的程度评估为一个范围,比如dmde2,我们认为损伤或者错配的程度是0.01%~0.05%,考虑到30ng input大约有9000个拷贝,那么检测到的损伤或者错配的原始分子可能在1~5个左右。同时,我们在大量的检测中发现,c》t和g》a的情况是最多的,也符合文献中介绍的胞嘧啶脱氨和g氧化后容易错配上t的情况。
[0122]
实施例2:从sanger结果中分析分子标签个数uminum的逻辑演示
[0123]
1、制备100种序列已知的分子标签,每个分子标签28nt,将每个碱基分开占位,如图2所示。
[0124]
2、假设pcr产物中含有如图3所示的5种分子标签序列,在一代测序之后,根据sanger结果,在每个位置的n-信息都可以获得,如图3所示。
[0125]
3、根据n-信息过滤分子标签的已知序列,比如在第16位碱基,需要排除该位置不含g和t的分子标签,在经过第1~16位的n-信息的排除之后,只剩下15种分子标签,如图4所示;
[0126]
4、继续根据n-信息进行排除,当进行到第28位碱基时,最终留下5个分子标签,恰好上之前假设存在的那5个分子标签,如图5所示;
[0127]
5、本实例描述了利用已知序列的分子标签在sanger测序之后获得n-信息来反推pcr产物中分子标签种类个数的逻辑,具体的实际分析由编写的软件完成。
[0128]
实施例3
[0129]
为了说明本发明方案具有更普遍的实用性,本实施例在不同于实施例1的其他人类基因组位置进行设计,与实施例1的引物设计原则相同,本实施例的引物设计原则参考本公司早期的专利cn110923325a《用于检测egfr基因突变的引物blocker组、试剂盒及方法》以及cn110982884a《用于检测aml相关基因突变的引物组、试剂盒及方法》;
[0130]
ssl3-fp:ccagaaaacaggcaggtctctc
[0131]
ssl3-bl:caggtctctctgctcttgaccgagc
[0132]
ssl3-rp:acagcaggcagttggga
[0133]
unitaq序列与实施例1相同,同时本实施例中的ssl3-rp序列只是特异性序列部分,在制备过程中,需要加上unitaq序列构建5-tgtaaaacgacggccagtaca(n28)-rp结构,其中,n28是如图6a和6b所示的100种umi序列,umi的设计部分借鉴了cn110060734b《一种高鲁棒性dna测序用条形码生成和读取方法》,不同之处在于cn110060734b设计的条形码用于样本的区别,读取的方式更为复杂,而本发明方案中用于区分样本中不同的原始分子,同时也具有更为简单的读取和识别方式。
[0134]
实验方法参考实施例1,根据hg19参考序列信息,定制合成阳性突变质粒模板,具体位置为图7中80位置附近的c》g,配置成0.1%的变异标准品,将pcr产物直接进行sanger测序,重复测序3次,实验结果如图7所示,横框中为umi所在的区域,竖框为c》g位置。
[0135]
从三次重复的sanger的结果可以清楚看到umi开始的前4位碱基是ctca的纯峰,利用与实施例2相同n-信息的思路进行排除,可以筛选到6种umi分子标签,如图8所示。
[0136]
第5位的n-信息为c和a,对umi的进一步筛选并没有帮助,第6位的n-信息为t、g和a,这是一个有用的信息,可以进一步筛选得到2种umi分子标签,如图9所示;
[0137]
随后位置的n-信息可以进一步明确,sanger结果就是由这两种umi序列组成的,同时两种umi序列在pcr产物中的比例接近1:1,各占50%,提示我们发生有碱基突变的原始dna分子至少有两条。
[0138]
以上仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。