1.本发明涉及药物化学技术领域,特别涉及一种喹啉-1,2,4-三嗪杂合体、制备方法及其应用。
背景技术:
2.疟疾是一种威胁人类生命的疾病,传统的疟疾治疗药主要包括奎宁,青蒿素等植物提取物,在此基础上开发的青蒿素、双氢青蒿素、蒿甲醚以及氯喹、伯喹等,是目前疟疾治疗中的重要药物。但随着抗疟药的长期使用,恶性疟原虫对抗疟药物氯喹,抗叶酸剂(磺胺多辛-乙胺嘧啶)和青蒿素等先后产生耐药性,并在全世界范围蔓延,阻碍了疟疾的消除。
3.目前处于临床研究阶段的抗疟疾新药屈指可数,显然,开发具有新型作用模式的抗疟新药迫在眉睫。
4.近年来,开发抗疟疾杂合体新药的研究在世界范围内亦呈现良好的发展势头。喹啉,作为一种通用的药效团,是一种作用杰出的稠合杂环化合物,具有广泛的药理学前景,例如抗癌,抗炎,抗菌,抗病毒以及优秀的抗疟疾活性。以喹啉为母核设计与其他具有抗疟活性的分子合成杂合体,由此设计的化合物分子可同时作用于多个抗疟靶点,具有两种以上的抗疟功能,以及比母体化合物更好的抗疟活性。
5.三嗪类化合物是一类重要的含氮杂环化合物,在化工染料、材料方面应用广泛,近年来发现其在抗炎、抗菌、抗肿瘤、缓解阿尔茨海默症等方面具有显著作用,因此可作为医药中间体进一步发展其应用。
技术实现要素:
6.本发明提供给了一种新的具有抗疟效果的化合物。
7.为了实现上述目的,本发明具体采用如下技术方案:
8.一种喹啉-1,2,4-三嗪杂合体,所述氯-喹啉与1,2,4-三嗪杂合体结构如式i所示:
[0009][0010]
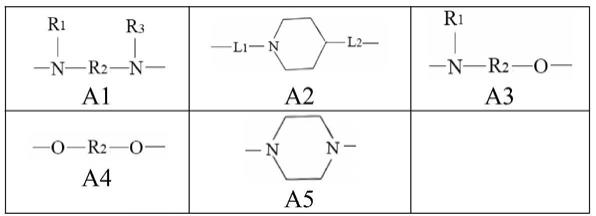
所述linker选自如下式a1~a5所示的基团:
[0011][0012]
其中:r1和r3选自h、c1~c3的烷基;
[0013]
r2选自-(ch2)
n-或其中n为2~8,n1为0~3,n2为0~3;
[0014]
l1和l2为单键或亚甲胺基。
[0015]
优选的,所述r1和r3选自h或甲基。
[0016]
优选的,式i所示结构选自如下:
[0017][0018]
喹啉-1,2,4-三嗪杂合体的制备方法为,式ii所示的化合物与式iii所示的化合物在碳酸钠或碳酸铯的存在下反应得到式i所示的喹啉-1,2,4-三嗪杂合体;
[0019][0020]
所述r选自如下式b1~b5所示的基团:
[0021][0022]
其中:r1和r3选自h、c1~c3的烷基;
[0023]
r2选自-(ch2)
n-或其中n为2~8,n1为0~3,n2为0~3;l1和l2为单键或亚甲胺基。
[0024]
优选的,所述的反应在乙腈或dmf中进行。
[0025]
优选的,所述式ii所示化合物与碳酸钠或碳酸铯的摩尔比为1:3~5。
[0026]
优选的,所述反应温度为15℃~35℃。
[0027]
优选的,式iii所示的化合物的制备方法如下:
[0028][0029]
3-甲硫基-1,2,4-三嗪经三氯过氧苯甲酸氧化得到式iii所示的化合物。
[0030]
优选的,式iii所示的化合物的制备的具体步骤为:将mcpba溶于二氯甲烷,后加入无水硫酸钠除水得到mcpba溶液,冰浴条件下,将mcpba溶液滴加至3-甲硫基-1,2,4-三嗪的二氯甲烷溶液,后室温反应至结束。
[0031]
喹啉-1,2,4-三嗪杂合体,式ii所示的化合物的制备方法如下:
[0032]
4,7-二氯喹啉与胺或醇在80~120℃反应得到;
[0033]
所述胺或醇选自如下式c1~c5所示的化合物
[0034][0035]
其中:r1和r3选自h、c1~c3的烷基;
[0036]
r2选自-(ch2)
n-或其中n为2~8,n1为0~3,n2为0~3;
[0037]
l1和l2为单键或亚甲胺基。
[0038]
前述所述的式i所示喹啉-1,2,4-三嗪杂合体在制备抗疟药物中的应用,所述式i中linker选自如下式a1~a3或a5所示的基团:
[0039][0040]
其中:r1和r3选自h、c1~c3的烷基;
[0041]
r2选自-(ch2)
n-或其中n为2~8,n1为0~3,n2为0~3;
[0042]
l1和l2为单键或亚甲胺基。
[0043]
有益效果
[0044]
本发明提供了一种新的化合物,且该化合物具有很好的抗疟效果。
附图说明
[0045]
图1实施例1最终产物的1h-nmr谱图;图2实施例1最终产物的
13
c-nmr谱图;
[0046]
图3实施例2最终产物的1h-nmr谱图;图4实施例2最终产物的
13
c-nmr谱图;
[0047]
图5实施例3最终产物的1h-nmr谱图;图6实施例3最终产物的
13
c-nmr谱图;
[0048]
图7实施例4最终产物的1h-nmr谱图;图8实施例4最终产物的
13
c-nmr谱图;
[0049]
图9实施例5最终产物的1h-nmr谱图;图10实施例5最终产物的
13
c-nmr谱图;
[0050]
图11实施例6最终产物的1h-nmr谱图;图12实施例6最终产物的
13
c-nmr谱图;
[0051]
图13实施例7最终产物的1h-nmr谱图;图14实施例7最终产物的
13
c-nmr谱图;
[0052]
图15实施例8最终产物的1h-nmr谱图;图16实施例8最终产物的
13
c-nmr谱图;
[0053]
图17实施例9最终产物的1h-nmr谱图;图18实施例9最终产物的
13
c-nmr谱图;
[0054]
图19实施例10最终产物的1h-nmr谱图;图20实施例10最终产物的
13
c-nmr谱图;
[0055]
图21实施例11最终产物的1h-nmr谱图;图22实施例11最终产物的
13
c-nmr谱图;
[0056]
图23实施例12最终产物的1h-nmr谱图;图24实施例12最终产物的
13
c-nmr谱图;
[0057]
图25实施例13最终产物的1h-nmr谱图;图26实施例13最终产物的
13
c-nmr谱图;
[0058]
图27实施例14最终产物的1h-nmr谱图;图28实施例14最终产物的
13
c-nmr谱图;
[0059]
图29实施例15最终产物的1h-nmr谱图;图30实施例15最终产物的
13
c-nmr谱图。
[0060]
图31实施例16最终产物的1h-nmr谱图;图32实施例16最终产物的
13
c-nmr谱图。
具体实施方式
[0061]
下面将结合本发明的具体实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。以下对至少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使用的任何限制。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0062]
式ii所示的化合物的制备方法采取下述方法:
[0063][0064]
4,7-二氯喹啉与二胺(c1、c2和c5)的反应:称取4,7-二氯喹啉(5.05mmol)溶解于20ml无水乙醇中,加入二胺(15.15mmol)混合均匀,110℃下加热搅拌,回流反应12~18h,tlc监测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除无水乙醇,加入乙酸乙酯和饱和食盐水萃取,取上层有机相加入无水硫酸钠除水,后用旋蒸仪将有机相液体减压旋转蒸发干燥,得到粗产物,用硅胶柱层析进行分离提纯,得到产品。
[0065]
4,7-二氯喹啉与醇胺(c3)的反应:称取4,7-二氯喹啉(5.05mmol),加入醇胺(40mmol)混合均匀,90℃下加热搅拌反应6h,tlc监测反应结束,停止反应,待反应体系降至室温,加入少量乙酸乙酯和蒸馏水,有固体析出,抽滤后放入烘箱中干燥,得到产品。
[0066]
4,7-二氯喹啉与羟基苯胺(c6)的反应:称取4,7-二氯喹啉(5.05mmol)溶解于20ml无水乙醇中,加入羟基苯胺(15.15mmol)混合均匀,120℃下加热搅拌,回流反应8h,tlc监测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除无水乙醇,反应体系加入蒸馏水,有固体析出,抽滤后放入烘箱中干燥,得到产品。
[0067]
4,7-二氯喹啉与二醇(c4)的反应:称取4,7-二氯喹啉(5.05mmol),加入二醇(176mmol)混合均匀,将叔丁醇钾(7.6mmol)缓慢加入到混合物中,通入氮气在80℃下反应16h,tlc检测反应结束,待反应体系降至室温,加入20ml饱和碳酸氢钠淬灭,加入二氯甲烷(25ml
×
3)萃取,加入无水硫酸钠干燥除水,最后将有机相减压旋蒸干燥后得到产品。
[0068]
式iii所示化合物的制备方法:
[0069]
取250ml烧瓶,称取3-甲硫基-1,2,4-三嗪(7mmol)加入烧瓶中,加入5ml二氯甲烷搅拌均匀,置于-10℃条件下。称取三氯过氧苯甲酸(21mmol)溶解于100ml二氯甲烷中,加入无水硫酸钠干燥除水,过滤,将滤镜缓慢滴加至烧瓶中使其混合,冰浴条件下反应1h,去除冰浴,于室温下继续反应2h,抽滤去除固体,滤液加入四丁基碘化铵(0.5mmol)搅拌10min后,使用旋蒸仪减压旋蒸干燥,得到粗产物,用硅胶柱层析分离提取,得到式iii所示化合物。
[0070]
本发明目标产物的制备方法:将式ii所示化合物(0.6mmol)与式iii所示化合物(0.6mmol)溶解于20ml干燥后的乙腈中,加入无水碳酸钠或碳酸铯(2.8mmol),室温下反应4h,tlc检测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除乙腈,加入乙酸乙酯和蒸馏水萃取,取上层有机相加入无水硫酸钠除水,后用旋蒸仪将有机相液体减压旋转蒸发干燥,得到粗产物,用硅胶柱层析进行分离提纯,得到产品。
[0071]
研究发现,游离血红素对疟原虫具有毒性,疟原虫通过将血红素转换成疟色素(β-羟高铁血红素)的方式解毒,红内期抗疟药通过与血红素络合从而抑制β-羟高铁血红素形成达到杀灭疟原虫的效果,是红内期抗疟药物的作用靶点之一。因此,本发明采用β-羟高铁血红素形成抑制实验对合成的15个目标化合物进行抗疟活性测定,并以此提供上述衍生物在制备抗疟疾药物中的应用。
[0072]
具体实施方式
[0073]
实施例1:n1-(7-氯喹啉-4-基)-n3-(1,2,4-三嗪-3-基)丙烷-1,3-二胺的制备
[0074][0075]
取50ml烧瓶,精确称取4,7-二氯喹啉(5.05mmol)加入烧瓶,加入20ml无水乙醇搅拌均匀至溶解,加入1,3丙二胺(15.15mmol)混合均匀,90℃下加热搅拌,回流反应12h,tlc监测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除无水乙醇,加入乙酸乙酯和饱和食盐水萃取,取上层有机相加入无水硫酸钠干燥除水,后用旋蒸仪减压旋蒸去除乙酸乙酯得到固体粗品,硅胶柱层析进行分离提纯(洗脱剂为二氯甲烷:甲醇=20:1),得到中间体ii。
[0076]
取250ml烧瓶,称取3-甲硫基-1,2,4-三嗪(7mmol)加入烧瓶中,加入5ml二氯甲烷搅拌均匀,置于-10℃条件下。称取三氯过氧苯甲酸(21mmol)溶解于100ml二氯甲烷中,加入无水硫酸钠干燥除水,过滤,将滤镜缓慢滴加至烧瓶中使其混合,冰浴条件下反应1h,去除冰浴,于室温下继续反应2h,抽滤去除固体,滤液加入四丁基碘化铵(0.5mmol)搅拌10min后,使用旋蒸仪减压旋蒸干燥,得到粗产物,用硅胶柱层析分离提取(洗脱剂为乙酸乙酯:石油醚=2:1),得到中间体iii。
[0077]
将ii(0.6mmol)与iii(0.6mmol)溶解于20ml干燥后的乙腈中,加入无水碳酸钠(2.8mmol),室温下反应4h,tlc检测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除乙腈,加入乙酸乙酯和蒸馏水萃取,取上层有机相加入无水硫酸钠干燥除水,后用旋蒸仪将有机相液体减压旋转蒸发干燥,得到粗产物,用硅胶柱层析进行分离提纯(洗脱剂为二氯甲烷:甲醇=40:1),得到目标化合物,白色固体,收率47.6%,m.p.169-171℃,1h nmr(500mhz,dmso-d6)δ8.56(d,j=2.3hz,1h),8.39(d,j=5.4hz,1h),8.30
–
8.22(m,2h),7.79(d,j=2.2hz,1h),7.46(dd,j=9.0,2.3hz,1h),7.38(t,j=5.4hz,1h),6.50(d,j=5.4hz,1h),3.36(d,j=6.6hz,4h),1.97(p,j=7.2hz,2h).
13
c nmr(126mhz,dmso-d6)δ152.23,150.58,150.50,149.37,133.91,127.82,124.56,124.51,117.90,117.85,99.14,99.11,55.36,29.48,27.68.
[0078]
实施例2:n1-(7-氯喹啉-4-基)-n4-(1,2,4-三嗪-3-基)丁烷-1,4-二胺的制备
[0079][0080]
制备方法参考实施例1。得到白色固体,收率50.6%,m.p.157-159℃,1h nmr(500mhz,methanol-d4)δ8.45(d,j=2.2hz,1h),8.35(d,j=5.7hz,1h),8.22(d,j=2.2hz,1h),8.11(d,j=9.0hz,1h),7.78(d,j=2.1hz,1h),7.40(dd,j=9.0,2.2hz,1h),6.54(d,j=5.7hz,1h),3.52(t,j=6.6hz,2h),3.44(t,j=6.7hz,2h),1.90
–
1.77(m,4h).
13
c nmr(126mhz,methanol-d4)δ151.43,150.79,150.54,148.05,139.77,134.99,129.44,129.43,
125.99,124.58,122.94,117.33,98.25,42.30,25.25
[0081]
实施例3:n1-(7-氯喹啉-4-基)-n8-(1,2,4-三嗪-3-基)辛烷-1,8-二胺的制备
[0082][0083]
制备方法参考实施例1。得到白色固体,收率90.2%,m.p.129-130℃,1h nmr(500mhz,methanol-d4)δ8.44(d,j=2.2hz,1h),8.35(d,j=5.7hz,1h),8.22(d,j=2.2hz,1h),8.10(d,j=9.0hz,1h),7.78(d,j=2.1hz,1h),7.39(dd,j=9.0,2.2hz,1h),6.50(d,j=5.7hz,1h),3.41(t,j=7.2hz,2h),3.35(d,j=7.3hz,4h),1.75(p,j=7.3hz,2h),1.64(q,j=7.0hz,2h),1.49
–
1.44(m,2h),1.40(d,j=3.9hz,6h).
13
c nmr(126mhz,methanol-d4)δ151.41,150.89,150.52,148.19,139.55,134.91,126.08,124.51,122.92,117.35,98.17,42.64,29.02,28.97,27.96,26.75,26.50.
[0084]
实施例4:n1-(7-氯喹啉-4-基)-n1,n2-二甲基-(1,2,4-三嗪-3-基)乙烷-1,2-二胺的制备
[0085][0086]
制备方法参考实施例1。得到无色油状液体,收率48.0%,1h nmr(500mhz,methanol-d4)δ8.43(d,j=2.3hz,1h),8.41(d,j=5.4hz,1h),8.16(d,j=2.2hz,1h),8.00(d,j=9.0hz,1h),7.81(d,j=2.1hz,1h),7.37(dd,j=9.1,2.2hz,1h),6.85(d,j=5.3hz,1h),4.00(d,j=6.5hz,2h),3.72(t,j=6.3hz,2h),3.13(s,3h),3.06
–
2.95(m,3h).
13
c nmr(126mhz,methanol-d4)δ161.11,157.34,150.54,149.70,149.32,139.14,134.76,126.64,126.13,124.94,120.85,107.95,52.38,46.21,39.97.
[0087]
实施例5:n-(4-((1,2,4-三嗪-3基)-胺基甲基)-苄基)-7-氯喹啉-4氨基的制备
[0088][0089]
制备方法参考实施例1。得到白色固体,收率89.0%,m.p.199-201℃,1h nmr(500mhz,dmso-d6)δ8.57(d,j=2.2hz,1h),8.32(dd,j=11.9,7.2hz,2h),8.24(d,j=2.3hz,1h),8.04(t,j=6.0hz,1h),7.80(d,j=2.2hz,1h),7.49(dd,j=9.0,2.2hz,1h),7.36
–
7.26(m,4h),6.35(d,j=5.5hz,1h),4.52(d,j=5.6hz,4h).
13
c nmr(126mhz,dmso-d6)δ152.22,150.40,150.32,149.44,138.65,137.50,133.93,127.96,127.73,127.37,124.74,124.74,124.47,117.98,99.91,45.82,45.70.
[0090]
实施例6:n-(3-((1,2,4-三嗪-3基)-胺基甲基)-苄基)-4氨基-7-氯喹啉的制备
[0091][0092]
制备方法参考实施例1。得到白色固体,收率81.0%,m.p.188-190℃,1h nmr(500mhz,dmso-d6)δ8.51(d,j=2.3hz,1h),8.34
–
8.26(m,2h),8.15(s,1h),8.04(t,j=6.0hz,1h),7.80(d,j=2.2hz,1h),7.48(dd,j=8.9,2.3hz,1h),7.34(s,1h),7.29
–
7.22(m,2h),7.20(d,j=7.3hz,1h),6.32(d,j=5.4hz,1h),4.52(d,j=6.0hz,4h).13c nmr(126mhz,dmso-d6)δ152.20,150.41,149.47,140.25,139.13,133.91,128.88,128.00,126.20,125.90,125.84,124.73,124.48,117.98,99.86,46.06,44.05.
[0093]
实施例7:4-(4-(1,2,4-三嗪-3-基)哌嗪-1-基)-7-氯喹啉的制备
[0094][0095]
制备方法参考实施例1。得到淡黄色固体,收率75.0%,m.p.171-173℃,1h nmr(500mhz,methanol-d4)δ8.69(d,j=5.2hz,1h),8.58(d,j=2.3hz,1h),8.37(d,j=2.2hz,1h),8.22(d,j=9.0hz,1h),7.98(d,j=2.2hz,1h),7.60(dd,j=9.0,2.2hz,1h),7.11(d,j=5.2hz,1h),4.22(t,j=5.1hz,4h),3.42(t,j=5.1hz,4h).
13
c nmr(126mhz,methanol-d4)δ161.36,157.70,151.49,150.05,149.19,139.99,135.25,126.95,126.20,125.72,121.77,109.22,51.59,43.15.
[0096]
实施例8:n-(7-氯喹啉-4-基)-哌啶-4-((1,2,4-三嗪-3基)-氨甲基)的制备
[0097][0098]
制备方法参考实施例1。得到淡黄色油状液体,收率48.0%,1h nmr(500mhz,chloroform-d)δ8.68(d,j=5.0hz,1h),8.57(d,j=2.2hz,1h),8.14(d,j=2.2hz,1h),8.02(d,j=2.1hz,1h),7.90(d,j=9.0hz,1h),7.40(dd,j=9.0,2.1hz,1h),6.81(d,j=5.1hz,1h),3.72
–
3.41(m,4h),2.95(s,1h),2.90
–
2.78(m,3h),2.17
–
2.03(m,1h),1.67(td,
j=12.2,3.7hz,2h).13c nmr(126mhz,chloroform-d)δ162.54,157.54,151.85,150.10,149.56
–
149.12(m),140.95,134.81,128.75,126.01,125.31,122.11,109.02,77.30,52.54,46.65(d,j=3.2hz),36.02,30.09.
[0099]
实施例9:n-(2(1,2,4-三嗪-3基)氧乙基)-4-氨基-7氯喹啉的制备
[0100][0101]
称取4,7-二氯喹啉(5.05mmol),加入乙醇胺(40mmol)混合均匀,90℃下加热搅拌反应6h,tlc监测反应结束,停止反应,待反应体系降至室温,加入少量乙酸乙酯和蒸馏水,有固体析出,抽滤后放入烘箱中干燥,得到式ii所示化合物。
[0102]
将式ii所示化合物(0.6mmol)与式iii所示化合物(0.6mmol)溶解于40ml干燥后的乙腈中,加入无水碳酸铯(2.8mmol),室温下反应4h,tlc检测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除乙腈,加入乙酸乙酯和蒸馏水萃取,取上层有机相加入无水硫酸钠干燥除水,后用旋蒸仪将有机相液体减压旋转蒸发干燥,得到粗产物,用硅胶柱层析进行分离提纯(洗脱剂为二氯甲烷:甲醇=28:1),得到化合物,白色固体,收率40.6%,m.p.88-90℃,1h nmr(500mhz,chloroform-d)δ9.06(d,j=2.2hz,1h),8.60(d,j=5.3hz,1h),8.51(d,j=2.2hz,1h),7.99(d,j=2.1hz,1h),7.75(d,j=8.9hz,1h),7.41(dd,j=8.9,2.1hz,1h),6.53(d,j=5.4hz,1h),5.82(s,1h),4.96(t,j=5.1hz,2h),3.89(q,j=5.2hz,2h).13c nmr(126mhz,chloroform-d)δ165.51,152.00,151.24,149.33,149.13,145.42,135.09,128.84,125.66,121.10,117.26,99.11,66.54,42.59.
[0103]
实施例10:n-(2(1,2,4-三嗪-3基)氧丙基)-4-氨基-7氯喹啉的制备
[0104][0105]
制备方法参考实施例9。得到白色固体,收率39.0%,m.p.72-75℃,1h nmr(500mhz,chloroform-d)δ9.00(d,j=2.3hz,1h),8.57
–
8.42(m,2h),7.94(d,j=2.1hz,1h),7.81(d,j=9.0hz,1h),7.38(dd,j=8.9,2.2hz,1h),6.44(d,j=5.3hz,1h),5.71(d,j=4.8hz,1h),4.77(t,j=5.8hz,2h),3.58(td,j=6.5,5.2hz,2h),2.39
–
2.28(m,2h).
13
c nmr(126mhz,chloroform-d)δ165.32,152.03,151.11,149.64,149.15,145.18,134.91,128.74,125.44,121.23,117.25,98.91,67.06,40.63,27.78.
[0106]
实施例11:n-(2(1,2,4-三嗪-3基)氧乙基)-4-甲氨基-7氯喹啉的制备
[0107][0108]
制备方法参考实施例9。得到无色油状物,收率45.0%,1h nmr(500mhz,methanol-d4)δ8.95(d,j=2.3hz,1h),8.54
–
8.48(m,2h),8.17(d,j=9.1hz,1h),7.85(d,j=2.2hz,
1h),7.44(dd,j=9.1,2.2hz,1h),7.00(d,j=5.3hz,1h),3.88(t,j=5.4hz,2h),3.15(s,3h).
13
c nmr(126mhz,methanol-d4)δ165.33,157.82,151.82,150.85,149.36,144.93,134.90,126.68,126.22,125.28,121.22,108.64,65.03,54.12,39.53.
[0109]
实施例12:n-(4-氧-(1,2,4-三嗪-3基)-苯基)-4氨基-7-氯喹啉的制备
[0110][0111]
称取4,7-二氯喹啉(5.05mmol)溶解于20ml无水乙醇中,加入对羟基苯胺(15.15mmol)混合均匀,120℃下加热搅拌,回流反应8h,tlc监测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除无水乙醇,反应体系加入蒸馏水,有固体析出,抽滤后放入烘箱中干燥,得到中间体ii。
[0112]
将化合物ii(0.6mmol)与化合物iii(0.6mmol)溶解于20ml干燥后的乙腈中,加入无水碳酸铯(2.8mmol),室温下反应4h,tlc检测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除乙腈,加入乙酸乙酯和蒸馏水萃取,取上层有机相加入无水硫酸钠除水,后用旋蒸仪将有机相液体减压旋转蒸发干燥,得到粗产物,用硅胶柱层析进行分离提纯(洗脱剂为二氯甲烷:甲醇=50:1),得到3l,黄色固体,收率79.6%,m.p.226-227℃,1h nmr(500mhz,dmso-d6)δ9.27(d,j=2.3hz,1h),9.16(s,1h),8.76(d,j=2.3hz,1h),8.48(d,j=5.3hz,1h),8.44(d,j=9.0hz,1h),7.91(d,j=2.2hz,1h),7.59(dd,j=9.0,2.3hz,1h),7.50
–
7.42(m,2h),7.41
–
7.33(m,2h),6.94(d,j=5.3hz,1h).
13
c nmr(126mhz,dmso-d6)δ166.37,152.85,152.47,149.98,148.88,148.55,147.16,138.19,134.44,128.12,125.46,124.88,124.54,122.90,118.73,102.16,49.07.
[0113]
实施例13:n-(4-氧-(1,2,4-三嗪-3基)-苯基)-4氨基-7-氯喹啉的制备
[0114][0115]
制备方法参考实施例12。得到白色固体,收率82.0%,m.p.219-220℃,1h nmr(500mhz,dmso-d6)δ9.27(d,j=2.4hz,1h),9.22(s,1h),8.75(d,j=2.3hz,1h),8.51(dd,j=5.3,1.6hz,1h),8.41(d,j=9.0hz,1h),7.91(d,j=2.2hz,1h),7.58(dd,j=9.0,2.2hz,1h),7.50(t,j=8.4hz,1h),7.31(dq,j=3.5,1.7hz,2h),7.10
–
7.04(m,2h).
13
c nmr(126mhz,dmso-d6)δ166.22,153.48,152.87,152.52,150.03,147.77,147.23,142.34,134.46,131.08,128.17,125.62,124.93,119.66,118.98,116.87,115.34,103.04.
[0116]
实施例14:4-(2-氧-(1,2,4-三嗪-3-基)-乙氧基)-7-氯喹啉的制备
[0117][0118]
称取4,7-二氯喹啉(5.05mmol),加入乙二醇(176mmol)混合均匀,将叔丁醇钾(7.6mmol)缓慢加入到混合物中,通入氮气在80℃下反应16h,tlc检测反应结束,待反应体系降至室温,加入20ml饱和碳酸氢钠淬灭,加入二氯甲烷(25ml
×
3)萃取,加入无水硫酸钠干燥除水,最后将有机相减压旋蒸干燥后得到式ii所示化合物。
[0119]
将式ii所示化合物(0.6mmol)与式iii所示化合物(0.6mmol)溶解于20ml干燥后的乙腈中,加入无水碳酸铯(2.8mmol),室温下反应4h,tlc检测反应结束,停止反应,待反应体系降至室温,减压旋蒸去除乙腈,加入乙酸乙酯和蒸馏水萃取,取上层有机相加入无水硫酸钠除水,后用旋蒸仪将有机相液体减压旋转蒸发干燥,得到粗产物,用硅胶柱层析进行分离提纯(洗脱剂为二氯甲烷:甲醇=50:1),得到目标产物,白色固体,收率50.4%,m.p.119-121℃,1h nmr(500mhz,methanol-d4)δ9.05(d,j=2.3hz,1h),8.69(d,j=5.4hz,1h),8.62(d,j=2.2hz,1h),8.12(d,j=8.9hz,1h),7.89(d,j=2.0hz,1h),7.47(dd,j=8.9,2.0hz,1h),7.07(d,j=5.4hz,1h),5.09
–
5.01(m,2h),4.76
–
4.69(m,2h).13c nmr(126mhz,methanol-d4)δ165.58,161.94,152.36,152.02,148.66,145.22,135.97,126.49,126.01,123.56,119.61,101.49,67.02,66.07.
[0120]
实施例15:4-(2-氧-(1,2,4-三嗪-3-基)-丙氧基)-7-氯喹啉的制备
[0121][0122]
制备方法参考实施例14。得到白色固体,收率57.0%,m.p.91-92℃,1h nmr(500mhz,methanol-d4)δ9.07(d,j=2.3hz,1h),8.71(d,j=5.4hz,1h),8.64(d,j=2.2hz,1h),8.14(d,j=8.9hz,1h),7.91(d,j=2.0hz,1h),7.49(dd,j=8.9,2.0hz,1h),7.09(d,j=5.4hz,1h),5.11
–
5.03(m,2h),4.78
–
4.71(m,2h).
13
c nmr(126mhz,methanol-d4)δ165.61,162.10,152.42,151.93,148.68
–
148.64(m),144.89,135.86,126.40,126.02,123.58,119.68,101.37,65.60,65.04,28.06.
[0123]
实施例16:n1-(7-氯喹啉-4-基)-n8-(1,2,4-三嗪-3-基)辛烷-1,8-二胺的制备
[0124][0125]
中间体ii、iii的制备方法同实施例1。将ii(0.6mmol)与iii(0.6mmol)溶解于5ml干燥后的dmf中,其他条件同实施例1,得到目标化合物,得到白色固体,收率85.5%,
m.p.129-130℃,1h nmr(500mhz,dmso-d6)δ8.52(d,j=2.3hz,1h),8.38(d,j=5.4hz,1h),8.28(d,j=9.0hz,1h),8.22(d,j=2.3hz,1h),7.77(d,j=2.2hz,1h),7.44(dd,j=9.0,2.3hz,1h),7.29(s,1h),6.45(d,j=5.4hz,1h),3.25(td,j=7.2,5.5hz,3h),1.65(t,j=7.3hz,2h),1.55(t,j=7.2hz,2h),1.40
–
1.29(m,8h).
13
c nmr(126mhz,dmso-d6)δ152.35,150.56,149.54,133.80,127.91,124.58,124.42,117.91,99.06,42.88,29.28,29.24,28.24,27.07,26.86.
[0126]
实施例17:n-(4-(1,2,4-三嗪-3-基)苯基-1-基)-4氨基-7-氯喹啉的制备
[0127][0128]
取50ml烧瓶,精确称取4,7-二氯喹啉(5.05mmol)加入烧瓶,加入20ml无水乙醇搅拌均匀至溶解,加入对苯二胺(15.15mmol)混合均匀,90℃下加热搅拌,回流反应9h,tlc监测反应结束,停止反应,待反应体系降至室温,过滤反应体系中的固体,干燥,得到中间体ii。
[0129]
中间体iii的制备方法同实施例1。将ii(0.6mmol)与iii(0.6mmol)溶解于5ml干燥后的dmf中,其他条件同实施例1,未得到目标化合物。
[0130]
实施例18:n-(3-(1,2,4-三嗪-3-基)苯基-1-基)-4氨基-7-氯喹啉的制备
[0131][0132]
制备方法同实施例17。未得到目标化合物。
[0133]
实施例19:n-(2-(1,2,4-三嗪-3-基)苯基-1-基)-4氨基-7-氯喹啉的制备
[0134][0135]
制备方法同实施例17。未得到目标化合物。
[0136]
实施例20:n-(2,5-二甲基-4-(1,2,4-三嗪-3-基)苯基-1-基)-4氨基-7-氯喹啉的制备
[0137][0138]
制备方法同实施例17。未得到目标化合物。
[0139]
实施例21:n-(4-(2-((1,2,4-三嗪-3-基)氧)乙基)苯基)-4氨基-7-氯喹啉的制备
[0140]
制备方法同实施例17。未得到目标化合物
[0141]
实施例22:n-(3-(((1,2,4-三嗪-3-基)氧)甲基)苯基)-4氨基-7-氯喹啉的制备
[0142]
制备方法同实施例17。未得到目标化合物
[0143]
实施例23:4-(2-氧-(1,2,4-三嗪-3-基)-丁氧基)-7-氯喹啉的制备
[0144][0145]
制备方法同实施例14。未得到目标化合物
[0146]
实施例24:4-(2-氧-(1,2,4-三嗪-3-基)-戊氧基)-7-氯喹啉的制备
[0147][0148]
制备方法同实施例14。未得到目标化合物
[0149]
实施例25:4-(2-氧-(1,2,4-三嗪-3-基)-己氧基)-7-氯喹啉的制备
[0150]
制备方法同实施例14。未得到目标化合物
[0151]
实施例26:本专利所述目标化合物的β-羟高铁血红素形成抑制活性测定
[0152]
选择实施例1~15制备的化合物,对其β-羟高铁血红素形成抑制活性进行测定。
[0153]
具体测试方法如下:以氯喹为阳性对照,取80μl不同浓度的目标化合物溶液(溶解于dmso中)与80μl氯化血红素储备液(1mmol/l,溶解于dmso中)混合于1.5ml的ep管中,一式三份,每管加入160μl醋酸盐缓冲溶液(4mol/l,ph=5.0),置于45℃条件下孵育8h,恢复至室温后每管加入180μl,30%(v/v)的吡啶-hepes缓冲溶液,使管中固体混悬后静置,移取上清液50μl至96孔板中,每孔加入150μl30%(v/v)的吡啶-hepes缓冲溶液,使用酶标仪在405nm处测定吸光度值,计算目标化合物对β-羟高铁血红素形成的抑制活性,活性测定ic
50
结果如下所示。
[0154]
本专利系列化合物对β-羟高铁血红素形成抑制活性ic
50
(μm)
[0155]
化合物实施例1实施例2实施例3实施例4实施例5ic
50
(μm)10.197.464.5310.409.89化合物实施例6实施例7实施例8实施例9实施例10ic
50
(μm)8.7412.6710.8810.4210.07化合物实施例11实施例12实施例13实施例14实施例15ic
50
(μm)7.337.339.38>117.98>112.75
[0156]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。