作为肾保护物质的对氨基马尿酸(pah)
1.本发明涉及对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物用于降低受试者中放射标记的或非放射标记的治疗性和诊断性(如,用于成像目的)化合物的肾毒性副作用的用途。其还涉及在使用放射标记的和/或非放射标记的化合物进行成像或疗法期间用于肾脏保护的药物组合物,其中组合物包括联合对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物的放射标记的和/或非放射标记的药物化合物,以及药学上可接受的赋形剂、稀释剂、载体或其组合。本发明还涉及降低受试者中放射标记的或非放射标记的治疗性和诊断性化合物的肾毒性副作用的方法,该方法包括联合施用对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物与放射标记的或非放射标记的治疗性或诊断性化合物,其中所述pah的施用在施用放射标记的或非放射标记的治疗性或诊断性化合物之前和/或期间和/或之后,以及包括在使用放射标记的和/或非放射标记的化合物进行成像或疗法期间向受试者施用根据本发明的药物组合物的方法。
2.具体地,本技术涉及pah或其药学上可接受的盐或羧酸衍生物用于抑制肾吸收以及改善放射标记的分子的体内生物分布的用途,该放射标记的分子潜在地损害肾脏,尤其用于治疗性放射药物,和/或涉及通过使用对氨基马尿酸(pah)或其盐或羧酸衍生物(如氨基马尿酸钠)在诊断放射药物的情况下改善对比度。
3.在先进医疗处理的背景下,患者被暴露于越来越多种的药物用于诊断和治疗目的。这些试剂中的一些引起与全身毒性相关的不利药物作用,包括肾功能的损害。这些化合物中大多数通过肾脏清除并重新吸收和部分保留在近端小管中,导致剂量限制性肾毒性。肾毒性导致严重的临床综合征,包括急性肾脏损伤(aki)。肾毒性试剂被认为是17-26%的住院aki的致病因素。药物诱导的肾损害涉及各种分类的药物,并且包括处方药剂以及常见的非处方药。
4.治疗剂和潜在诊断剂的毒性可能是药理化合物本身固有的,并且在肾脏微环境中毒性的潜力可能会升高。例如,化疗的目的是通过旨在阻止细胞分裂的各种机制杀死恶性细胞。由于细胞周期在非恶性细胞中正常运行,因此包括肾实质细胞在内的健康组织也会受到影响。肾脏作为过滤器官尤其暴露为有毒化合物的靶。由于它接受了很大比例的心输出量,通过肾脏的强劲血流使肾脏暴露于药物和药物代谢物。这些药剂中的一些具有在肾小球过滤所需的电荷和大小,并随后通过胞饮作用或内吞作用进入肾小管上皮细胞。其他药物通过管周毛细血管运输并进入基底外侧表面的肾小管上皮细胞,在那里它们被有机阴离子和有机阳离子转运蛋白(分别为oat和oct)吸收并流出到管腔中,在那里它们可能导致有临床意义的肾毒性。
5.一些抗癌治疗剂,如顺铂,已知具有肾毒性。顺铂(sp-4-2)-二氨二氯合铂(ii))是由nh3和cl络合的铂原子(方形平面络合物)。它的肾毒性是基于它被肾细胞吸收并与细胞的dna结合,从而抑制细胞机制,特别是细胞复制。natochin等报道了顺铂的肾毒性(comp.biochem.physiol vol.94c,no.1pp115-120,1989)。氯化胆碱、pah、呋塞米和依他尼酸被描述为降低大鼠的顺铂肾毒性作用。
6.肾毒性也被称为在施用基于放射性核素的治疗剂/诊断剂时的不良副作用。
neuroendocrine tumor patients treated with targeted radiopeptide therapy.giovacchini g et al.,eur j nucl med mol imaging.2011sep;38(9):1675-82)。因此,用于施用的氨基酸的量通常限于25g赖氨酸和25g精氨酸。rolleman ej等人报道了肾脏吸收降低,如下:“(1)市售氨基酸溶液(aa)(21% /-14%,p《0.02),(2)25g(17% /-9%,p《0.04)、50g(15% /-13%,p《0.04)或75g的赖氨酸(44% /-11%,p《0.001)和(3)25g的赖氨酸加25g的精氨酸(lysarg)的组合(33% /-23%,p《0.01)。单独流体输注(500、1,000或2,000ml的盐水/葡萄糖)不改变放射性的肾吸收,在以75g的赖氨酸(lys75)和lysarg进行研究的患者中,血清钾水平显著升高”(safe and effective inhibition of renal uptake of radiolabeled octreotide by a combination of lysine and arginine.rolleman ej et al.,eur j nucl med mol imaging.2003jan;30(1):9-15)。高钾血症仍然是该肾脏保护方法的未解决问题,因为高浓度的赖氨酸和精氨酸被用作混合物的一部分(giovacchini g et al.,同上)。
14.赖氨酸和精氨酸被近端小管中的钠非依赖性氨基酸转运蛋白(scl3a1)重吸收。如[
177
lu-dota
°‑
tyr3]-奥曲肽等的放射性药物的清除机制尚未完全阐明。公开的数据表明,scl3a1氨基酸转运蛋白可能在放射性药物的清除中发挥作用,但很明显,这一观察结果并不代表主要,更谈不上涉及的唯一机制。
[0015]
因此,本发明的目的是提供一种抑制肾吸收的创新方法,以用作更广谱(放射性)药物的联合疗法,而不管其肾积累的潜在机制如何,从而减少(放射性)药物作为治疗性或更罕见的诊断性化合物的肾毒性副作用。然而,高度期望医学成像中的诊断来改善它们在体内的生物分布并通过增加它们在待成像部位的吸收(例如任何组织的吸收)来增强对比度。
[0016]
这些目的由本文公开的主题解决,特别是由权利要求限定的。
[0017]
本发明基于以下发现:对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物可以合适地用于例如改善治疗性和诊断性化合物(特别是包括放射性核素)的生物分布。受试者中潜在损伤肾脏的试剂例如化疗剂或放射药物,诸如[
177
lu-dota
°‑
tyr3]-奥曲肽、
177
lu psma-抑制剂等的清除可以通过根据本发明的创新方法进行改进。而且,已经发现,pah施用可以用于下调肾药物再吸收。由此,可以(但非必须)增加药物的血清水平,从而导致增强的药物生物利用度。
[0018]
因此,本发明特别适用于抑制所有种类的蛋白质分子的肾吸收,例如表现出固有的肾毒性的蛋白和肽或其片段或抗体片段。无论这种蛋白质分子与毒素、放射性核素、细胞抑制剂或其他潜在的细胞毒性剂结合在一起,甚至更是这样。尤其是,预料之外地发现,观察到本发明与pah的共同施用还降低放射性药物由于其放射性而产生的肾毒性。这一发现甚至更令人惊讶,因为基于放射性核素的治疗/诊断的潜在机制是与在抗癌药物(如顺铂)中观察到的其他机制独一无二的(放射性)并且截然不同的。
[0019]
此外,本发明允许成像剂(特别是用于spect和pet目的)在待鉴定的组织中(例如在肿瘤靶位点)积聚,并因此减少其在脱靶组织中的存在。因此,本发明允许改进包含放射性核素的给定分子例如缀合物分子的图像对比度,因为其被肾脏的清除减少。
[0020]
对氨基马尿酸(pah)
[0021]
氨基马尿酸或对氨基马尿酸(pah)——马尿酸的衍生物——是(a)氨基酸甘氨酸
和(b)对氨基苯甲酸的酰胺衍生物,其并不天然存在于人类中。它们通过酰胺键共价连接。pah的钠盐——氨基马尿酸钠——是一种诊断试剂,其广泛用于肾功能的诊断测试,特别是用于测量肾血浆流量。在下面,氨基马尿酸、对氨基马尿酸和氨基马尿酸盐(特别是,碱或碱土盐),特别是钠盐,同义使用并且称为“pah”。
[0022]
对氨基马尿酸(pah):
[0023][0024]
对氨基马尿酸钠:
[0025][0026]
通常,pah以无菌、未防腐的20%注射用水溶液提供。pah被肾小球过滤并由近端小管主动分泌。在低血浆浓度(1.0至2.0mg/100ml)下,受试者的肾脏通过单次循环从肾血流中清除平均90%的pah。pah还用于测量肾小管分泌机制的功能能力或转运最大值(tm pah)。这是通过将血浆浓度升高到足以使肾小管细胞分泌pah的最大能力达到饱和的水平(40-60mg/100ml)来实现的。pah基本上没有任何副作用并且毒性可以忽略不计(雌性小鼠的静脉注射ld50为7.22g/kg)。像呕吐和恶心或高钾血症这样的现象没有,或者如果有的话,仅很少报告。
[0027]
与现有技术的联合用药(如,通过使用氨基酸混合物)相反,对氨基马尿酸(氨基马尿酸盐)允许非特异性地调节(放射性)药物的清除,因为它是各种转运蛋白的底物和/或功能抑制剂。pah被转运蛋白主动分泌到尿液中。迄今为止,pah被认为是14种不同转运蛋白的底物或抑制剂,包括有机阳离子跨膜转运蛋白(如,oct1、oct1a、oct2、oct3、octn1、octn2、octn3)和有机阴离子跨膜转运蛋白(如,oat1、oat2、oat3、oat4、oat5、oatp、urat1)。pah的底物还是hmrp、atp依赖性流出转运蛋白。
[0028]
因此,对氨基马尿酸或其药学上可接受的盐或羧酸衍生物可以用于与广谱的放射标记的或非放射标记的治疗性或诊断性化合物联合用药,无论载体分子的性质如何(即,肽、抗体片段、拟肽、小分子等)。pah对肾脏清除率的调节被确定导致施用药物的血液循环延长和其生物利用度增加。例如通过输注或注射,可在肾脏将要清除的(放射性)药物施用前和/或期间和/或后施用氨基马尿酸或其盐。
[0029]
一般评论
[0030]
尽管下面详细描述了本发明,但应当理解,本发明不限于本文所述的特定方法、方案和试剂,因为它们可以变化。还应当理解的是,这里使用的术语并非旨在限制本发明的范围,本发明的范围仅由所附权利要求来限制。除非另有定义,本文使用的所有技术和科学术语与本领域普通技术人员通常理解的含义相同。
[0031]
下面将描述本发明的要素。这些要素与特定实施方式一起列出,然而,应当理解,它们可以以任何方式和以任何数量组合以形成另外的实施方式。不同描述的实例和优选实施方式不应被解释为将本发明仅限于明确描述的实施方式。该描述应当被理解为支持和包含将明确描述的实施方式与任何数量的公开和/或优选要素组合的实施方式。此外,除非上
下文另有说明,否则本技术中所有描述的要素的任何排列和组合都应被认为是本技术的说明书所公开的。
[0032]
贯穿本说明书和所附权利要求,除非上下文另有要求,否则术语“包括”和诸如“包含”和“含有”的变体将被理解为暗示包括所陈述的成员、整数或步骤,但不排除任何其他非陈述的成员、整数或步骤。术语“由
……
组成”是术语“包括”的特定实施方式,其中排除了任何其他未陈述的成员、整数或步骤。在本发明的上下文中,术语“包括”涵盖术语“由
……
组成”。因此,术语“包括”涵盖“包含”以及“由
……
组成”,例如,“包括”x的组合物可以仅由x组成,也可以包含其他内容,例如x y。
[0033]
除非本文另有说明,或与上下文明显矛盾,否则在描述本发明的上下文中(尤其是在权利要求的上下文中)使用的术语“一个”和“一种”和“该”以及类似的参考将被解释为涵盖单数和复数。此处对值范围的引用仅旨在用作单独提及落入该范围内的每个单独值的速记方法。除非在本文中另有说明,否则每个单独的值都被并入说明书中,就好像它在本文中被单独叙述一样。说明书中的任何语言都不应被解释为表示对本发明的实践必不可少的任何未要求保护的元素。
[0034]
与数值x相关的术语“约”是指x
±
10%。
[0035]
本文使用的术语“受试者”通常包括人和非人动物,优选地哺乳动物(如,非人灵长类动物,包括狨猴、绢毛猴、蜘蛛猴、猫头鹰猴、长尾猴、松鼠猴和狒狒、猕猴、黑猩猩、猩猩、大猩猩、牛、马、羊、猪、鸡、猫、狗、小鼠、大鼠、兔、豚鼠等),包括嵌合和转基因动物和疾病模型。在本发明的上下文中,术语“受试者”优选地是指非人灵长类动物或人,最优选地是人。
[0036]
在第一方面,本发明涉及对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物降低用该化合物进行治疗的受试者中放射标记的或非放射标记的治疗性和诊断性化合物的不期望的肾毒性性质的用途。
[0037]
如上所述,pah或其盐是肾脏中各种转运蛋白的底物和/或抑制剂。因此,其可以用于广谱的放射标记的或非放射标记的治疗性和诊断性化合物,该化合物预期进入肾小管细胞,如经由通过有机阴离子转运蛋白(oat)和有机阳离子转运蛋白(oct)吸收,因此发挥其潜在肾毒性。其还可以用于保护肾细胞免受放射性标记的治疗剂或诊断剂的放射性,如当由于放射性引起氧化应激时。此外,针对肾毒性的保护可能特别意味着针对肾小球毒性的保护。
[0038]
治疗性肾毒性化合物的实例包括但不限于广泛用于缓解疼痛和炎症迹象的非甾体抗炎药。尽管如此,它们仍会引发更多种类的肾脏并发症,诸如,肾前性氮血症、急性肾小管坏死、急性乳头状坏死、急性间质性肾炎、慢性小管间质性肾炎(镇痛剂肾病)微小病变、膜性肾病、高钾血症和代谢性酸中毒(低肾素性醛固酮减少症、低钠血症、高血压);血管紧张素转换酶抑制剂和血管紧张素ii受体阻滞剂,其用于治疗高血压和充血性心力衰竭,以及延缓糖尿病性肾病的进展,并且其可导致急性肾脏损伤(aki)和高钾血症的风险更高;抗微生物剂,诸如新霉素、庆大霉素、妥布霉素、阿米卡星和链霉素,它们常用于治疗革兰氏阴性细菌感染,以及其细胞内积聚导致肾小管细胞死亡或细胞膜转运蛋白功能改变,导致电解质异常(低钾血症、低镁血症和低钙血症);磺胺类抗生素,诸如磺胺甲噁唑-甲氧苄氨嘧啶,其可通过抑制远曲小管上皮钠通道而导致高钾血症,其为钾排泄提供动力,以及磺胺嘧啶,其可引起急性间质性肾炎和结晶性肾病;糖肽类抗生素,诸如万霉素,其可导致急性肾
小管坏死;氟喹诺酮类抗生素,诸如环丙沙星,其可引起急性间质性肾炎和结晶尿;其他抗生素,诸如青霉素和头孢菌素,其可引起急性间质性肾炎和急性肾小管坏死;和多粘菌素,诸如粘菌素和多粘菌素b,其通过中毒性肾小管损伤引起急性间质性肾炎;抗病毒药物,诸如阿昔洛韦,其可诱发继发于肾小管结晶沉淀的急性肾损伤,除膦甲酸(fosarnet),其可导致急性肾小管坏死、急性肾损伤、电解质异常,诸如低钙血症、低镁血症、低钾血症和低磷或高磷血症;抗逆转录病毒药物,诸如替诺福韦,其对肾小管细胞有毒性,导致急性肾损伤,伴有或不伴有近端肾小管病变,其可导致慢性肾病,以及蛋白酶抑制剂,诸如茚地那韦、阿巴卡韦、利托那韦和阿扎那韦,其可在肾小管中结晶,导致晶体相关肾脏损伤和肾结石;抗真菌药,诸如两性霉素b,其可引起急性肾小管坏死和肾小管功能障碍,其表现为肾小管酸中毒、尿浓度缺陷和电解质紊乱;免疫抑制剂,诸如钙调神经磷酸酶抑制剂(如他克莫司、环孢素),其特别用于实体器官移植后的免疫抑制治疗,可能会导致急性肾脏损伤;锂(lithium),其是双相情感障碍患者的主要治疗方法,并且可导致各种形式的肾毒性,诸如肾源性尿崩症(ndi)、慢性肾病和慢性肾小管肠病。导致急性肾脏损伤的其他治疗剂是质子泵抑制剂;对乙酰氨基酚;hmg-coa还原酶抑制剂;和渗透剂。
[0039]
治疗性肾毒性化合物还包括化疗剂,它们在治疗各种肿瘤中起核心作用,并可导致广谱的肾并发症,诸如与急性肾脏损伤或慢性肾脏疾病相关的肾综合征,以及与电解质紊乱相关的肾综合征。这些化疗剂的实例包括顺铂(急性肾小管坏死、近端肾小管病、高钠血症、低镁血症、低钙血症、远端肾小管酸中毒、血栓性微血管病)、培美曲塞(急性肾小管坏死)、链佐星(也称为链脲佐菌素)(急性肾小管坏死、近端肾小管病)、光神霉素(急性肾小管坏死)、唑来膦酸盐(急性肾小管坏死)、干扰素(急性间质性肾炎)、别嘌呤醇(急性间质性肾炎)、吉西他滨(血栓性微血管病)、丝裂霉素c(血栓性微血管病)、抗血管生成剂(血栓性微血管病)、甲氨蝶呤(结晶性肾病)、异环磷酰胺(近端肾小管病、高钠血症)、环磷酰胺(低钠血症)、长春新碱(低钠血症)、西妥昔单抗(低镁血症)、甲氨蝶呤(急性肾脏损伤)。
[0040]
例如,顺铂和异环磷酰胺,它们是各种实体器官肿瘤(包括影响儿童的实体器官肿瘤)的治疗方案中的标准组成部分,已知通过有机阳离子转运蛋白(oct2)在近端小管处进行细胞吸收(shirali a,perazella m,advances in chronic kidney disease,vol 21,no 1(january),2014:pp56-63)。因此,推测与pah共注射可有效降低这些化疗药物的肾毒性副作用。
[0041]
另外的肾毒性化合物包括放射性造影剂,诸如碘化放射性造影剂,这是几种诊断和介入放射学程序所必需的,并且可导致造影剂肾病(cin)和急性肾损伤。碘化离子造影剂的实例是泛影酸盐(hypaque 50)、甲硝唑(isopaque 370)、碘酞酸盐(conray)、碘克酸(hexabrix);非离子造影剂包括碘帕醇(isovue 370)、碘海醇(omnipaque 350)、碘昔兰(oxilan 350)、碘普罗胺(ultravist 370)、碘克沙醇(visipaque 320)、碘佛醇。
[0042]
在本发明的优选实施方式中,pah用于减少用作诊断剂和治疗剂的放射性药物的肾毒性副作用。
[0043]
放射性药物可以包括非金属(有机)放射性核素(
18
f、
11
c、
13
n、
15
o、
124
i等)或放射性金属(如
90
y、
99m
tc、
111
in、
131
l、
67
ga、
68
ga、
64
cu、
161
tb、
225
ac、
44
sc、
47
sc、
67
cu、
89
zr、
177
lu等)。尽管一些放射性金属可以以金属盐或金属络合物的形式靶向特定组织,但通常需要将放射性核素/放射性金属与靶向生物分子结合,以便将放射性核素以靶向方式递送至靶位,如肿瘤组
织。生物分子可以是如小有机分子、肽、单克隆抗体(mab)或mab片段。它们作为载体(“运载体”)将放射性核素运送到靶组织。
[0044]
市售小络合物放射性药物包括例如
99m
tc
–
sestamibi其用于心肌灌注成像;
99m
tc
–
tetrofosmin其用于心肌灌注成像;
99m
tc
–
pentetate(dtpa)其用于肾成像和功能研究;
99m
tc
–
bicisate(ecd)其用于脑灌注成像;
99m
tc
–
mdp其用于骨骼闪烁扫描;
99m
tc
–
teboroxime其用于心肌灌注成像;
111
in
–
oxyquinoline(indium-111),其用于白细胞闪烁扫描;
111
in
–
pentetate(indium-111),其用于csf动力学成像;
153
sm
–
edtmp其用于骨痛的治疗(疗法);
188
re
–
hedp,其用于转移性骨痛疗法。
[0045]
建立放射性核素和靶向生物分子(载体)稳定缀合的最简洁的方法是使用合适的双功能螯合剂或螯合试剂,该螯合剂或螯合试剂紧密结合或配位放射性核素,同时提供其与生物分子的缀合的功能部分。
[0046]
市售的肽或免疫缀合物的实例包括如
99m
tc
–
depreotide(neo),其用于评估某些肺部病变;
99m
tc
–
arcitumumab(cea-tc-mab),其用于结肠直肠癌成像;
111
in
–
capromab pendetide其用于前列腺癌成像;
111
in
–
pentetreotide其用于神经内分泌肿瘤成像;
111
in
–
imciromab pentetate其用于怀疑由心肌梗塞引起的胸痛的成像;
111
in
–
satumomab pendetide其用于与结肠直肠癌和卵巢癌相关的转移性疾病的成像;
90y–
ibritumomab tiuxetan其用于非霍奇金淋巴瘤(nhl)的治疗;
68
ga-edotreotide或
68
ga-dotate,其用于神经内分泌肿瘤的成像;
177
lu-b dotatete(lutathera),其用于神经内分泌肿瘤的疗法。
[0047]
因此,在本发明的特别优选的实施方式中,pah用于减少放射性药物的肾毒性副作用,如上文所述,在放射化学/配体疗法和/或诊断中。
[0048]
在放射配体疗法(也称为prrt
–
肽-受体放射核素疗法)中,放射性药物通过放射配体进行标记,其特异性结合至(肿瘤)细胞靶,如,肿瘤细胞表面蛋白或标记。在化合物与肿瘤靶(例如与受体)结合后,放射性核素释放高能β粒子辐射,以精确靶向目标部位的细胞。
[0049]
在放射配体诊断中,放射标记的化合物与(肿瘤)靶细胞结合,如受体。放射性同位素的衰变可以通过正电子发射断层扫描(pet)或单光子发射计算机断层扫描(spect)来测量。
[0050]
因此,在本发明的优选实施方式中,pah或其药学上可接受的盐或羧酸衍生物用于减少放射标记的药物(“放射性药物”)的肾毒性副作用。放射性药物优选是缀合物分子,其包括(i)与(肿瘤)细胞靶结构(如,受体或抗原)结合的载体,(ii)螯合试剂(或螯合剂),和(iii)放射性核素。螯合试剂通常与放射性核素配位,由此形成放射标记的络合物,其与载体分子缀合。
[0051]
放射性核素
[0052]
术语“放射性核素”(或“放射性同位素”)指具有不稳定的中子与质子比的天然或人工来源的同位素,其随着微粒(即质子(α-辐射)或电子(β-辐射))或电磁辐射(γ-辐射)的发射而崩解)。换句话说,放射性核素经历放射性衰变。在放射性药物的放射标记的络合
物中,任何已知的放射性核素可以通过螯合试剂进行络合。这类放射性核素可以包括单不限于
18
f、
131
i、
94
tc、
99m
tc、
90
in、
111
in、
67
ga、
68
ga、
86
y、
90
y、
177
lu、
161
tb、
186
re、
188
re、
64
cu、
67
cu、
55
co、
57
co、
43
sc、
44
sc、
47
sc、
225
ac、
213
bi、
212
bi、
212
pb、
227
th、
153
sm、
166
ho、
152
gd、
153
gd、
157
gd或
166
dy,特别是
68
ga、
177
lu或
99m
tc。
[0053]
合适的放射性核素的选择取决于螯合试剂的化学结构和螯合能力,等等,最重要的是,取决于所得(络合)缀合物分子的预期应用(如诊断与治疗,以及如对疾病的治疗)。比如,β-发射器,诸如
90
y、
131
i、
161
tb和
177
lu可以用于同步全身性放射性核素疗法。提供dota、dotaga或dotam作为螯合剂,可以有利地能够使用任一
68
ga、
43,44,47
sc、
177
lu、
161
tb、
225
ac、
213
bi、
212
bi、
212
pb作为放射性核素。
[0054]
在一些优选实施方式中,放射性核素可以是
177
lu。在一些优选实施方式中,放射性核素可以是
111
in。在一些优选实施方式中,放射性核素可以是
90
y。在一些优选实施方式中,放射性核素可以是
68
ga。
[0055]
螯合剂
[0056]
如上所述,载体分子,如靶向(癌)细胞的化合物,优选地链接至配位放射性核素的螯合试剂或螯合剂。
[0057]
术语“螯合剂”或“螯合试剂”在本文中可互换使用。它们是指能够与(“配位”)中心(金属)离子形成两个或多个单独配位键的多齿(多键合)配体。具体而言,此类分子或共享一个电子对的分子也可称为“路易斯碱”。中心(金属)离子通常通过两个或多个电子对与螯合试剂配位。术语“双齿螯合试剂”、“三齿螯合试剂”和“四齿螯合试剂”是本领域公认的,并且是指分别具有两个、三个和四个电子对的螯合试剂,这些电子对易于同时提供给由螯合试剂配位的金属离子。通常,螯合试剂的电子对与单个中心(金属)离子形成配位键;然而,在某些实例中,螯合试剂可以与一种以上的金属离子形成配位键,多种多样结合方式是可能的。
[0058]
术语“配位”(“coordinating”、“coordination”)是指一种相互作用,其中一个多电子对供体配位地键合(“配位”)至优选一个中心(金属)离子,即与优选一个中心(金属)离子共享两个或多个未共享的电子对。
[0059]
螯合剂或螯合试剂优选为在一端具有金属螯合基团而在另一端具有反应性官能团的大环双官能螯合剂,其能够结合其他部分,如肽。优选地,可以选择螯合剂,使得螯合剂形成方形双锥络合物,用于络合放射性核素。在另一个实施方案中,螯合剂不是来自平面或方形平面络合物。
[0060]
优选地根据其配位所需中心(金属)离子的能力来选择螯合试剂,所需中心(金属)离子通常是本文指定的放射性核素。
[0061]
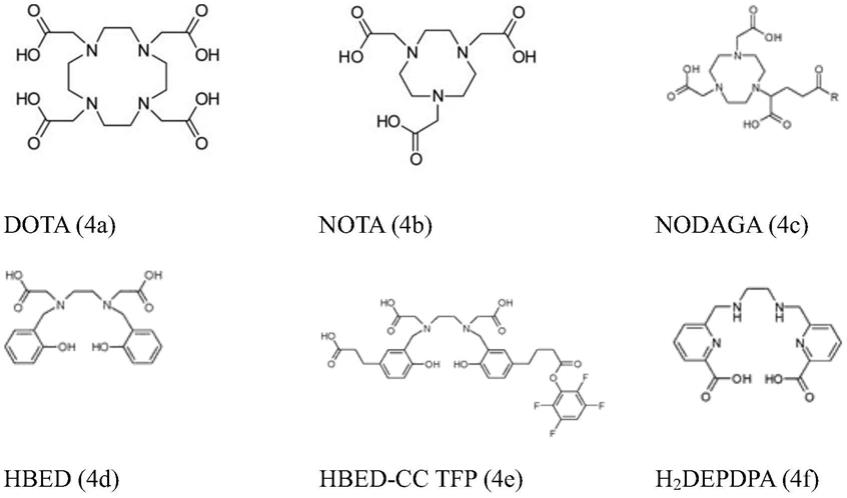
因此,螯合剂的特征可以是下式(4a)-(4jj)之一:
[0062]
[0063]
[0064]
[0065][0066]
优选地,螯合剂可以是dota(1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸,其特征可以在于式(4a)),hbed-cc(n,n"-双[2-羟基-5-(羧乙基)苄基]乙二胺-n,n"-二乙酸,其特征可以在于式(4e)),dotaga(2-[1,4,7,10-四氮杂环十二烷-4,7,10-三(乙酸酯)]-戊二酸),dotam(1,4,7,10-四(氨基甲酰甲基)-1,4,7,10-四氮杂环十二烷)或其衍生物。
[0067]
有利地,dota与诊断性(如
68
ga)和治疗性(如
90
y或
177
lu)放射性核素有效地形成络合物,并由此能够使用相同的缀合物进行成像(诊断性)和治疗目的,即,作为治疗诊断剂(theragnostic agent)。能够络合钪放射性核素(
43
sc、
44
sc、
47
sc)的dota衍生物——包括do3ap(其特征在于式(4hh))、do3ap
pra
(其特征在于式(4ii))或do3ap
abn
(其特征在于式(4jj))——也可以是优选的,并且在kerdjoudj等人的(dalton trans.,2016,45,1398-1409)中进行描述。
[0068]
有利地,hbed-cc与诊断性放射性核素(如
68
ga、
99m
tc)有效形成络合物。
[0069]
本发明的上下文中其他优选的螯合剂包括(2-(4,7-二(羧甲基)-1,4,7-三氮壬-1-基-戊二酸(nodaga)、1,4,7-三氮杂环-壬烷-1,4,7-三乙酸(nota)、2-(4,7,10-三(羧甲基)-1,4,7,10-四-氮杂环十二烷-1-基)-戊二酸(dotaga)、1,4,7-三氮杂环壬烷次膦酸(trap)、1,4,7-三氮杂环-壬烷-1-[甲基(2-羧乙基)-次膦酸]-4,7-二-[甲基(2-羟甲基)-次膦酸](nopo)、3,6,9,15-四-氮杂双环[9,3,1]-十五烷-1(15),11,13-三烯-3,6,9-三乙酸(pcta)、n'-{5-[乙酰基(羟基)氨基]-戊基}-n-[5-({4-[(5-氨基戊基)(羟基)氨基]-4-氧代丁酰基}-氨基)戊基]-n-羟基琥珀酰胺(dfo)、二乙烯-三胺五乙酸(dtpa)、和肼烟酰胺(hynic)。
[0070]
螯合剂基团,例如dota基团,可以优选与中心(金属)离子络合,特别是与如本文所定义的放射性核素。可选地,螯合剂基团,例如dota,可能不与中心(金属)离子络合,特别是本文定义的放射性核素,并因此可以以未络合的形式存在。如果螯合剂(如dota)没有与所述金属离子络合,则螯合剂的羧酸基团可以是游离酸的形式,也可以是盐的形式。
[0071]
在本领域技术人员的技能和知识范围内选择螯合剂和放射性核素的合适组合。比如,在一些优选实施方式中,螯合剂可以是dota和放射性核素可以是
177
lu。在其他优选实施方式中,螯合剂可以是dota和放射性核素可以是
68
ga。在其他优选实施方式中,螯合剂可以是hynic和放射性核素可以是
99m
tc。
[0072]
载体分子
[0073]
载体分子可以是与(肿瘤)细胞靶标结合的任何分子,如受体或另一细胞(表面)分子。在本发明的优选实施方式中,载体分子选自肽、拟肽、抗体片段、抗体模拟物、小分子和结。
[0074]
细胞靶可以是任何存在于放射性核素缀合物分子旨在结合以进行放射疗法或诊断的靶细胞中,或优选地存在于其上。
[0075]
例如,载体分子可以针对存在于疾病细胞(如肿瘤细胞)上的受体或细胞表面分子。在下文中,详细描述了存在于肿瘤细胞上的受体和细胞表面分子的实例,它们可以是载体分子的靶结构。然而,靶结构不限于下面描述的受体和细胞表面分子。存在于癌症或其他疾病细胞上的其他受体和细胞表面分子被考虑作为载体分子的靶结构。此外,考虑了靶向存在于癌症或其他疾病细胞上的受体和细胞表面分子的其他载体分子。
[0076]
促生长素抑制剂受体靶向化合物
[0077]
根据本发明,pah或其盐或羧酸盐衍生物可适当地用于减少靶向促生长素抑制剂受体(sstr)的(放射性)药物的肾毒性副作用。
[0078]
为了良好至中度分化的神经内分泌肿瘤(net)的治疗,例如,可以使用靶向促生长素抑制剂受体(sstr)的肽。在net中,放射配体疗法已经确立,并且可以实现高比率的长期肿瘤缓解和稳定。靶向促生长素抑制剂受体的肽例如是促生长素抑制剂类似物tyr3-奥曲肽(d-phe-c(cys-tyr-d-trp-lys-thr-cys)-thr(ol))以及tyr3-奥曲酸盐(d-phe-c(cys-tyr-d-trp-lys-thr-cys)-thr)(capello a et al.:tyr3-octreotide and tyr3-octreotate radiolabeled with 177
lu or 90
y:peptide receptor radionuclide therapy results in vitro,cancer biother radiopharm,2003oct;18(5):761-8)。促生长素抑制剂激动剂的进一步实例是肽奥曲肽(d-phe-环(cys-phe-d-trp-lys-thr-cys)thr(ol))和noc(d-phe-环(cys-1-nal-d-trp-lys-thr-cys)thr(ol))。
[0079]
靶向促生长素抑制剂-受体的化合物的其他实例是促生长素抑制剂拮抗肽,诸如jr10(p-no
2-phe-c(d-cys-tyr-d-aph(cbm)-lys-thr-cys)d-tyr-nh2);jr11(cpa-c(d-cys-aph(hor)-d-aph(cbm)-lys-thr-cys)d-tyr-nh2);bass(p-no
2-phe-环(d-cys-tyr-d-trp-lys-thr-cys)d-tyr-nh2;lm3(p-cl-phe-环(d-cys-tyr-d-aph(cbm)-lys-thr-cys)d-tyr-nh2。
[0080]
在本发明的特别优选的实施方式中,pah用于减少基于促生长素抑制剂类似物的(放射性)药物的肾毒性副作用;其实例包括:
177
lu-dotatoc(
177
lu-dota
°‑
[tyr3]-奥曲肽)(
177
lu-dota-d-phe-环(cys-tyr-d-trp-lys-thr-cys]-thr(ol)、
177
lu-dotanoc(
177
lu-dota-d-phe-环(cys-1-nal-d-trp-lys-thr-cys)thr(ol))、
177
lu-dotatate(
177
lu-dota-d-phe-环(cys-tyr-d-trp-lys-thr-cys)thr)、
68
ga-dotatoc(
68
ga-dota-d-phe-环(cys-tyr-d-trp-lys-thr-cys)thr(ol))、
68
ga-dotanoc(
68
ga-dota-d-phe-环(cys-1-nal-d-trp-lys-thr-cys)thr(ol))、
90
y-dotatoc(
90
y-dota-d-phe-环(cys-tyr-d-trp-lys-thr-cys)thr(ol))、
90
y-dotatate(
90
y-dota-d-phe-环(cys-tyr-d-trp-lys-thr-cys)thr)、
111
in-dtpa-奥曲肽(
111
in-dtpa-d-phe-环(cys-phe-d-trp-lys-thr-cys)thr(ol))。
[0081]
在另一优选实施方式中,pah或其盐或羧酸衍生物用于减少基于促生长素抑制剂拮抗化合物的(放射性)药物的肾毒性副作用;实例包括:
111
in-dota-bass(
111
in-dota-p-no
2-phe-环-(d-cys-tyr-d-trp-lys-thr-cys)d-tyr-nh2、
111
in-dota-jr11(
111
in-dota-cpa-环[d-cys-aph(hor)-d-aph(cbm)-lys-thr-cys]d-tyr-nh2)、
68
ga-dota-jr11(ga-ops201)(
68
ga-dota-cpa-环[d-cys-aph(hor)-d-aph(cbm)-lys-thr-cys]d-tyr-nh2)、
68
ga-dodaga-jr11(ga-ops202)(
68
ga-nodaga-cpa-环[d-cys-aph(hor)-d-aph(cbm)-lys-thr-cys]d-tyr-nh2)、
177
lu-dota-jr11(lu-ops201)(
177
lu-dota-cpa-环[d-cys-aph(hor)-d-aph(cbm)-lys-thr-cys]d-tyr-nh2)。
[0082]
靶向psma的化合物
[0083]
pah或其盐或羧酸衍生物还可以合适地用于减少靶向前列腺特异性膜抗原(psma)的(放射性)药物的肾毒性副作用。
[0084]
人前列腺特异性膜抗原(psma)(也称为谷氨酸羧肽酶ii(gcpii)、叶酸酯水解酶1、叶酰聚(folypoly)-γ-谷氨酸羧肽酶(fgcp)和n-乙酰化-α-连接的酸性二肽酶i(naaladase i))是一种ii型跨膜锌金属肽酶,其在神经系统、前列腺、肾脏和小肠中高度表达。它通常也被认为是前列腺癌的肿瘤标志物。
[0085]
在较早的技术中,已经开发了各种携带治疗或诊断部分的psma靶向剂。
[0086]
最近,能够结合psma的胞外结构域的各种小分子psma靶向剂被开发用于pet/ct和spect/ct成像,包括放射标记的n-[n-[(s)-1,3-二羧基丙基]氨基甲酰基]-s-[11c]甲基-l-半胱氨酸(dcfbc)和若干基于脲的肽模拟psma-抑制剂(参见bouchelouche et al.discov med.2010jan;9(44):55
–
61),包括mip-1095(hillier et al.cancer res.2009sep1;69(17):6932-40)、目前在临床评估中的psma配体,和等人开发的dota-缀合的psma-抑制剂psma-617(jnm 2015,56:914
–
920和ep2862857a1)。
[0087]
基于脲的psma配体通常包括三个组分:结合基序(glu-脲-lys)、接头和放射性标记承载部分(用于放射性标记的螯合剂分子或用于氟化试剂的假体基团)。最常见使用的低分子量psma配体的实例是
123
i-mip-1072和
123
i-mip-1095(barrett ja et al.j nucl med.2013;54:380-387);
99m
tc-mip-1404和
99m
tc-1405(hillier sm et al.j nucl med.2013;54:1369-1376),其在临床试验中用于spect成像。n-[n-[(s)-1,3-二羧基丙基]氨基甲酰基]-4-18
f-氟苄基-l-半胱氨酸(
18
f-dcfbc)(cho sy et al.,j nucl.med.2012;53:1883-1891)和
68
ga-psma-11(
68
ga-psma-n,n
’‑
二-[2-羟基-5-(羧乙基)苄基]乙二胺-n,n
’‑
二乙酸)(eder m et al,pharmaceuticals(basel)2014;7:779-796)是用于pet成像的试剂。进一步的治疗诊断剂(threranostic agents)是例如
131
i-mip-1095(zechmann et al.,eur j nucl med mol imaging.2014;41:1280-1292)、基于螯合剂的psma-617(afshar-oromieh a et al.,j nucl med.2015;56:1697-1705)和psma-i&t(weineisen m et al.,j nucl med.2015;56:1169-1176)、psma-i&s(robu s et al.,j nucl med.2017;58:235-242)。进一步,提到
18
f-标记的小分子脲衍生物
18
f-dcfpyl(chen y et al.,clin cancer res.2011;17:7645-7653)和
18
f-psma-1007(giesel fl et al.,eur j nucl med molecular imaging.2017;44:678-688)。
[0088]
近年,kelly等人(j nucl med.2017pii:jnumed.116.188722.doi:10.2967/jnumed.116.188722.[epub ahead of print])评估了对psma和对人血清白蛋白(hsa)都表现亲和力的试剂。由kelly等人开发的配体包括用于has结合的p-(碘苯基)丁酸实体和基于脲的psma结合实体。在由kelly等人开发的化合物中,放射性治疗碘(
131
i)共价地附接至has结合部分,其又经由烃基链直接连接至psma结合实体。另一实例是具有白蛋白结合实体的
177
lu-标记的氨基磷酸酯类psma抑制剂(choy et al.theranostics 2017;7(7):1928-1939)。络合
177
lu放射性核素的dota螯合剂醚连接至不可逆psma抑制剂ctt1298(ep2970345a1)。
[0089]
因此,在本发明的优选实施方式中,pah或其盐或羧酸衍生物被用于减少(放射性)药物的肾毒性副作用,该药物包括结合至如上所定义的螯合剂分子的psma靶向配体,该螯
合剂分子与如上所述的放射性核素(如选自
68
ga、
177
lu、
225
ac、
111
in、
99m
tc)络合。
[0090]
在特别优选的实施方式中,pah用于减少
68
ga-psma-11的肾毒性副作用。
[0091]
叶酸酯缀合物
[0092]
fr-α作为用于成像目的和靶向疗法概念的肿瘤相关靶引起了最大的兴趣。许多研究小组将使用具有各种治疗探针的叶酸缀合物在体外和体内靶向fr阳性肿瘤细胞作为例子。因此,叶酸酯受体(fr)已被证明是使用叶酸放射性缀合物进行核成像的有价值靶。
[0093]
然而,长期以来,使用基于叶酸酯的放射性药物进行治疗一直被认为是无法实现的目标,因为它们的大量肾积累。至于其他放射性缀合物,如本文所述,本发明允许通过本发明的策略减少体内放射性药物的异位积累,从而改善肿瘤与肾脏的比率。
[0094]
叶酸酯缀合物放射性药物的优选实例使用
99m
tc(guo et al.,j nucl med.1999;40:1563-1569;mathias et al.,bioconjug chem.2000;11:253-257;leamon et al.,bioconjug chem.2002;13:1200-1210;reddy et al.,j nucl.med.2004;45:857-866;m
ü
ller et al.,j nucl med mol imaging 2006;33:1007-1016;m
ü
ller et al.,bioconjug chem.2006;17:797-806)、
111
in(siegel et al.,j nucl med.2003;44:700-707)、
66/67/68
ga(mathias et al.,nucl med biol.1999;26:23-25;mathias et al.,nucl med biol.2003;30:725-731)和
18
f(bettio et al.,j nucl med.2006;47:1153-1160)。
[0095]
代表性叶酸酯缀合物是例如
111
in-dtpa-叶酸酯、
177
lu-ec0800、
177
lu-cm09、
149/161
tb-cm09、
99m
tc(co)3、
99m
tc-ec20、
111
in-dtpa-叶酸酯、
111
in/
177
lu-dota-点击-叶酸酯、
67
ga-dota-bz-叶酸酯(
67
ga-ec0800),
68
ga-nodaga-叶酸酯和
[0096][0097]
cck2受体靶向化合物
[0098]
pah或其盐或羧酸衍生物还可以合适地用于减少靶向cck2受体的(放射性)药物的肾毒性副作用。
[0099]
cck2受体(胆囊收缩素)位于中枢和外周神经系统区域,在多种类型的人类癌症中过度表达,如甲状腺髓样癌、小细胞肺癌和间质卵巢癌。已对开发用于体内靶向cck2受体的合适放射性配体进行了广泛研究。已经合成并表征了多种放射标记的cck/胃泌素-相关肽。所有的肽都具有共同的c-末端cck受体结合四肽序列trp-met-asp-phe-nh2或其衍生物。肽可以基于其母肽(胃泌素或cck)的序列及其形式(即,线性、环、多倍体)进行分类。
[0100]
cck受体配体的实例是胃泌素类似物,诸如sargastrin(gln-gly-pro-trp-leu-glu-glu-glu-glu-glu-ala-tyr-gly-trp-nle-asp-phe-nh2)、minigastrin0(mg-0)d-glu-(glu)
5-ala-tyr-gly-trp-met-asp-phe-nh2)、minigastrin 11(mg-11)(d-glu-ala-tyr-gly-trp-met-asp-phe-nh2)、环-minigastrin 1(cyclo-mg1)(cyclo[γ-d-glu-ala-tyr-d-lys]-trp-met-asp-phe-nh2)、环-minigastrin 2(cyclo-mg2)(cyclo[γ-d-glu-ala-tyr-d-lys]-trp-nle-asp-phe-nh2、demogastrin 1(d-glu-(glu)
5-ala-tyr-gly-trp-met-asp-phe-nh2)、demogastrin 2(d-glu-(glu)
5-ala-tyr-gly-trp-met-asp-phe-nh2、h2-met(his-his-glu-ala-tyr-gly-trp-met-asp-phe-nh2)、h2-nle(his-his-glu-ala-tyr-gly-trp-nle-asp-phe-nh2)、h6-met(his)
6-glu-ala-tyr-gly-trp-met-asp-phe-nh2);和cck8类似物,诸如cck8(d-asp-tyr-met-gly-trp-met-asp-phe-nh2)、cck8(nle)(d-asp-tyr-nle-gly-trp-nle-asp-phe-nh2)、scck8(d-asp-tyr(oso3h)-met-gly-trp-met-asp-phe-nh2)、scck8[phe2(p-ch2so3h)、nle
3,6
](d-asp-phe(p-ch2so3h)-nle-gly-trp-nle-asp-phe-nh2)、scck8[phe2(p-ch2so3h)、hpg
3,6
](d-asp-phe(p-ch2so3h)-hpg-gly-trp-hpg-asp-phe-nh2)。
[0101]
cck受体靶向肽优选被放射性核素放射标记,用于成像或治疗应用。合适的放射性核素包括以上指定的放射性核素,并且特别是包括放射性核素
99m
tc、
111
in、
18
f、
68
ga、
131
i、
90
y和
177
lu。为了使用放射性核素进行放射标记,优选地使用缀合至肽的螯合剂。作为螯合剂,可以使用以上指定的螯合剂,其中dota、dotaga、dotam、dtpa和hynic是优选的。
[0102]
因此,在本发明的优选实施方式中,pah被用于减少cck2受体靶向(放射性)药物的肾毒性副作用,包括但不限于
177
lu-dota-sargastrin、
111
in-dtpa-mg0、
111
in-dota-mg11、
111
in-dota-mg11(nle)、
111
in-dota-h2-met、
111
in-dota-h2-nle、
111
in-dota-h6-met、[
99m
tc]2n
40
、d-glu
1-mg(
99m
tc-demogastrin 1)、[
99m
tc]2n
40-1
,gly0,d-glu
1-mg(
99m
tc-demogastrin 2)、
99m
tc-hynic-mg11、
99m
tc-hynic-环-mg1、
99m
tc-hynic-环-mg2;和cck8类似物,诸如
111
in-dtpa-cck8、
111
in-dtpa-cck8(nle)、
99m
tc-hynic-cck8、
99m
tc-hynic-scck8、
111
in-dota-scck8[phe2(p-ch2so3h),nle
3,6
]、和
111
in-dota-scck8[phe2(p-ch2so3h),hpg
3,6
]。
[0103]
整联蛋白-结合分子
[0104]
pah或其盐或羧酸衍生物还可以合适地用于减少靶向整联蛋白的(放射性)药物的肾毒性副作用。
[0105]
整联蛋白是由α-和β-亚基组成的异二聚体糖蛋白。已知的八个β-单位和十八个α-单位有24种不同的组合。整联蛋白介导细胞-细胞和细胞-基质相互作用,并通过内向外(insight-out)和外向内(outside-in)信号传导转导跨质膜的信号。一些整联蛋白在肿瘤诱导的血管生成和肿瘤转移过程中在内皮细胞和肿瘤细胞的迁移过程中起重要作用。血管生成,即从先前存在的脉管系统中形成新血管,是各种人类肿瘤发展和传播的关键步骤。肿瘤学中的各种治疗策略都集中在抑制肿瘤诱导的血管生成上。关于整联蛋白,整联蛋白αvβ3和αvβ5的作用受到了显著的关注,因为它们在增殖血管内皮细胞方面很突出。因此,用于开发用于对血管生成成像的放射性药物的最突出的靶结构之一是整联蛋白αvβ3。
[0106]
通过用含有arg-gly-asp(rgd)氨基酸序列的小肽拮抗αvβ3整联蛋白,可以在体内阻断肿瘤诱导的血管生成。这种天然存在于细胞外基质蛋白中的三肽序列是αvβ3整联蛋白
的主要结合位点。由于αvβ3整联蛋白在肿瘤中的选择性表达,放射标记的rgd肽是肿瘤中αvβ3整联蛋白靶向的有吸引力的候选物。在过去的十年中,许多放射标记的线性和环状rgd肽已被评估为用于通过spect或pet以及治疗剂对肿瘤成像的放射性示踪剂。
[0107]
因此,pah或其盐或羧酸衍生物可以特别合适用于减少包括放射标记的rgd肽的(放射性)药物的肾毒性副作用。
[0108]
rgd肽优选用放射性核素进行放射标记,用于成像或治疗应用。合适的放射性核素包括以上指定的放射性核素,和特别是包括放射性核素
18
f、
99m
tc、
68
ga、
111
in、
131
i、
90
y、
67
cu、和
177
lu。为了使用放射性核素进行放射标记,优选地使用缀合至肽的螯合剂。作为螯合剂,可以使用以上指定的任何合适的螯合剂,其中nota、dota、dotaga、dotam、dtpa、hynic是优选的。
[0109]
例如,pah或其盐或羧酸衍生物可以合适地用于减少以下药物的肾毒性副作用:
18
f-galacto-rgd、
99m
tc-nc100692(
99m
tc-maracilatide)、
18
f-ah11185(
18
f-fluciclatide)、
18
f-rgd-k5、
68
ga-nota-rgd、
18
f-fpprgd2、
18
f-alf-nota-prgd2(
18
f-alfatide)、
18
f-nota-e[peg4-c(rgdfk)]2(
18
f-alfatide ii)、
68
ga-nota-prgd2、
67
cu-cyclam-raft-c(-rgdfk-)4、
111
in-dota-e-[c(rgdfk)]2、
99m
tc-hynic-e-[c(rgdfk)]2。
[0110]
神经降压肽受体靶向化合物
[0111]
神经降压肽受体1(ntr1)在胰腺导管腺癌中过度表达,该癌是最致命的癌症之一。已经开发了几种ntr1拮抗剂,诸如sr142948a和sr48692,和
177
lu-3bp-2273,其是基于sr142948a已经开发的
177
lu-标记的dota-缀合的ntr1拮抗剂。其已经用于治疗胰腺导管腺癌(baum rp et al.,the journal of nuclear medicine,vol.59,no.5,may 2018)。
[0112]
因此,pah或其盐或羧酸衍生物还可以合适地用于减少靶向神经降压肽受体1的(放射性)药物的肾毒性副作用,特别是用于癌症诊断或疗法的放射标记的ntr1拮抗剂,优选地
177
lu-或
68
ga-标记的ntr1拮抗剂,更优选地
177
lu-3bp-2273,尽管可以考虑其他放射性核素,例如以上提到的放射性核素,以及其他螯合剂,例如以上提到的螯合剂。
[0113]
胰高血糖素-样肽-1(glp-1)受体靶向化合物
[0114]
glp-1受体在基本上所有良性胰岛素瘤和胃泌素瘤上过表达。从胰腺的β细胞中出现并以小结节形式存在的良性胰岛素瘤会分泌胰岛素,导致可能危及生命的低血糖症。
[0115]
因此,pah或其盐或羧酸衍生物也可以合适用于减少靶向glp-1受体的(放射性)药物的肾毒性副作用。其非限制性实例包括基于39-mer肽毒蜥外泌肽-4的
111
in-、
99m
tc-和
68
ga-标记的肽,诸如lys
40
(ahx-dota-111
in)nh
2-毒蜥外泌肽-4。然而,可以考虑其他放射性核素,例如,以上提到的放射性核素,以及其他螯合剂,例如以上提到的螯合剂。
[0116]
胃泌素释放肽(grp)受体靶向化合物
[0117]
pah或其盐或羧酸衍生物还可以合适地用于减少靶向grp受体的(放射性)药物的肾毒性副作用。
[0118]
grp受体已在主要的人类肿瘤中得到证实,诸如乳腺癌和前列腺癌。铃蟾肽是一种十四肽神经激素和哺乳动物grp(一种27mer肽)的两栖动物同系物。已经开发了各种铃蟾肽类似物,用于
99m
tc标记和spect。特别地,经由gly-5-氨基戊酸间隔区(
99m
tc-rp527)将截短的铃蟾肽偶联至n3s-螯合剂。此外,已经开发了若干铃蟾肽类似物和铃蟾肽拮抗剂,并使用不同的螯合剂用不同的放射性同位素(如
68
ga、
64
cu、
18
f)进行标记。其实例包括pan-铃蟾肽
类似物
68
ga-bzh3(zhang h et al.,cancer res 2004;64:6707-6715),和经由gly-4-氨基苯甲酰基间隔区偶联至dota的
177
lu-标记的铃蟾肽(7-14)衍生物(bodei l et al.,eur j nucl med mol imaging 2007:34(suppl 2):s221。
[0119]
然而,pah或其盐或羧酸衍生物还可以合适地用于减少grp受体靶向(放射性)药物的肾毒性副作用,该grp受体靶向(放射性)药物包括其他放射性核素,例如以上提到的放射性核素,以及其他螯合剂,例如以上提到的螯合剂。
[0120]
神经激肽1型受体靶向化合物
[0121]
神经激肽1型受体在神经胶质瘤细胞和肿瘤血管上持续过度表达(hennig im et al.,int j cancer 1995;61:786-792)。经由神经激肽1型受体作用的放射标记的11-氨基酸肽物质p(arg pro lys pro gln gln phe phe gly leu met)可以合适地用于靶向恶性胶质瘤。特别地,物质p已经缀合至螯合剂dotaga,和90y-标记的dotaga-物质p已经用于临床研究(kneifel s et al.,eur j nucl med mol imaging.2007;34:1388-1395。在另一研究中,使用发射α-放射的缀合物213bi-dota-[thi8,met(o2)11]-物质p评估了靶向α-放射核素疗法用于脑肿瘤的可行性和有效性(cordier et al.,eur j nucl med mol imaging.2010;37:1335-1344)。
[0122]
因此,pah或其盐或羧酸衍生物还可以合适地用于减少靶向神经激肽1型受体的治疗性和诊断性化合物的肾毒性副作用,特别是物质p缀合物,其包含用于诊断或治疗的放射性核素以及配位所述放射性核素的螯合剂。
[0123]
affilins
[0124]
pah或其盐或羧酸衍生物也可适当地用于减少用作治疗性和诊断性化合物的抗体模拟物的肾毒性副作用。
[0125]
affilins是人工蛋白质,设计来选择性地结合抗原。affilin蛋白在结构上分别来源于人泛素或γ-b晶体蛋白。affilin蛋白是通过修饰这些蛋白质的表面暴露氨基酸来构建,并通过展示技术(诸如噬菌体展示和筛选)进行分离。它们在对抗原的亲和力和特异性方面类似于抗体,但在结构上却不同,这使它们成为一种抗体模拟物。由scil proteins gmbh开发,作为潜在的生物制药药物、诊断和亲和配体。affilin分子可以容易地被修饰,并适合标记肿瘤细胞用于诊断目的或通过辐射特异性杀死肿瘤细胞。
[0126]
可以生成多特异性affilin分子,其同时结合不同的目标。放射性核素或细胞毒素可以与affilin蛋白结合,使它们成为潜在的肿瘤治疗剂和诊断剂。放射性核素-螯合剂-affilin缀合物,如
177
lu-dota-affilin和
68
ga-dota-affilin,已被设计用于成像和治疗目的。pah可有效减少这些affilin缀合物的肾毒性副作用。它还可以用于降低分别包含其他放射性核素(例如以上所指定)和螯合剂(例如以上所指定)的其他affilin缀合物的肾毒性副作用。
[0127]
因此,在优选的实施方式中,pah或其药学上可接受的盐或羧酸衍生物被用于减少用于癌症疗法或成像的(放射标记的)治疗性和诊断性化合物的肾毒性副作用,所述癌症诸如例如神经内分泌肿瘤、前列腺癌、胰腺癌、肾癌、膀胱癌、甲状腺髓样癌、小细胞肺癌、间质卵巢癌、胰腺导管腺癌、胰岛素瘤、胃泌素瘤、乳腺癌等。
[0128]
例如,pah或其药学上可接受的盐或羧酸衍生物用于减少用于前列腺癌疗法或成像的放射标记的治疗性和诊断性化合物的肾毒性副作用,所述化合物例如靶向促生长素抑
制剂受体的(放射性)药物,或靶向前列腺特异性膜抗原(psma)的(放射性)药物。在优选实施方式中,pah或其药学上可接受的盐或羧酸衍生物被用于减少
177
lu-dotatoc(
177
lu-dota
°‑
[tyr3]-奥曲肽)的肾毒性副作用。
[0129]
在本发明的实施方式中,pah或其药学上可接受的盐或羧酸衍生物用于与减少放射性标记的和非放射性标记的治疗性和诊断性化合物的肾毒性副作用的其他物质联合使用。减少放射性标记的和非放射性标记的治疗性和诊断性化合物的肾毒性副作用的其他物质的实例是氨基酸,诸如赖氨酸和精氨酸及其混合物、明胶、氨磷汀、白蛋白衍生肽、胰蛋白酶化白蛋白、psma-结合分子(诸如pmpa)、维生素、gelofusine、或fralb-c(溴化氰裂解的牛血清白蛋白)。
[0130]
在本发明的特别优选的实施方式,pah的药学上可接受的盐是氨基马尿酸钠。优选地,pah或其药学上可接受的盐或羧酸衍生物,优选地氨基马尿酸钠,在盐溶液中使用,优选地,在注射用水(wfi)或nacl溶液中,更优选地,在20%nacl溶液中。
[0131]
在本发明中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以足以有效减少治疗性和/或诊断性化合物的肾毒性副作用的量使用。pah的该有效量可以通过常规实验进行确定,如通过使用动物模型。此类模型包括但不限于兔、绵羊、小鼠、大鼠、狗和非人灵长类动物模型。
[0132]
比如,pah的施用量范围可以是(每千克体重)约0.1mg/kg至10g/kg,优选地约0.5mg/kg至5g/kg,更优选地约1mg/kg至1g/kg。
[0133]
在本发明的优选实施方式中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以每千克体重约5mg至约500mg的量,例如以每千克体重约5、10、20、30、40、50、60、70、80、90、100、150、200、250、300、350、400、450、500mg上至每千克体重500mg的量使用。更优选地,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以每千克体重约50mg至约500mg,更优选地每千克体重约50mg至约250mg的量,例如以每千克体重约50、55、60、65、70、75、80、85、90、95、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240或250mg的量使用。更优选地,pah或其药学上可接受的盐或羧酸衍生物以每千克体重约75mg至约200mg的量,例如以每千克体重约75、80、85、90、95、100、105、110、115、120、125、130、135、140、145、150、155、160、165、170、175、180、185、190、195、上至200mg,或每千克体重200mg的量使用。更优选地,pah或其药学上可接受的盐或羧酸衍生物以每千克体重约80mg至约160mg的量,例如以每千克体重约80、85、90、95、100、105、110、115、120、125、130、135、140、145、150、155、上至160mg的量,或每千克体重160mg的量使用。
[0134]
通常,pah或其药学上可接受的盐或羧酸衍生物以比(联合施用)治疗性或诊断性化合物更大(摩尔和/或w/w)量使用。
[0135]
比如,治疗性或诊断性化合物与pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以约1/1.000.000至1/10(w/w)的比例使用,优选地约1/500.000至1/100(w/w),更优选地约1/250.000至约1/500(w/w)。
[0136]
在本发明的优选实施方式中,治疗性或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例使用:约1/250.000至约1/5.000(w/w)的比例,例如约1/250.000、1/200.000、1/150.000、1/100.000、或1/50.000至约1/5.000(w/w)的比例,更优选地约1/240.000至约1/8.000(w/w)的比例,例如约1/240.000、1/230.000、
1/220.000、1/210.000、1/200.000、1/190.000、1/180.000、1/170.000、1/160.000、1/150.000、1/140.000、1/130.000、1/120.000、1/110.000、1/100.000、1/90.000、1/80.000、1/70.000、1/60.000、1/50.000、1/40.000、1/30.000、1/20.000、1/19.000、1/18.000、1/17.000、1/16.000、1/15.000、1/14.000、1/13.000、1/12.000、1/11.000、1/10.000、1/9.000、或1/8.000(w/w)的比例。在进一步优选实施方式中,治疗性和/或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例使用:约1/100.000至约1/10.000(w/w)的比例,例如约1/100.000、1/95.000、1/90.000、1/85.000、1/80.000、1/75.000、1/70.000、1/65.000、1/60.0001/55.000、1/50.000、1/45.000、1/40.000、1/35.000、1/30.000、1/25.000、1/20.000、1/15.000、1/10.000(w/w)的比例。在进一步优选实施方式中,治疗性和/或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例使用:约1/50.000至约1/40.000(w/w)的比例,例如约1/50.000、1/49.000、1/48.000、1/47.000、1/46.000、1/45.000、1/44.000、1/43.000、1/42.000、1/41.000、1/40.000(w/w)的比例。
[0137]
在第二方面,本发明涉及药物组合物,其包括与对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物联合的(a)放射标记的和/或非-放射标记的药物化合物,以及药学上可接受的赋形剂、稀释剂、载体或其组合。
[0138]
放射标记的或非放射标记的药物化合物可以是以上指定的潜在表现肾毒性副作用的任何肾毒性治疗性或诊断性化合物,优选地放射标记的诊断性和/或治疗性化合物。优选地,药物组合物包括放射标记的药物化合物,其是与载体分子缀合的放射性核素络合物,由此包括载体分子、螯合剂和放射性核素。
[0139]
优选地,放射性核素络合物的载体分子选自以上指定的载体分子,特别是选自小有机分子、肽、拟肽、抗体片段、抗体模拟物、小分子和结,并且更优选地选自以上指定的促生长素抑制剂类似物、psma-抑制剂、胃泌素类似物、整联蛋白结合分子和抗原结合蛋白,如选自tyr3-奥曲肽、tyr3-奥曲酸盐、jr11、psma-11、sargastrin、rgd、affilin或叶酸酯缀合物。在特别优选的实施方式中,载体分子选自tyr3-奥曲肽和tyr3-奥曲酸盐。
[0140]
优选地,放射性核素络合物的螯合剂选自以上指定的螯合剂,更优选地选自dota、dotam、dotag、hbed-cc、nota、nodaga、dotaga、trap、nopo、pcta、dfo、dtpa、do3ap、do3ap
pra
、do3ap
abn
、hynic或其衍生物。
[0141]
优选地,放射性核素络合物的放射性核素选自以上指定的放射性核素,并且更优选地选自
94
tc、
99m
tc、
90
in、
111
in、
67
ga、
68
ga、
86
y、
90
y、
18
f、
131
i 177
lu、
161
tb、
186
re、
188
re、
64
cu、
67
cu、
55
co、
57
co、
43
sc、
44
sc、
47
sc、
225
ac、
213
bi、
212
bi、
212
pb、
227
th、
153
sm、
166
ho、
225
ac和
166
dy。甚至更优选地,药物组合物中包括的放射性核素络合物的放射性核素选自
177
lu、
68
ga、
111
in、
90
y、
99m
tc、
18
f、
131
i、
225
ac和
161
tb,或最优选地选自
177
lu、
68
ga、
111
in、
90
y、
99m
tc、
225
ac和
161
tb。在一个实施方式中,放射性核素具体地选自二价放射性核素,特别是选自
64
cu、
67
cu和
212
pb,选自三价放射性核素,特别是
177
lu、
90
y、
67
ga、
68
ga、
111
in、
225
ac、
161
tb、
44
sc和
47
sc,或选自四价放射性核素,特别是
227
th。在另一实施方式中,放射性核素是
99m
tc,其可以是二、四、或五价的。更具体地,放射性核素可以选自三价放射性核素。
[0142]
还优选的是,放射性核素适于通过dotatoc(dota)进行络合。特别地,可以使
90
in、
111
in、
67
ga、
68
ga、
86
y、
90
y、
177
lu、
161
tb、
64
cu、
67
cu、
55
co、
57
co、
43
sc、
44
sc、
47
sc、
225
ac、
213
bi、
212
bi
、
212
pb、
153
sm、
166
ho、
225
ac和
166
dy与作为螯合剂的dotatoc结合。
[0143]
如上所述,载体分子、螯合剂和放射性核素的合适结合可以通过本领域技术人员适当选择。例如,放射性核素络合物可以选自[
177
lu-dota
°‑
tyr3]-奥曲肽、
177
lu-dota-ja11、
177
lu-rgd、
177
lu-dota-affilin 2、
177
lu-dota-sargastrin、
68
ga-hbed-cc-psma-11。优选地,放射性核素络合物选自[
177
lu-dota
°‑
tyr3]-奥曲肽和[
177
lu-dota
°‑
tyr3]-奥曲酸盐。
[0144]
药物组合物优选地包括安全且有效量的放射标记的或非放射标记的治疗性或诊断性化合物。
[0145]
如本文所用,“安全且有效量”是指足以允许诊断和/或显著诱导待治疗疾病的阳性改变的试剂的量。然而,与此同时,“安全且有效量”足够小以避免严重的副作用,也就是说,允许优势和风险之间存在合理的关系。此外,“安全且有效量”将根据待诊断或治疗的特定病症以及待治疗患者的年龄和身体状况、病症的严重程度、治疗持续时间、伴随疗法的性质、所使用的特定药学上可接受的赋形剂或载体的性质、以及类似因素而改变。
[0146]
在本发明的特别优选实施方式中,药物组合物包括氨基马尿酸钠,作为pah的药学上可接受的盐。
[0147]
通常,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以比药物组合物中含有的(共施用的)治疗性或诊断性化合物更大的量存在。
[0148]
比如,治疗性或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下的比例存在于药物组合物中:约1/1.000.000至1/10(w/w),优选地约1/500.000至1/100(w/w),更优选地约1/250.000至约1/500(w/w)。
[0149]
在本发明的优选实施方式中,治疗性或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例存在于药物组合物中:约1/250.000至约1/5.000(w/w),例如约1/250.000、1/200.000、1/150.000、1/100.000、或1/50.000至约1/5.000(w/w)的比例,更优选地约1/240.000至约1/8.000(w/w)的比例,例如约1/240.000、1/230.000、1/220.000、1/210.000、1/200.000、1/190.000、1/180.000、1/170.000、1/160.000、1/150.000、1/140.000、1/130.000、1/120.000、1/110.000、1/100.000、1/90.000、1/80.000、1/70.000、1/60.000、1/50.000、1/40.000、1/30.000、1/20.000、1/19.000、1/18.000、1/17.000、1/16.000、1/15.000、1/14.000、1/13.000、1/12.000、1/11.000、1/10.000、1/9.000或1/8.000(w/w)的比例。更优选地,治疗性和/或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例存在于药物组合物中:约1/100.000至约1/10.000(w/w),例如约1/100.000、1/95.000、1/90.000、1/85.000、1/80.000、1/75.000、1/70.000、1/65.000、1/60.000 1/55.000、1/50.000、1/45.000、1/40.000、1/35.000、1/30.000、1/25.000、1/20.000、1/15.000、1/10.000(w/w)的比例。在进一步优选的实施方式中,治疗性和/或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例存在于药物组合物中:约1/50.000至约1/40.000(w/w),例如约1/50.000,1/49.000,1/48.000,1/47.000,1/46.000,1/45.000,1/44.000,1/43.000,1/42.000,1/41.000,1/40.000(w/w)的比例。
[0150]
在本发明的实施方式中,除了pah外,药物组合物还包括另外的物质,其减少放射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用,其中除了pah外的减少放
射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用的物质优选地选自氨基酸,如赖氨酸和精氨酸、明胶、氨磷汀、白蛋白衍生肽、psma-结合分子(诸如pmpa)、维生素。
[0151]
包括pah或其药学上可接受的盐或羧酸衍生物、(a)放射标记的和/或非放射标记的药物化合物,和任选地包括减少放射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用的另外的物质的药物组合物进一步包括药学上可接受的赋形剂、稀释剂、载体或其组合。药物组合物优选为液体或半液体组合物,其更优选地为液体或半液体组合物,其更优选地是水溶液,其可以被缓冲和/或表现出等渗特性。
[0152]
术语“药学上可接受的”是指这样的化合物或试剂,其与本发明药物组合物的组分相容,特别是与诊断性或治疗性药物化合物相容,并且不干扰和/或实质降低其诊断或治疗活性。药学上可接受的载体优选地具有足够高的纯度和足够低的毒性以使其适合施用于待治疗的受试者。
[0153]
制剂、载体和赋形剂
[0154]
药学上可接受的赋形剂可以表现出不同的功能作用,并且包括但不限于稀释剂、填料、填充剂、载体、崩解剂、粘结剂、润滑剂、助流剂、涂料、溶剂和共溶剂、缓冲剂、防腐剂、佐剂、抗氧化剂、润湿剂、消泡剂、增稠剂、甜味剂、调味剂和湿润剂。
[0155]
合适的药学上可接受的赋形剂通常基于药物组合物的制剂进行选择。
[0156]
对于液体形式的药物组合物,有用的药学上可接受的赋形剂通常包括溶剂、稀释剂或载体,诸如(无热原)水、(等渗)盐水溶液诸如磷酸盐或柠檬酸盐缓冲盐水、不挥发油、植物油,例如花生油、棉籽油、芝麻油、橄榄油、玉米油、乙醇、多元醇(例如甘油、丙二醇、聚乙二醇等);卵磷脂;表面活性剂;防腐剂,如苄基醇、对羟基苯甲酸酯、氯丁醇、苯酚、抗坏血酸、硫柳汞等;等渗剂,诸如糖类;多元醇,诸如甘露醇、山梨糖醇、氯化钠;单硬脂酸铝或明胶;抗氧化剂,如坏血酸、亚硫酸氢钠;螯合试剂,如乙二胺四乙酸(edta);缓冲剂,如乙酸盐、柠檬酸盐或磷酸盐,以及调节张力的试剂,如氯化钠或葡萄糖。ph可以用酸或碱调节,例如盐酸或氢氧化钠。缓冲液相对于特定参比介质可以是高渗、等渗或低渗的,即缓冲液相对于特定参比介质可以具有更高、相同或更低的盐含量,其中优选地可以使用上述盐的这种浓度,其不导致因渗透或其他浓度效应引起的细胞损伤。参比介质是体内方法中出现的液体,如血液、淋巴液、细胞溶质液或其他体液,或如可用作体外方法中参比介质的液体,诸如普通缓冲液或液体。这种常见的缓冲液或液体是技术人员已知的。
[0157]
经由注射,尤其是通过血管内、更优选地静脉内(i.v.)注射施用的液体药物组合物在制造和储存条件下应该优选地是无菌和稳定的。此类组合物通常被配制成肠胃外可接受的水溶液,其无热原、具有合适的ph、是等渗的并且保持活性成分的稳定性。
[0158]
对于液体药物组合物,合适的药学上可接受的赋形剂和载体包括水,通常是无热原的水;等渗盐水或缓冲(水性)溶液,如磷酸盐、柠檬酸盐等缓冲溶液。特别地,对于本发明(药物)组合物的注射剂,可以使用水或优选地缓冲液,更优选地水性缓冲液,其可以包含钠盐,例如至少50mm的钠盐,钙盐,例如至少0.01mm的钙盐,以及任选的钾盐,例如至少3mm的钾盐。
[0159]
钠盐、钙盐和任选的钾盐可以以其卤化物的形式出现,例如氯化物、碘化物或溴化物,以其氢氧化物、碳酸盐、碳酸氢盐或硫酸盐等的形式出现。不受其限制,钠盐的实例包括如nacl、nai、nabr、na2co3、nahco3、na2so4,钾盐的实例包括如kcl、ki、kbr、k2co3、khco3、
k2so4,和钙盐的实例包括如cacl2、cai2、cabr2、caco3、caso4、ca(oh)2。此外,此外,在缓冲液中可以包含上述阳离子的有机阴离子。
[0160]
适用于如上限定的注射目的的缓冲液可以包含选自氯化钠(nacl)、氯化钙(cacl2)和任选地氯化钾(kcl)的盐,其中除了氯根之外还可以存在其他阴离子。cacl2也可以用另一种盐代替,如kcl。通常,注射缓冲液中的盐以至少50mm氯化钠(nacl)、至少3mm氯化钾(kcl)和至少0.01mm氯化钙(cacl2)的浓度存在。注射缓冲液相对于特定参比介质可以是高渗的、等渗的或低渗的,即缓冲液相对于特定的参比介质可以具有更高、相同或更低的盐含量,其中优选地可以使用上述盐的这种浓度,其不导致因渗透或其他浓度效应引起的细胞损伤。
[0161]
对于(半)固体形式的药物组合物,合适的药学上可接受的赋形剂和载体包括粘结剂,诸如微晶纤维素、黄蓍胶或明胶;淀粉或乳糖;糖,例如乳糖、葡萄糖和蔗糖;淀粉,例如玉米淀粉或马铃薯淀粉;纤维素及其衍生物,例如羧甲基纤维素钠、乙基纤维素、乙酸纤维素;崩解剂,例如海藻酸;润滑剂,例如硬脂酸镁;助流剂,例如硬脂酸、硬脂酸镁;硫酸钙、胶体二氧化硅等;甜味剂,例如蔗糖或糖精;和/或调味剂,例如薄荷、甲基水杨酸盐或橙子调味剂。
[0162]
通常,用于局部施用的药物组合物可以配制成霜剂、软膏剂、凝胶剂、糊剂或粉末剂。用于口服施用的药物组合物可以配制成片剂、胶囊、液体、粉剂或缓释形式。然而,根据优选的实施方式,本发明的药物组合物胃肠外施用,特别是通过静脉内或肿瘤内注射,并因此被配制成液体或冻干形式用于胃肠外施用,如本文别处所讨论的。肠胃外制剂通常储存在小瓶、iv袋、安瓿、药筒或预装注射器中,并可作为注射剂、吸入剂或气溶胶施用,注射剂是优选的。
[0163]
药物组合物可以冻干形式提供。冻干的药物组合物在施用前优选地在合适的缓冲液中重构,有利地基于水性载体。
[0164]
本发明的药物组合物还提供用于制备用于减少放射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用的药物。
[0165]
该药物组合物或药物优选地用于减少放射标记的治疗性和诊断性化合物的肾毒性副作用,用于成像或治疗疾病,特别是肿瘤疾病,这类神经内分泌肿瘤、前列腺癌、胰腺癌、肾癌、膀胱癌、脑癌、胃肠道癌、甲状腺髓样癌、小细胞或非小细胞肺癌、和间质卵巢癌、胰腺导管腺癌、胰岛素瘤、胃泌素瘤、乳腺癌、或肉瘤。
[0166]
试剂盒
[0167]
根据进一步方面,本发明提供了试剂盒,其包括根据本发明使用的药物成分,如对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物、如上指定的放射标记的或非放射标记的治疗性或诊断性化合物、和/或根据本发明的药物组合物。例如,在实施方式中,在试剂盒的一部分中,试剂盒可以包括对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物,并且在试剂盒的另一部分中可以包括如上所指定的根据本发明的药物组合物,或在试剂盒的另一部分中包括用于可溶性pah的溶液或药学上可接受的盐或羧酸衍生物的溶液。溶液可以是等渗的或高渗的,它可以是缓冲的,如任选地缓冲水溶液,如nacl水溶液或注射用水(wfi)。在本发明的另一实施方式,试剂盒在试剂盒的一部分中可以包括对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物,和在试剂盒的另一部分中包括如上指定的放
射标记的和/或非放射标记的治疗性或诊断性化合物。
[0168]
任选地,试剂盒可包括至少一种如本文在药物组合物上下文中定义的其他试剂,包括如氨基酸,诸如赖氨酸和精氨酸及其混合物、明胶、氨磷汀、白蛋白衍生的肽、psma结合分子(如pmpa)、维生素、放射性核素、抗微生物剂、增溶剂等。
[0169]
试剂盒可以是包含在合适容器中的以上例举的任何组分的两部分或更多部分的试剂盒。例如,每个容器可以是小瓶、瓶子、挤压瓶、广口瓶、密封套管、包膜或囊、管或泡罩包装的形式或任何其他合适的形式,条件是容器优选地防止组分过早混合。每个不同的组分可以单独提供,或者一些不同的组分可以一起提供(即在同一容器中)。
[0170]
容器也可以是小瓶、管子、广口瓶、或包膜、或套管、或泡罩包装或瓶子内的隔室或腔室,条件是一个隔室的内容物不能在药剂师或医生有意混合之前与另一隔室的内容物物理相关联。
[0171]
试剂盒或部分试剂盒还可包含技术说明书,其中包含有关其任何组分的施用和剂量的信息。
[0172]
治疗和诊断方法和用途
[0173]
根据进一步方面,本发明涉及对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物,和/或如上所述的药物组合物,和/或如上所述的试剂盒用于制备对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物,和/或如上所述的药物组合物用于减少受试者中放射标记的和非放射标记的治疗和诊断化合物的肾毒性副作用的药物的用途。
[0174]
还涉及如上所述的药物组合物或如上所述的试剂盒,其用于减少受试者中放射标记的和非放射标记的治疗和诊断化合物的肾毒性副作用的方法中。
[0175]
在进一步方面,本技术还提供了减少受试者中放射标记的和非放射标记的治疗和诊断化合物的肾毒性副作用的方法,该方法包括在使用放射标记的和/或非放射标记的化合物的成像或疗法期间,向受试者施用如上所述的药物组合物或如上所述的试剂盒。
[0176]
在进一步的方面,本技术还提供了减少受试者中放射标记的和非放射标记的治疗和诊断化合物的肾毒性副作用的方法,该方法包括在受试者中施用对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物联合放射标记的或非放射标记的治疗或诊断化合物,其中pah的施用在放射标记的或非放射标记的治疗或诊断化合物的施用之前和/或期间和/或之后。
[0177]
在优选的实施方式中,该方法用于减少在放射配体疗法或诊断中的受试者中放射药物的肾毒性副作用。优选地,该放射药物室是包括以上指定的载体分子、螯合剂和放射性核素的放射性核素络合物。在更优选的实施方式中,载体分子选自肽、拟肽、抗体片段、抗体模拟物、小分子、结,其可具有细胞受体,特别是细胞表面受体的激动或拮抗配体的性质。在特别优选的实施方式中,载体分子选自以上指定的促生长素抑制剂类似物、psma-抑制剂、胃泌素类似物、整联蛋白结合分子,并且例如选自tyr3-奥曲肽、tyr3-奥曲酸盐、jr11、psma-11、sargastrin、rgd。
[0178]
在进一步实施方式中,用于本发明的方法中的放射药物化合物的螯合剂选自dota、hbed-cc、nota、nodaga、dotaga、dotam、trap、nopo、pcta、dfo、dtpa、do3ap、do3ap
pra
、do3ap
abn
、hynic或其衍生物。
[0179]
在进一步优选的实施方式中,用于本发明的方法中的放射药物化合物的放射性核
素选自
94
tc、
99m
tc、
90
in、
111
in、
67
ga、
68
ga、
86
y、
90
y、
177
lu、
161
tb、
186
re、
188
re、
64
cu、
67
cu、
55
co、
57
co、
43
sc、
44
sc、
47
sc、
225
ac、
213
bi、
212
bi、
212
pb、
227
th、
153
sm、
166
ho、
166
dy、
18
f和
131
i,并且更优选地选自
177
lu、
225
ac和
68
ga。
[0180]
在本发明的进一步优选实施方式中,用于本发明的方法中的包含缀合物分子的放射性核素选自[
177
lu-dota
°‑
tyr3]-奥曲肽、
177
lu-dota-ja11、
177
lu-dota-rgd、
177
lu-dota-sargastrin、
68
ga-hbed-cc-psma-11、psma11、
177
lu-psmai&t和
99m
tc-etarforlatide。
[0181]
在本发明的特别优选的实施方式中,用于本发明的方法中的pah的药学上可接受的盐是氨基马尿酸钠。
[0182]
在本发明的方法中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)优选地以足以有效减少治疗性和/或诊断性化合物的肾毒性副作用的量施用,该化合物通常平行施用至受试者。可以通过常规实验确定pah的有效量,如通过使用动物模型。此类模型包括但不限于兔、绵羊、小鼠、大鼠、狗和非人灵长类动物模型。
[0183]
比如,pah的施用量的范围可以(每千克体重)约0.1mg/kg至10g/kg,优选地约0.5mg/kg至5g/kg,更优选地约1mg/kg至1g/kg。
[0184]
在本发明的特别优选的方法中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以每千克体重约5mg至约500mg的量进行施用,通常平行施用(如在施用肾毒性治疗或诊断化合物之前、并行或之后),例如以每千克体重约5、10、20、30、40、50、60、70、80、90、100、150、200、250、300、350、400、450、500mg的量。因此,本文举例说明的量可以在同一天给出(如,与诊断/治疗肾毒性化合物在同一天给出)。pah或其盐或羧酸衍生物的施用方案通常遵循肾毒性化合物的施用方案。更优选地,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下的量进行施用:每千克体重约50mg至约500mg,更优选地,每千克体重约50mg至约250mg,例如每千克体重约50、55、60、65、70、75、80、85、90、95、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240或250mg。更优选地,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下量进行施用:每千克体重约75mg至约200mg,例如每千克体重约75、80、85、90、95、100、105、110、115、120、125、130、135、140、145、150、155、160、165、170、175、180、185、190、195、或200mg的量。最优选地,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下量进行施用:每千克体重约80mg至约160mg,例如每千克体重约80、85、90、95、100、105、110、115、120、125、130、135、140、145、150、155、160mg的量。
[0185]
一般地,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以比(共施用的)治疗或诊断化合物更大的量进行施用。
[0186]
比如,治疗性或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例进行施用:约1/1.000.000至1/10(w/w),优选地约1/500.000至1/100(w/w),更优选地约1/250.000至约1/500(w/w)。
[0187]
在本发明的优选方法中,治疗性或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例进行施用:约1/250.000至约1/5.000(w/w),例如约1/250.000、1/200.000、1/150.000、1/100.000、或1/50.000至约1/5.000(w/w)的比例,更优选地约1/240.000至约1/8.000(w/w)的比例,例如约1/240.000、1/230.000、1/220.000、1/210.000、1/200.000、1/190.000、1/180.000、1/170.000、1/160.000、1/
150.000、1/140.000、1/130.000、1/120.000、1/110.000、1/100.000、1/90.000、1/80.000、1/70.000、1/60.000、1/50.000、1/40.000、1/30.000、1/20.000、1/19.000、1/18.000、1/17.000、1/16.000、1/15.000、1/14.000、1/13.000、1/12.000、1/11.000、1/10.000、1/9.000、或1/8.000(w/w)的比例。在进一步优选方法中,治疗性和/或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例进行施用:约1/100.000至约1/10.000(w/w),例如约1/100.000、1/95.000、1/90.000、1/85.000、1/80.000、1/75.000、1/70.000、1/65.000、1/60.000 1/55.000、1/50.000、1/45.000、1/40.000、1/35.000、1/30.000、1/25.000、1/20.000、1/15.000、1/10.000(w/w)的比例。在进一步优选的方法中,治疗性和/或诊断性化合物和pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)以如下比例进行施用:约1/50.000至约1/40.000(w/w),例如约1/50.000、1/49.000、1/48.000、1/47.000、1/46.000、1/45.000、1/44.000、1/43.000、1/42.000、1/41.000、1/40.000(w/w)的比例。
[0188]
在本发明的另一实施方式中,对氨基马尿酸(pah)或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)在本发明的方法中联合减少放射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用的进一步物质进行施用。在优选实施方式中,除了pah外的减少放射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用的物质选自氨基酸、明胶、氨磷汀、白蛋白衍生的肽、psma-结合分子(诸如pmpa)、维生素。除了pah外的减少放射标记的和非放射标记的治疗性和诊断性化合物的肾毒性副作用的物质可以在pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)的施用之前和/或期间和/或之后进行施用。
[0189]
本发明的药物组合物或试剂盒可以一天、每天、每隔一天、每周或每月几次施用于有需要的受试者。优选地,治疗、诊断或预防是用有效剂量的本发明药物组合物或试剂盒实现的。
[0190]
在本发明的方法中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)可以分别在放射标记的或非放射标记的治疗性或诊断性化合物、药物组合物或试剂盒的施用之前和/或期间和/或之后施用至受试者。例如,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)在放射标记的或非放射标记的治疗性或诊断性化合物、药物组合物或试剂盒的施用之前施用。在本发明的优选方法中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)在如上定义的本发明的药物组合物或试剂盒的施用之前施用,即,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)。pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)可以在放射标记的或非放射标记的治疗性或诊断性化合物的施用之前和/或期间和/或之后交替地施用。可以在治疗性或诊断性化合物、药物组合物或试剂盒的施用之前60min、30min、10min或5min进行pah的预施用,优选地pah分别在治疗性或诊断性化合物、药物组合物或试剂盒的施用之前约0.5-5h或10-60分钟进行施用。在某些实施方式中,pah可以在治疗性或诊断性化合物的施用之前以及其后进行施用,如在其后0.5-5h或10-60min,或在之前、期间和其后。
[0191]
在本发明的方法中,pah或其药学上可接受的盐或羧酸衍生物(优选地氨基马尿酸钠)优选地以缓冲的水溶液施用,如等渗或高渗溶液,例如注射用水(wfi)或nacl溶液,如,20%nacl溶液。
[0192]
在本发明的方法中,pah、药物组合物或试剂盒通常肠胃外施用。可以优选地全身性完成施用,例如通过血管内、静脉内(i.v.)、皮下、肌肉内或皮内注射。可选地,可以局部完成施用,例如通过肿瘤内注射。
附图说明
[0193]
图1显示了利用氨基酸和pah的共输注随时间的[
177
lu-dota
°‑
tyr3]-奥曲肽的肾脏吸收,其中使用0.9%nacl作为对照。
[0194]
图2显示了利用氨基酸和pah的共输注随时间的相比基线(0.9%nacl输注)的肾脏中[
177
lu-dota
°‑
tyr3]-奥曲肽吸收的百分比减少。
[0195]
图3显示了在0.5h p.i.的早期时间点处与0.9%nacl、lys/arg和pah的共注射在肾脏中[
177
lu-dota
°‑
tyr3]-奥曲肽的百分比注射剂量。
[0196]
图4显示了在24h p.i.的晚时间点处与0.9%nacl、lys/arg和pah的共注射在肾脏中[
177
lu-dota
°‑
tyr3]-奥曲肽的百分比注射剂量。
[0197]
图5显示了5min和60min p.i.时[
177
lu-dota
°‑
tyr3]-奥曲肽的离体器官吸收(n=5)。pah显著地减少肾脏中的吸收,即使在早期时间点(**p≤0.005,****p≤0.0005)。
[0198]
图6显示了分别利用与pah(200mg/ml)和0.9%nacl的共注射,在不同的放射标记的(lu-177)试剂(dota-rgd、affilin、dota-sargastrin、dota-jr11)注射(0.5、1、4、24h)后不同的时间点处在肾脏中存在的注射的放射性的百分比。
[0199]
图7显示了分别利用与pah(200mg/ml)和0.9%nacl的共注射,在不同的放射标记的(lu-177)试剂(dota-rgd、affilin、dota-sargastrin、dota-jr11)注射(0.5、1、4、24h)后不同的时间点处在血液中存在的注射的放射性的百分比。
[0200]
图8显示了分别利用与pah(200mg/ml)和0.9%nacl的共注射,在
68
ga-标记的hbed-cc-psma-11注射(0.1,0.5,1,2,4h)后在不同时间在肾脏(上图)和血液(下图)中存在的注射的放射性的百分比。
[0201]
图9图形地显示了在腹膜内注射pah或生理盐水溶液后静脉内施用至小鼠的
177
lu-dotatoc的生物分布分析结果。
[0202]
图10图形地显示了在腹膜内注射pah或生理盐水溶液后静脉内施用至小鼠的
99m
tc-etarfolatide的生物分布分析结果。
[0203]
图11显示了注射了
177lu-psmai&t、注射了盐水ve、4-(二丙基氨磺酰基)苯甲酸kp2或pah kp1溶液的三组大鼠的比较柱状图,以肾脏中注射的放射性的百分比表示。
[0204]
图12显示了注射了
177lu-psmai&t、注射了盐水ve、4-(二丙基氨磺酰基)苯甲酸kp2或pah kp1溶液的三组大鼠的比较柱状图,以左心室中注射的放射性的百分比表示。
[0205]
图13显示了注射了
177lu-psma i&t、注射了盐水ve、4-(二丙基氨磺酰基)苯甲酸kp2或pah kp1溶液的三组大鼠的比较柱状图,以左肾髓质中注射的放射性的百分比表示。
[0206]
图14显示了注射了
177lu-psmai&t、注射了盐水ve、4-(二丙基氨磺酰基)苯甲酸kp2或pah kp1溶液的三组大鼠的比较柱状图,以肾皮质中注射的放射性的百分比表示。
实施例
[0207]
以下数据表明,与氨基马尿酸钠溶液(pah)联合用药对减少各种放射性药物(分别
包括各种载体分子、各种螯合剂和各种放射性核素)的肾潴留和吸收具有显著作用,并且pah施用比赖氨酸和精氨酸输注更有效。此外,发现放射性药物的生物利用度提高,并且通过与pah联合用药增加了放射性药物的肿瘤吸收。
[0208]
实验方案(实施例1和2):
[0209]
spect/ct实验:
[0210]
通过单光子发射计算机断层扫描(spect)在健康wistar大鼠中使用[177lu-dota
°‑
tyr3]-奥曲肽在每肾脏保护剂的每组3只动物的六个组中获得的三维放射性图像中的目标体积(voi)的肾脏生物分布确定。50mbq的[177lu-dota
°‑
tyr3]-奥曲肽与注射用水中的200mg/ml氨基酸溶液(l-arg和l-lys)、注射用水中的200mg/ml的对氨基马尿酸钠溶液(pah)和作为参比的nacl 0.9%溶液共施用。在0.5h,1h,4h,8h,24h p.i.之后进行成像采集。
[0211]
离体器官分布:
[0212]
所有动物在放射性示踪剂注射前10min预先注射1.0ml nacl、arg-lys或pah。用于生物分布的注射溶液制备为12μl的[
177
lu]lu-dota-toc、1.5ml的nacl或arg-lys或pah的混合物。
[0213]
用于spect动物的注射溶液制备为80μl[177lu]lu-dota-toc、1.5ml nacl或arg-lys或pah的混合物。5min或60min后注射并处死动物。收集目标器官和组织并测量其活性。雄性wistar大鼠的平均体重为210
±
12g(5min)和218
±
13g(60min)。平均注射活性为对于spect,4.49
±
0.38mbq/kg体重(5min)和159
±
13mbq/kg体重(5min),和对于spect动物,4.19
±
0.57mbq/kg体重(60min)或139
±
8mbq/kg体重(60min)。
[0214]
使用microsoft excel 2010和graphpad prism 6.05进行统计分析。数据表示为中位数和[25%和75%百分位数]以及平均值和标准偏差(sd)或平均值的标准误差(sem)。为了比较,采用普通单向方差分析和tukey多重比较检验,两样本双尾t检验。以p《0.05为接受的显著性。
[0215]
实施例1:[
177
lu-dota
°‑
tyr3]-奥曲肽的肾脏吸收
[0216]
[
177
lu-dota
°‑
tyr3]-奥曲肽的肾脏吸收通过定量分别共输注0.9%nacl(对照)、lys/arg(250mg lys/250mg arg)和pah(500mg)的大鼠中的小动物spect来确定。在表1至3中呈现了结果。
[0217]
表1:用0.9%nacl(对照)共输注的肾脏放射性[以注射的放射性百分比表示,衰减校正数据]
[0218][0219]
表2:用lys/arg(250mg lys/250mg arg)共输注的肾脏放射性[以注射的放射性百分比表示,衰减校正数据]
[0220][0221]
表3:用pah(500mg)共输注的肾脏放射性[以注射的放射性百分比表示,衰减校正数据]
[0222][0223]
表1至3中显示的值在图1至4中以图形方式示出。
[0224]
从所呈现的结果中可以明显看出,lys/arg的共同输注导致肾脏吸收在8小时和24小时后分别减少21%和26%,并且个体之间差异大(表2、图1、2和4)。氨基酸输注的结果与文献中报道的结果相似(33% /-23%,rolleman ej等人,同上)。相比之下,pah的共同输注导致在同一时间点降低56%和50%,标准偏差低(表3,图1和2)。输注pah还导致早期时间点肾脏吸收降低。在注射后0.5小时,lys/arg混合物仅显示10%的轻微减少,相比之下pah显示50%的减少(表2和3,图1-3)。
[0225]
实施例2:[
177
lu-dota
°‑
tyr3]-奥曲肽的离体器官吸收
[0226]
根据上述方案,在5min和60min p.i.的早期时间点确定[
177
lu-dota
°‑
tyr3]-奥曲肽的离体器官浓度。结果在图5中以图形表示。即使在早期时间点,与lys/arg相比,当使用pah-输注作为联合药物时,[
177
lu-dota
°‑
tyr3]-奥曲肽的吸收相对于nacl输注显著降低(p 0.005)。
[0227]
综上所述,上述结果表明,与现有技术中已知的赖氨酸和精氨酸的联合相比,氨基马尿酸钠溶液在减少肾潴留和[177lu-dota
°‑
tyr3]-奥曲肽的吸收方面具有显著更高的效果。
[0228]
实验方案(实施例3-5)
[0229]
成像研究旨在评估pah在减少由不同结构的螯合剂和结合支架配位的
177
lu或
68
ga标记的肽的肾吸收方面的有效性。放射标记的测试化合物的示踪剂分布在两组不同的健康wistar大鼠中进行,重点是肾吸收/清除和血液水平。一组施用
177
lu/
68
ga示踪剂与盐水组合作为对照(单独限定mbq/kg和mg/kg),而另一组首先接受pah腹腔注射(注射前10min)。然后施用
177
lu/
68
ga-肽以及两次静脉注射pah溶液。使用pet或spect评估每种
177
lu/
68
ga-肽(在有或没有预先给药pah的情况下施用)的肾脏清除率和总体药代动力学。每组包括至少3只大鼠用于统计事项。
[0230]
177
lu-肽:由itg提供的
177
lu标记的化合物在收到时使用itlc进行放射化学纯度测试。为每种化合物提供了测试参数,rcp》95%。表4总结了要研究的每种
177
lu-肽的标称剂量
水平、浓度和体积。
[0231]
表4
[0232][0233]
68
ga-肽:
68
ga标记的化合物在应用前直接制备,并使用itlc测试其放射化学纯度。(为每种化合物提供了标记和qc测试参数,rcp》95%)。表5总结了要研究的每种
68
ga-肽的标称剂量水平、浓度和体积。
[0234]
表5
[0235][0236]
实施例3:通过同时施用pah减少各种放射标记的化合物的肾脏吸收
[0237]
给小鼠注射四种不同的分子,这些分子与环状螯合剂dota缀合并放射标记有治疗性放射性核素镏-177:分别为
177
lu-dota-rgd、
177
lu-dota-affilin、
177
lu-dota-sargastrin和
177
lu-dota-jr11,同时共注射pah(200mg/ml)或作为对照的盐水(0.9%nacl)。根据上述方案确定随着时间的推移存在于肾脏中的注射放射性的百分比。结果在图6中图形显示。
[0238]
根据图6,pah与所有测试的化合物,尤其是小肽,诸如dota-rgd、dota-jr11和dota-sargastrin同时施用,在减少肾脏吸收方面有显著效果。从图6还可以明显看出,在施用后0.5至1小时的早期时间点,肾脏吸收减少的效果已经存在。
[0239]
因此,上述实验结果表明,pah的施用降低了通过不同机制在肾脏中积累的各种放射标记的化合物即dota连接的肽、拟肽等的肾脏吸收。
[0240]
实施例4:pah对不同放射性标记化合物的血液活性的影响
[0241]
给小鼠注射四种不同的分子,这些分子与环状螯合剂dota缀合并放射标记有治疗性放射性核素镏-177:分别为
177
lu-dota-rgd、
177
lu-dota-affilin、
177
lu-dota-sargastrin、和
177
lu-dota-jr11,同时共注射pah(200mg/ml)或作为对照的盐水(0.9%nacl)。根据上述方案确定随着时间的推移存在于血液中的注射放射性的百分比。结果在图7中图形显示。
[0242]
根据图7,通过对affilin共注射pah血液活性显著增加。因此,虽然与其他测试的化合物相比,通过pah,affilin的肾脏吸收没有那么显著降低(参见图6),但数据表明,同时通过共注射pah会增加affilin的血液活性,从而提高放射性药物的生物利用度。
[0243]
实施例5:pah对
68
ga-标记的psma-11的肾脏吸收和血液活性的影响
[0244]
给小鼠注射与环状螯合剂hbed-cc缀合的诊断性
68
ga-标记的psma-11,并且共注射pah(200mg/ml)或作为对照的盐水(0.9%nacl)。根据上述方案确定随着时间的推移存在于肾脏(上图)和血液(下图)中的注射放射性的百分比。结果在图8中图形显示。
[0245]
如图8所示,通过pah的同时施用存在
68
ga-标记的hbed-cc缀合的psma-11的肾脏吸收的显著减少。从图8也可以明显看出,在施用后0.1、0.5和1小时的早期时间点,肾脏吸收减少的效果已经存在。
[0246]
因此,上述实施例3-5的实验结果表明,pah的施用减少了各种放射标记的化合物(分别具有不同载体分子(肽、拟肽、重组蛋白)、不同螯合剂(环状螯合剂、无环螯合剂)、和不同放射性核素(治疗性放射性核素、诊断性放射性核素))的肾脏吸收,并因此可适用于减少多种放射标记和非放射标记的诊断性和治疗性化合物的肾毒性副作用。
[0247]
实施例6:腹腔注射pah或生理盐水溶液后向小鼠静脉给予
177
lu-dotatoc的生物分布比较分析
[0248]
促生长素抑制剂受体阳性胰腺肿瘤荷载cd1裸鼠(ar2j)接收i.p.注射50μl nacl0.9%(a组)或50μl pah 20%(b组),精确地10分钟后通过眶后静脉窦i.v.注射
177
lu-dotatoc/nacl(a组)或
177
lu-dotatoc/pah(b组)。表6中总结了标称剂量水平、浓度和体积
[0249]
表6
[0250][0251]
*使用治疗当天记录的个体体重计算个体剂量体积,记录个体体积和确切治疗时
间并保存在研究文件中。
[0252]
**考虑到
177
lu从递送之日起3天内的放射性衰变的浓度和剂量范围。
[0253]
小鼠(每组3只动物)分别在0.5小时、1小时、2小时和4小时处死。器官在0.9%nacl中快速冲洗并干燥,然后称重和计数以消除可能的污染血液。对以下器官/组织进行取样或取材,称重并计数
177
lu:血液、肿瘤、肾脏、肝脏、膀胱(空)、心脏、脾脏、肺、脑、肌肉、胃(无内容物)、小肠(无内容物)、结肠(无内容物)残留尸体。来自器官/组织计数的数据表示为注射剂量的百分比(%id/g))。
177
lu-dotatoc在0.9%nacl中的器官/组织分布结果(a组),以及
177
lu-dotatoc与pah的器官/组织分布结果(b组)在图9中图形化呈现。
[0254]
根据图9,相比于接收nacl中的
177
lu-dotatoc的a组(上图),接收
177
lu-dotatoc与pah的b组(下图)中肾脏中的
177
lu-dotatoc吸收显著减少。在注射
177
lu-dotatoc后的早期阶段(0.5小时、1小时),放射标记化合物的肾吸收减少尤为明显。同时,与a组相比,肿瘤和血液中b的
177
lu-dotatoc水平显著地增加。不希望受具体理论的束缚,假设通过pah经由肾脏清除
177
lu-dotatoc的阻断导致
177
lu-dotatoc的血液循环延长,并从而导致放射标记的化合物在肿瘤部位的积累也增加。综上所述,结果显示了,相比于接收nacl中的
177
lu-dotatoc的a组,接收
177
lu-dotatoc与pah的
177
lu-dotatoc的生物利用度增强。
[0255]
实施例7:
99m
tc-etarfolatide与pah或生理盐水联合给予的生物分布比较分析
[0256]
7.1etarfolatide的放射性标记
[0257]
99m
tc-etarfolatide具有以下化学结构:
[0258][0259]
放射性标记是通过源自kim等人的教导的方法进行的(ann nucl med 2016;30:369-379)。通过使用酒石酸盐作为共配体采用配体交换方法。在eppendorf管中,添加100μg etarfolatide、50μl酒石酸盐溶液(在微孔水中20mg/50μl)和80μl的sncl2二水合物溶液(在0.01m hcl溶液中1mg/ml)。在铅屏蔽通风橱中,添加约750mbq(20mci)新洗脱的
99m
tc-高锝酸盐,并将反应瓶在100℃的水浴中加热30分钟,然后冷却至室温。
[0260]
使用以下确定放射性标记效率和稳定性:(i)即时薄层色谱——硅胶(itlc-sg),以水(
99m
tc-etarfolatide和游离高锝酸盐的rf=0.9
–
1.0;胶体的rf=0.0
–
0.1)以及丙酮(游离高锝酸盐的rf=0.9
–
1.0;胶体和
99m
tc-etarfolatide的rf=0.0
–
0.1)作为流动相,和(ii)rp-hplc:溶剂a:h2o中0.1%tfa。溶剂b:accn中0.1%tfa;梯度洗脱,流速1ml/min。
[0261]
在实验的每一天,都使用大约相同的
99m
tc活性进行新的放射性标记程序。rp-hplc显示rcp=~100%,而tlc显示最大胶体形成为5%或更少。在制备后24小时通过hplc评估
99m
tc-etarfolatide的稳定性,并显示放射性标记没有损失。
[0262]
放射标记的叶酸酯的体积测量为1300μl,而活性为734mbq(实验日:3月4日)。制备了相当于300mbq(即15mbq x 20只小鼠)的样品。531μl取自放射标记的叶酸酯,其用1469μl
生理盐水或1469μl pah(2000μl 99mtc-etarfolatide,100μl/15mbq/小鼠)稀释。注射前,样品通过0.22m无菌过滤器进行过滤。注射的15mbq/小鼠对应于2.04μg etarfolatide/小鼠(在整个实验过程中etarfolatide的注射量保持稳定)。
[0263]
用生理盐水或pah稀释的
99m
tc-etarfolatide的样品在制备后立即以及稀释后24小时进行评估,并且当通过hplc(100%rcp)评估时,没有显示放射性标记丢失的迹象,但是tlc评估表明
99m
tc-etarfolatide/pah是稳定的(100%rcp),而
99m
tc-etarfolatide/盐水显示14%胶体形成。
[0264]
7.2pah溶液的制备
[0265]
在corning管中称取2g pah钠盐;添加4ml h2o;涡旋
–
pah部分溶解;添加20μl naoh(itg提供),涡旋
–
pah部分溶解;添加20μl naoh,涡旋
–
pah完全溶解;添加2ml h2o,ph 10;添加80μl hcl 37%(以20μl为增量),ph~6。测量所得溶液的最终体积,发现为7100μl,我们向其中添加了2900μl(所有这些都在另一corning管中),最终得到在10ml h2o中的2g pah,即200mg pah/ml。通过0.22μm过滤器过滤后,将corning管用铝箔覆盖并冷却。
[0266]
7.3生物分布分析
[0267]
30小鼠(雄性和雌性)随机指定为两组。
[0268]
a组:在已经接受ip注射pah 10min,然后示踪剂注射后,将小鼠注射pah(2.04μg etarfolatide/小鼠)中稀释的
99m
tc etarfolatide。评估5个时间点:5min、30min、1h、2h、4h(3小鼠/时间点)。
[0269]
每个时间点注射的剂量:
[0270][0271]
b组:在已经接受ip注射盐水10min,然后示踪剂注射后,将小鼠注射在盐水(2.04μg etarfolatide/小鼠)中稀释的
99m
tc etarfolatide。评估5个时间点:5min、30min、1h、2h、4h(3小鼠/时间点)。
[0272]
每个时间点注射的剂量:
[0273][0274]
生物分布分析的结果如图10所示。30分钟后,通过pah的保护作用,
99m
tc-etarfolatide的肾吸收减少变得突出。因此,对于具有不同于三价
177
lu的另一种化合价状态的
99m
tc的
99m
tc螯合缀合物分子,证实了对基于
177
lu的缀合物分子所观察到的影响。
[0275]
实施例8:
177
lu-标记的psma i&t的生物分布分析
[0276]“psma i&t”的化学结构如下:
[0277][0278]
用于成像实验的测试制品
[0279][0280]
成像实验的测量系统和参数设置
[0281][0282]
方案执行
[0283]
该方案以如下方式执行:测试一组注射ta以及在三个独立的组中注射不同kp试剂的动物组的肾脏中的放射性浓度。因此,形成了以下实验组矩阵
[0284][0285]
1.psma i&t-4-(二丙基氨磺酰基)苯甲酸成像实验的数据
[0286]
1.1.大鼠1
[0287]
[0288][0289]
1.2.大鼠2
[0290][0291]
1.3.大鼠3
[0292]
[0293][0294]
2.psma i&t-pah成像实验的数据
[0295]
2.1大鼠4
[0296][0297]
2.2大鼠5
[0298][0299][0300]
2.3大鼠6
[0301][0302]
3.psma i&t-盐水成像实验的数据
[0303]
3.1大鼠7
[0304][0305]
3.2大鼠8
[0306][0307]
3.3大鼠9
[0308][0309]
结果
[0310]
pe1:毒性观察
[0311]
结果:没有观察到急性立即和迟发急性毒性的迹象。
[0312]
pe2:在器官中随时间的放射性积累
[0313][0314]
结果:下表每个时间点包含两个肾脏总和中衰减校正的放射性浓度比,以注射全身放射性浓度的%表示。
[0315]
结果-表i:
[0316]
在注射
177lu-psma i&t并接受4-(二丙基氨磺酰基)苯甲酸悬浮液po的组中以注射的放射性的百分比表示的肾脏放射性
[0317][0318]
结果-表ii:在注射
177lu-psmai&t并接受pah溶液ip.和iv.的组中以注射的放射性的百分比表示的肾脏放射性
[0319][0320]
结果-表iii:在注射
177lu-psmai&t并接受盐水ip.和iv.的组中以注射的放射性的百分比表示的肾脏放射性
[0321][0322]
结果-表iv:在注射
177lu-psma i&t并接受4-(二丙基氨磺酰基)苯甲酸悬浮液po.的组中以注射的放射性的百分比表示的血液(左心室)放射性
[0323][0324]
结果-表v:在注射
177lu-psmai&t并接受pah溶液ip.和iv.的组中以注射的放射性的百分比表示的血液(左心室)放射性
[0325][0326]
结果-表vi:在注射
177lu-psmai&t并接受盐水ip.和iv.的组中以注射的放射性的百分比表示的血液(左心室)放射性
[0327][0328]
结果-表vii:
[0329]
在注射
177lu-psmai&t并接受4-(二丙基氨磺酰基)苯甲酸悬浮液po.的组中以注射的放射性的百分比表示的肾髓质放射性
[0330][0331]
结果-表viii:
[0332]
在注射
177lu-psmai&t并接受pah ip.和iv.的组中以注射的放射性的百分比表示的肾髓质放射性
[0333][0334]
结果-表ix:
[0335]
在注射
177lu-psmai&t并接受盐水ip.和iv.的组中以注射的放射性的百分比表示的肾髓质放射性
[0336][0337]
结果-表x:
[0338]
在注射
177lu-psmai&t并接受4-(二丙基氨磺酰基)苯甲酸悬浮液po.的组中以注射的放射性的百分比表示的肾皮质放射性
[0339][0340]
结果-表xi:
[0341]
在注射
177lu-psmai&t并接受pah溶液ip.和iv.的组中以注射的放射性的百分比表示的肾皮质放射性
[0342][0343]
结果-表xii:
[0344]
在注射
177lu-psmai&t并接受盐水ip.和iv.的组中以注射的放射性的百分比表示的肾皮质放射性
[0345][0346]
结果由图11至14描述。与盐水对照相比以及与4-(二丙基氨磺酰基)苯甲酸对照实验相比,所有实验显示在施用pah后肾脏细胞中的放射性显著降低(图11)。在施用pah后,肾皮质(图13)和肾髓质(图14)在实验过程中的所有时间点都显示出降低的放射性。
[0347]
测试了4-(二丙基氨磺酰基)苯甲酸作为进一步的对照。4-(二丙基氨磺酰基)苯甲酸被认为是肾脏的近端肾小管细胞oa
–
(有机阴离子)分泌的强抑制剂。它抑制细胞基底外侧的1型有机阴离子转运蛋白(oat1)。oat1以其从血液中吸收有机阴离子进入肾小管细胞与二羧酸盐(如琥珀酸盐或2-氧代戊二酸盐)交换而闻名。根据实施例8的实验(参见图11至14),均显示基于4-(二丙基氨磺酰基)苯甲酸的抑制机制对oat1没有影响。因此,可以得出结论,降低肾脏细胞的放射性的基于pah的作用是基于另一种机制,而不是针对4-(二丙基氨磺酰基)苯甲酸的已知机制。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。