1.本发明涉及煤化工领域,具体而言,是一种采用固相萃取方法将煤基柴油样品分离为饱和烃(烷烃 环烷烃)、烯烃、芳烃、杂原子化合物,以便为后续测定柴油族组成提供实验样品。
背景技术:
2.2019年我国原油对外依存度达到了70%以上,造成我国的能源安全存在较大的风险,对我国经济的稳定持续发展造成了威胁,因此我国加大了原油开采力度并提出了可行的原油替代策略。我国能源资源存在“富煤、贫油、少气”的特点,全力开发现代煤化工,大力发展基于煤炭的煤制油、煤制化工品工艺路线,生产满足工业生产所需要的清洁油品、天然气等清洁能源产品,延伸发展烯烃、芳烃、乙二醇等石油替代产品,是保障我国能源安全的重要安全举措[顾宗勤.中国现代煤化工产业进展[j].煤炭加工与综合利用,2016,199(04):6-10.]。
[0003]
虽然煤基油品与石油基油品在化合物种类上没有本质区别,但是由于工艺路线的差异,两者在诸如辛烷值与十六烷值等物性、化合物组成上存在较大差别[公磊,吴秀章,卢卫民,等.煤基高温费托合成技术进展[j].化工进展,2016,35(s1):122-129.]。为了更好地将煤基油品用作清洁燃料,需要首先对其族组成、分子组成进行研究,然后针对性开发加工工艺,调整油品分子组成,生产出质量合格的清洁油品。
[0004]
目前分析煤基油品族组成表征方法基本借鉴石油基油品族组成测定方法,没有考虑到两者分子组成的差异,因而造成了测定结果不能完整反映煤基油品实际组成,对于反应原理研究、工艺条件优化造成了困难。例如煤基柴油常采用基于石油基柴油制定的标准sh/t 0606测定其族组成,但是标准sh/t 0606要求柴油样品的非烃化合物含量不高于0.2wt%,否者会造成测定结果中芳烃化合物含量的可靠性变差;柴油样品的烯烃含量不高于5wt%,否则会造成测定结果中环烷烃含量偏高。因此标准sh/t 0606不适合直接用于煤基柴油族组成分析过程。
技术实现要素:
[0005]
本发明的目的是提供一种利用固相萃取技术将煤基柴油样品同时分离为饱和烃(烷烃 环烷烃)、烯烃、芳烃、杂原子化合物的预处理方法,可以用于煤基柴油样品中烯烃和杂原子化合物的富集纯化,排除其他烃类化合物对定性和定量分析的干扰。本发明也可以解决尚无煤基柴油样品族组成的预处理方法问题,为应用石化标准sh/t 0606准确测定煤基柴油样品族组成提供切实可行的预处理手段,扩大该标准的使用范围。
[0006]
本发明提供一种固相萃取分离柴油族组成的方法,包括如下步骤:
[0007]
步骤一:将固相萃取柱a与b串联,其中固相萃取柱a在上,采用第一洗脱剂润湿固相萃取柱a,从固相萃取柱a上部加入柴油样品,然后用第一洗脱剂冲洗固相萃取柱;
[0008]
步骤二:将固相萃取柱a与b分开,继续用第一洗脱剂冲洗固相萃取柱b,得到饱和
烃组分;
[0009]
步骤三:用第二洗脱剂冲洗固相萃取柱b,得到烯烃组分;
[0010]
步骤四:用第三洗脱剂冲洗固相萃取柱a,得到芳烃组分;
[0011]
步骤五:用第四洗脱剂冲洗固相萃取柱a,得到杂原子化合物;
[0012]
其中,所述第一洗脱剂为选自由正戊烷、正己烷、正庚烷和石油醚组成的群组中的至少一种;
[0013]
所述第二洗脱剂为a与b的混合物,其中所述a为选自由二氯甲烷、氯仿、乙醚、苯和甲苯组成的群组的至少一种,所述b为选自由甲醇、乙醇、异丙醇、丙酮和丁酮组成的群组中的至少一种;
[0014]
所述第三洗脱剂为c与d的混合物,其中所述c为选自由正戊烷、正己烷、正庚烷和石油醚组成的群组中的至少一种,所述d为选自由二氯甲烷、氯仿、乙醚、苯和甲苯组成的群组中的至少一种;
[0015]
所述第四洗脱剂为选自由甲醇、乙醇、异丙醇、丙酮、丁酮组成的群组中的至少一种。
[0016]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述固相萃取柱a内装有固定相,所述固定相为硅胶和氧化铝的混合物,其中氧化铝的含量为80-100wt%。
[0017]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述固相萃取柱b内装有固定相,所述固定相为负载银的硅胶,其中银的负载量为5-15wt%。
[0018]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述硅胶的比表面积为500-850m2/g,孔体积为0.1-0.6ml/g,平均孔径为1-4nm。
[0019]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述氧化铝的比表面积为200-450m2/g,孔体积为0.05-0.4ml/g,平均孔径为1-3nm。
[0020]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述第二洗脱剂中a与b的体积比为1.5-19:1。
[0021]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述第三洗脱剂中c与d的体积比为1.5-9:1。
[0022]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所属步骤一种,柴油样品的加入量与固相萃取柱a的质量比0.07-0.7:1,柴油样品的加入量与固相萃取柱b的质量比为0.07-0.7:1。
[0023]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述步骤一中,润湿固相萃取柱a的第一洗脱剂的体积用量为0.5ml,冲洗固相萃取柱a与b的第一洗脱剂的体积用量为1.5-5ml,所述步骤二中,冲洗固相萃取柱b的第一洗脱剂的体积用量为2-4ml。
[0024]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述步骤三中,第二洗脱剂的体积用量为2-6ml。
[0025]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述步骤四中,第三洗脱剂的体积用量为2-6ml。
[0026]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述步骤五中,第四洗脱剂的体积用量为2-6ml。
[0027]
本发明所述的固相萃取分离柴油族组成的方法,其特征在于,所述柴油样品为煤
化工工艺的柴油。
[0028]
本发明所述的固相萃取分离煤基柴油族组成的方法,其特征在于,所述柴油样品为煤直接液化柴油、煤间接液化柴油。
[0029]
固相萃取是利用不同化合物在固定相上吸附能力的差异,通过调整洗脱剂极性将物质分离为极性不同的组分。因此,用于固相萃取的洗脱剂的选择非常重要,但即使将相同的溶剂以不同比例混合也会得到极性不同且溶剂选择性不同的洗脱剂,从而造成最终的分离结果有很大差别。煤基柴油中环烷烃与烯烃、芳烃与醚类化合物极性相近,很难将两组化合物很好地分离。此外,目前尚未有利用固相萃取来分离煤基柴油中的芳烃和杂原子化合物的相关报道。
[0030]
本发明还可详述如下:
[0031]
为实现上述目的,本发明提供一种固相萃取方法,通过固相萃取柱a与b的配合实现柴油样品中不同组分的分离。固相萃取柱a固定相为硅胶和氧化铝混合物,其中氧化铝的含量为80-100wt%;固相萃取柱b固定相为负载银的硅胶,其中银的负载量为5-15wt%。本发明可用于富集煤基柴油样品中的烯烃和杂原子化合物,解决其他烃类化合物对两种定性和定量的干扰,也可以解决尚无分离煤基柴油样品族组成预处理方法的问题,为应用石化标准sh/t0606准确定量煤基柴油样品族组成提供切实可行的预处理手段,扩大该标准的使用范围。
[0032]
上述硅胶和中性氧化铝固定相的制备方法为:将硅胶于80-160℃干燥2-6小时后放入干燥器冷却,得到活化的硅胶;将中性氧化铝于350-550℃焙烧1-5小时后放入干燥器冷却,得到活化的中性氧化铝。按照银的负载量为5-15wt%,将上述活化后的硅胶与硝酸银溶液等体积混合,静置24h后于80-160℃干燥2-6小时后放入干燥器冷却,得到活化负载银的硅胶。硅胶与氧化铝按照一定比例混合后得到固相萃取柱a的固定相,载银硅胶与氧化铝按照一定比例混合后得到固相萃取柱b的固定相。
[0033]
本发明固相萃取分离得到的溶液,经溶剂挥发浓缩后即可得到柴油的各组分,优先选用氮气吹扫进行产品浓缩工作。将溶液挥发浓缩至0.5ml左右即可进气相色谱(gc)进行相关分析,部分条件下也可不经过浓缩直接进色谱进行分析。优选采用气相色谱(gc)-质谱(ms)-氢火焰离子化检测器(fid)进行分析。质谱是对化合物结构定性的主要研究工具之一,通过谱图检索等手段可以对富集物中各种化合物进行定性分析。一般认为氢火焰离子化检测器对化合物的影响因子差别不大,因此通过gc-fid色谱图可对组分进行定量分析,判断分离交叉量的高低。取富集液注入气相色谱,气相色谱内色谱柱根据化合物的沸点和极性进行分离,然后进入检测器得到化合物的单体含量信息。定量可采用内标法,内标物优选氘代三联苯、氯苯。
[0034]
本发明采用固相萃取法将煤基柴油分离为不同组成的组分,通过调整洗脱剂极性与用量,将柴油样品分离为饱和烃(烷烃 环烷烃)、烯烃、芳烃、杂原子化合物四部分,组分间分离交叉较小。本发明可以解决采用石化标准0606分析煤基柴油时烯烃、杂原子化合物对烯烃、芳烃定量的干扰。与传统的柱色谱法相比,本发明所采用的固相萃取法柱效高、萃取柱装填重复性高、带压洗脱溶剂且溶剂用量少,因此方法重复性高、速度快。本发明一次可以同时分离富集柴油样品中烯烃与杂原子化合物,一次分离的时间仅仅为30min,大大提高了分离效率。
附图说明
[0035]
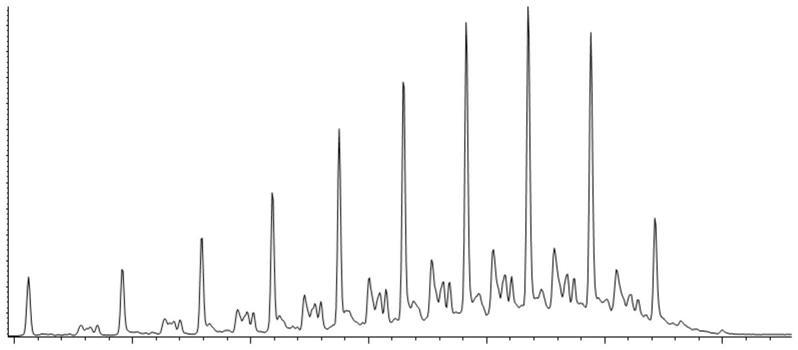
图1为应用本发明实施例1的预处理方法所获得的饱和烃(烷烃 环烷烃)组分的gc-ms图;
[0036]
图2为应用本发明实施例1的预处理方法所获得的烯烃组分的gc-ms图;
[0037]
图3为应用本发明实施例1的预处理方法所获得的芳烃组分的gc-ms图;
[0038]
图4为应用本发明实施例1的预处理方法所获得的杂原子化合物组分的gc-ms图。
具体实施方式
[0039]
以下对本发明的实施例作详细说明。本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
[0040]
下面结合具体实施例对本发明作进一步的具体说明,但本发明不受下述实施例的限制。任何不超出本发明的构思和范畴的改动,都在本发明的范围之内。
[0041]
实施例中所用的硅胶为国药集团化学试剂有限公司生产的层析用硅胶,颗粒度≥70.0%,比表面积为511.9m2/g,孔体积为0.468ml/g。中性氧化铝为国药集团化学试剂有限公司生产的层析用中性氧化铝,灼烧失重≤8.0%,比表面积为177.8m2/g,孔体积为0.255ml/g。
[0042]
将硅胶于140℃干燥5小时,得到活化的硅胶;中性氧化铝于500℃焙烧3小时,得到活化的中性氧化铝。按上述比例将活化的硅胶和中性氧化铝混合均匀,即得固相萃取柱a所用的中性氧化铝-硅胶固定相。按照银的负载量为5-15wt%,将上述活化后的硅胶与硝酸银溶液等体积混合,静置24h后于140℃干燥5小时后放入干燥器冷却,得到活化负载银的硅胶。载银硅胶与氧化铝按照一定比例混合后得到固相萃取柱b的固定相。
[0043]
分析所用的gc-ms仪器型号为7890agc-5975ms,带fid检测器。gc条件:hp-pona毛细管色谱柱,50m
×
0.2mm
×
0.5μm;程序升温初温80℃,保持5min后以40℃/min速率升温,终温300℃,保持9min;载气为高纯氦,恒压操作,压力为35.374psi;进样口温度320℃,分流比20:1,进样量1μl。msd条件:ei电离源(70ev),离子源温度230℃,四级杆温度130℃,全扫描质量范围30-500u,接口温度310℃,溶剂延迟5min。fid条件:检测器温度350℃,空气流量为300ml/min,氢气流量为30ml/min,尾吹气25ml/min。
[0044]
实施例1
[0045]
在固相萃取柱a内装填1.2g氧化铝固定相,固相萃取柱b内装填1.2g银含量为5wt%的载银硅胶固定相,用0.5ml第一洗脱剂(正戊烷)润湿固相萃取柱a。
[0046]
用移液枪取0.08g某公司高温费托合成1#柴油样品加入固相萃取柱a上部并被固定相完全吸附。用1.5ml第一洗脱剂冲洗固相萃取柱a b,然后分开固相萃取柱a与b,向固相萃取柱b继续加入2ml第一洗脱剂,得到饱和烃,记为组分1。用6ml第二洗脱剂(二氯甲烷与乙醇的体积比为90:10)冲洗固相萃取柱b,得到烯烃组分,记为组分2。用4ml第三洗脱剂(正戊烷与二氯甲烷的体积比为90:10)冲洗固相萃取柱a得到芳烃,记为组分3。用6ml第四洗脱剂甲醇冲洗固相萃取柱a,得到杂原子化合物,记为组分4。各组分加入一定量氘代三联苯作为内标,采用氮气吹扫去除各组分中的溶剂。
[0047]
采用gc-ms-fid分析组分1、组分2、组分3、组分4,结果表明,组分1主要为饱和烃,
占柴油样品的12.3wt%,其中杂质烯烃组分含量为0.3wt%;组分2主要为烯烃,占柴油样品的58.4wt%,其中杂质饱和烃含为0.1wt%,基本不含芳烃;组分3主要为芳烃,占柴油样品13.2wt%,杂质烯烃含量0.5wt%,含氧化合物含量0.2wt%;组分4主要为含氧化合物,占柴油样品14.9wt%,杂质芳烃含量0.1wt%。
[0048]
以癸烯-1、正辛醇的加标回收率考察固相萃取法分离柴油各组分的效果。取两份某公司低温费托合成1#柴油样品,其中一份加入一定量的癸烯-1、正辛醇作为加标样品。两份样品均按照上述固相萃取法将柴油分离为四部分,各加入一定量的氘代三联苯作为内标,分别进gc-fid分析各化合物含量。加标样品中癸烯-1、正辛醇含量减去未加标样品癸烯-1、正辛醇含量,其差值同加入癸烯-1、正辛醇的理论值之比即为两化合物的加标回收率,其值依次为96.7%、97.8%。
[0049]
上述结果表明,本发明方法分离组分中各组分回收率高、组分间交叉量低,具有较好的富集效果。
[0050]
实施例2
[0051]
在固相萃取柱a内装填1.2g氧化铝固定相,固相萃取柱b内装填1.2g银含量为8wt%的载银硅胶固定相,用0.5ml第一洗脱剂(正己烷)润湿固相萃取柱a。
[0052]
用移液枪取0.24g某公司高温费托合成生产的2#柴油样品加入固相萃取柱a上部并被固定相完全吸附。用4ml第一洗脱剂冲洗固相萃取柱a b,然后分开固相萃取柱a与b,向固相萃取柱b继续加入3ml第一洗脱剂,得到饱和烃,记为组分1。用6ml第二洗脱剂(二氯甲烷与乙醇的体积比为85:15)冲洗固相萃取柱b,得到烯烃组分,记为组分2。用6ml第三洗脱剂(正己烷与二氯甲烷的体积比为90:10)冲洗固相萃取柱a得到芳烃,记为组分3。用6ml第四洗脱剂甲醇冲洗固相萃取柱a,得到杂原子化合物,记为组分4。各组分加入一定量氘代三联苯作为内标,采用氮气吹扫去除各组分中的溶剂。
[0053]
以癸烯-1、正辛醇的加标回收率考察固相萃取法预处理高温费托合成柴油的分离效果,结果两者的加标回收率依次为94.5%、95.2%。
[0054]
实施例3
[0055]
在固相萃取柱a内装填1.2g氧化铝含量为90wt%的硅胶-氧化铝固定相,固相萃取柱b内装填1.2g银含量为10wt%的载银硅胶固定相,用0.5ml第一洗脱剂(正己烷)润湿固相萃取柱a。
[0056]
用移液枪取0.56g某公司直接液化3#柴油样品加入固相萃取柱a上部并被固定相完全吸附。用4ml第一洗脱剂冲洗固相萃取柱a b,然后分开固相萃取柱a与b,向固相萃取柱b继续加入2ml第一洗脱剂,得到饱和烃,记为组分1。用2ml第二洗脱剂(二氯甲烷与乙醇的体积比为70:30)冲洗固相萃取柱b,得到烯烃组分,记为组分2。用3ml第三洗脱剂(正己烷与二氯甲烷的体积比为75:25)冲洗固相萃取柱a得到芳烃,记为组分3。用4ml第四洗脱剂甲醇冲洗固相萃取柱a,得到杂原子化合物,记为组分4。各组分加入一定量氘代三联苯作为内标,采用氮气吹扫去除各组分中的溶剂。
[0057]
采用gc-ms-fid分析组分1、组分2、组分3、组分4,结果表明,组分1主要为饱和烃,占柴油样品的55.1wt%,其中杂质烯烃组分含量为0.1wt%;组分2主要为烯烃,占柴油样品的8.7wt%,其中杂质饱和烃含为0.1wt%,芳烃含量0.0wt%;组分3主要为芳烃,占柴油样品的20.7wt%,其中杂质烯烃含量0.2wt%,杂原子化合物含量1.1wt%;组分4为含氮、含氧
化合物,占柴油样品的13.7wt%,其中杂质芳烃含量0.3wt%。
[0058]
以癸烯-1、正辛醇的加标回收率考察固相萃取法预处理直接液化柴油的分离效果,结果两者的加标回收率依次为97.9%、98.1%。
[0059]
实施例4
[0060]
在固相萃取柱a内装填1.2g氧化铝含量为80wt%的硅胶-氧化铝固定相,固相萃取柱b内装填1.2g银含量为15wt%的载银硅胶固定相,用0.5ml第一洗脱剂(正己烷)润湿固相萃取柱a。
[0061]
用移液枪取0.80g某公司低温费托合成4#柴油样品加入固相萃取柱a上部并被固定相完全吸附。用5ml第一洗脱剂冲洗固相萃取柱a b,然后分开固相萃取柱a与b,向固相萃取柱b继续加入4ml第一洗脱剂,得到饱和烃,记为组分1。用4ml第二洗脱剂(二氯甲烷与乙醇的体积比为60:40)冲洗固相萃取柱b,得到烯烃组分,记为组分2。用2ml第三洗脱剂(正己烷与二氯甲烷的体积比为60:40)冲洗固相萃取柱a得到芳烃,记为组分3。用2ml第四洗脱剂甲醇冲洗固相萃取柱a,得到杂原子化合物,记为组分4。各组分加入一定量氘代三联苯作为内标,采用氮气吹扫去除各组分中的溶剂。
[0062]
采用gc-ms-fid分析组分1、组分2、组分3、组分4,结果表明,组分1主要为饱和烃,占柴油样品的75.7wt%,其中杂质烯烃组分含量为0.0wt%;组分2主要为烯烃,占柴油样品的15.4wt%,其中杂质饱和烃含为0.5wt%,基本不含芳烃;组分3主要为芳烃,占柴油样品5.6wt%,杂质烯烃含量0.0wt%,含氧化合物含量0.1wt%;组分4主要为含氧化合物,占柴油样品2.6wt%,杂质芳烃含量0.1wt%。
[0063]
以癸烯-1、正辛醇的加标回收率考察固相萃取法预处理低温费托合成柴油的分离效果,结果两者的加标回收率依次为99.1%、96.5%。
[0064]
对比例1
[0065]
按照石化标准sh/t 0606附录b所述的方法分离富集某公司高温费托合成1#柴油样品。
[0066]
取0.1ml试样滴入石化标准sh/t 0606专用固相萃取柱上部并被完全吸附。依次用2ml正戊烷和0.5ml二氯甲烷冲洗固定相,萃取得到饱和烃。再用2ml二氯甲烷冲洗固定相,得到芳烃。加入一定量正三十二烷作为内标,进gc-ms-fid分析族组成,结果表明饱和烃组分含量占柴油样品总量的70.5wt%,其杂质芳烃含量为2.4wt%;芳烃组分含量为22.6wt%,其杂质饱和烃含量为4.5wt%。
[0067]
由此可见,相比于本发明提供的固相萃取法,采用石化标准sh/t 0606的固相萃取法只能得到饱和烃、芳烃组分,其中烯烃会影响饱和烃定量,含氧化合物会影响芳烃组分定量,造成测定结果中饱和烃、芳烃含量结果偏高,影响测定结果的准确性。
[0068]
对比例2
[0069]
层析柱装填20g硅胶。
[0070]
加入10ml正戊烷活化层析柱。待正戊烷液面低于层析柱上部时,用移液枪移取2g某公司高温费托合成1#柴油加入层析柱上部。加入2g石英砂固定柴油样品。加入35ml正戊烷洗脱饱和烃组分,加入60ml甲苯洗脱芳烃,加入50ml甲醇洗脱杂原子化合物,用双链球调节洗脱液流量为1.5ml/min。采用氮气吹扫去除各组分中的溶剂。整个过程耗时160min。
[0071]
采用gc-ms-fid分析组分1、组分2、组分3,结果表明,组分1主要为饱和烃,其中杂
质芳烃组分含量8.9wt%,组分2主要为芳烃,其中杂质饱和烃含量10.5wt%,杂原子化合物含量8.5wt%,组分3主要为含氧化合物,其中杂质芳烃含量12.5wt%。
[0072]
由此可见,采用柱色谱法分离煤基柴油样品时耗时长、溶剂用量大、固定相用量大,无法分离链烷烃与烯烃,不适用于测定煤基柴油族组成的预处理。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。