一种表达pcv2d cap蛋白的载体及方法
技术领域
1.本发明属于杆状病毒表达载体系统技术领域,具体涉及一种表达pcv2d cap蛋白的载体及方法。
背景技术:
2.1993年出现了bacmid技术,bacmid是组装有杆状病毒dna的细菌人工染色体(bacterial artificial chromosomes,bacs),因此bacmid可以维持于大肠杆菌细胞内并在大肠杆菌中增殖,bac-to bac具有“bacteria”到“baculovirus”之意。杆状病毒表达载体系统包含3个部分:转移质粒、杆状病毒表达载体和昆虫细胞。转移质粒由ph或p10启动子、启动子下游的多克隆位点、polya序列、需要可在外源基因n端加入gp64、蜂毒素或几丁质酶等信号肽促使蛋白分泌表达,在开放阅读框n端或c端加入6x组氨酸标签利于蛋白纯化,tev酶切位点可用于后期组氨酸标签的移除。感受态大肠杆菌dh10bac中包含两个质粒:穿梭质粒和辅助质粒。穿梭质粒(bacmid)具有在细菌中低拷贝mini-f复制子、mini-atttn7转座位点、卡纳霉素抗性基因的杆状病毒基因组,不含有杆状病毒多角体蛋白;辅助质粒编码转座酶,可维持在大肠杆菌中,使转移质粒上的表达盒通过转座作用定向转移到杆状病毒表达载体上,含有四环素抗性基因。
3.转移质粒转化到感受态细胞中后,在转座酶催化作用下,将tn7转座子左右臂定向转移到bacmid的tn7位点,形成具有外源基因的bacmid。转移载体中携带有lacz基因,其产生的a-肽与宿主菌的a-肽互补可编码β半乳糖苷酶,经过iptg诱导分解底物x-gal形成蓝色噬菌斑。当外源基因插入到转移载体中lacz的多克隆位点,导致lacz失活,破坏了a.-肽的互补形成,在含有x-gal和iptg的平板中生长白色噬菌斑。因此重组质粒可在相应抗性、iptg和x-gal(或bluo-gal)底物的琼脂糖平板上通过蓝白斑筛选获得,然后经过碱裂解方法获得病毒dna,纯化的杆粒dna用于转染昆虫细胞,生产具有感染性的bv。
4.第二代商品化bevs为invitrogen公司生产的bac-to-bactm该表达系统,杆状病毒表达载体优势:(i)昆虫细胞是真核细胞,具有完备的翻译后加工修饰功能,重组蛋白与天然蛋白具有相似的生物活性;(ii)外源基因容量大,杆状病毒表达载体大小约为134kbp,适合较大的基因克隆和同时多个基因的克隆;(iii)生物安全性好,杆状病毒不感染包括人在内的哺乳动物;(iv)在强启动子ph或p10的调控下,重组蛋白具有很高的表达水平。该系统生产的杆状病毒不需要蚀斑纯化过程,然而,需要通过抗性基因从亲代病毒dna中筛选重组病毒dna,含有重组病毒dna的细菌增殖过程以及重组病毒dna的抽提纯化过程,均可能会影响杆状病毒粒子的稳定性,甚至导致重组病毒表达丢失。
5.autographa california multiple nucleopolyhedrovirus,acmnpv(苜蓿银纹夜蛾核型多角体病毒)是研究最广泛且应用较多的一种杆状病毒,以acmnpv为基础的杆状病毒表达载体系统(baculovirus expression vector system,bevs)发展迅速,已成为应用最普遍的表达系统之一。第一代商品化bevs是clontech生产的bacpak6tm,该表达系统对acmnpv基因组进行以下修饰,即经过bsu361酶切,病毒dna线性化,除去了lacz基因和
orf1629的部分基因,导致病毒在昆虫细胞中无法复制的病毒dna与转移载体混合后转染到昆虫细胞进行同源重组,表达盒插入到病毒dna,修复了orf1629基因,病毒dna恢复环形,同时恢复了感染性。但由于bsu361酶切效率并不是100%,转染出的病毒是亲代病毒和重组病毒的混合物,需要进一步的蚀斑纯化实验,将重组病毒从亲代病毒中筛选出来。
6.flashbac
tm bevs对acmnpv基因组进行了改造,acmnpv病毒dna缺失orf1629基因部分序列和bac取代多角体蛋白基因。缺失了orf1629部分基因序列的病毒dna不能在昆虫细胞内复制,bac取代多角体蛋白基因后,病毒dna能在细菌细胞内维持并进行低拷贝复制。改造后病毒dna在细菌中增殖后,经碱裂解和氯化铯梯度纯化,获得用于共转染的flashbacm
tm
病毒dna,与带有外源基因的转移质粒共转染于昆虫细胞,通过细胞内同源重组,重组病毒的orf1629基因完整,病毒恢复复制功能,外源基因同时插入到病毒dna中,重组杆状病毒获得拯救。该系统中,去除了蓝白斑筛选过程,亲代病毒不可能在昆虫细胞中复制。
7.flashbac
tm
gold bevs是在flashbac
tm
基础上缺失了几丁质酶基因(chia)和组织蛋白酶基因(v-cath)。chia是一个辅助基因,编码的酶能够促进细胞内切酶和外切酶的活性。对于感染的昆虫细胞,chia表达于感染的晚期,与组织蛋白酶一起促进宿主表皮分解和组织溶解,从而释放病毒,感染更多的宿主细胞。已有文献报道,缺失病毒基因组中chia和(或)v-cath基因,可提高昆虫细胞和昆虫幼虫蛋白稳定性。possee等将chia基因从杆状病毒基因组缺失后,分泌性重组蛋白表达水平提高;lee等在b.mori幼虫中表达纤维素酶,表达量提高了17%;suzuki等用缺失v-cath的杆状病毒表达萤火虫荧光素酶和人生长因子,发现蛋白稳定性大大增加;park等使用缺失chia/v-cath的病毒在b.mori中表达gfp融合蛋白,血淋巴中几丁质酶和组织蛋白酶的活性分别降低了95%和50%,与未修饰的病毒相比,表达的gfp(uv)-beta、3-n-葡萄糖胺乙酰转移酶降解量减少,活性增加了2.8倍。flashbac
tm
gold杆状病毒表达载体不仅增加了分泌蛋白的表达量,同时提高了重组蛋白的稳定性。
8.flashbac
tm
ultrabevs是在flash
tm
bacgold基础上,缺失了p10、p26和p74基因。
9.silvia等将bevs的双表达盒载体进行了改造,polh启动子后克隆杆状病毒源激活因子ie0、iel;p6.9p10双启动子代替原来的p10启动子,与杆状病毒源的增强子hrl顺式连接,启动外源蛋白的表达,polhac-ie-01/hr1p6.9p10称为tb表达盒,该表达盒延长了细胞感染后保持完整性的时间,提高了蛋白的完整性,且egfp表达量是原表达盒的4倍,tb表达盒可显著提高bevs的蛋白表达水平。随后,javier等使用新的杆状病毒表达盒tb分别高效表达了pcv2cap蛋白和rhdvvp60蛋白,两种蛋白的表达量比原表达盒相比均提高了300%,均组装形成了pcv2 vlps和rhdvvlps。
10.猪圆环病毒是一种无囊膜、单股、负链、环状的dna病毒,它属于圆环病毒科圆环病毒属。猪圆环病毒主要有3种基因型,圆环病毒1型、圆环病毒2型和圆环病毒3型。pcv2致病性较强,可引起多种猪圆环病毒相关性疾病(porcine circovirus-as sociated diseases,pcvads),其中最主要的是断奶仔猪多系统衰弱综合征(postweaning multisystemic wasting syndrome,pmws),pcv2主要破坏淋巴系统,导致机体免疫功能缺陷,且容易与其它病原发生混合感染,该病在我国乃至世界猪群中广泛存在,已成为一种常见病和多发病,给养猪业带来了巨大的经济损失。pcv2又被分为pcv2a、pcv2b、pcv2c、pcv2d、pcv2e和pcv2f这6个基因亚型,其中pcv2d在中国的流行率最高,大部分的养猪场都
能够检测到pcv2d病毒。
11.cap蛋白是pcv2d唯一的衣壳蛋白和主要的免疫原性蛋白,杆状病毒表达系统具有可插入较大的外源基因片段、目的蛋白表达量高和表达的蛋白生物学活性较好的优点被广泛应用。目前已知pcv2基因组中含有11个开放阅读框(orfs),其中最主要的为orf1和orf2。orf1位于单链dna 51~985nt,编码病毒的复制相关蛋白(rep和rep');orf2位于单链dna1034~1735nt,该区域的互补链编码病毒的结构蛋白(cap蛋白)。mahe等发现pcv2 cap蛋白65-87aa、113-139aa、169-183aa和193-207aa 4个区域具有免疫原性。cap蛋白前41个氨基酸为核定位信号肽(nls),关文献报道,缺失或突变nls的cap蛋白表达量高于未改变nls的cap蛋白。liu等在大肠杆菌中表达了pcv2 orf2编码的完整cap蛋白,表达产量约为l mg/l;之后,zhoul和marial分别将完整的orf2基因和缺失nls的orf2基因在大肠杆菌中表达,发现缺失nls的重组菌表达量高达20mg/l,
12.吉林农业大学的张艳艳等人根据h5细胞对密码子的偏好性,对cap蛋白密码子进行了优化并合成,同时突变了cap蛋白的nls序列,此外还在杆状病毒转移载体中克隆了增强子、双启动子,显著提高了目的蛋白在杆状病毒载体中表达水平,cap蛋白的表达含量从20-50ug/ml提高到了150ug/ml左右。
13.虽然市面上已经有杆状病毒表达的pcv2d cap蛋白疫苗,但是运用新的技术和方法可以对其表达量进一步提高,有助于提高生产效率和降低生产成本。
技术实现要素:
14.本发明使用bac-to-bac杆状病毒表达系统,通过对pfast bac dual载体进行改造,使用cag、sv40启动子等顺式作用元件、增加acmnpv基因中同源重复序列(hr1)、增加转录后调控元件(wpre)来提高pcv2 cap重组蛋白的表达量。对bac-to-bac杆状病毒表达系统表达的目的蛋白浓度进一步提高,有助于提高生产效率和降低生产成本。
15.一种表达pcv2d cap蛋白的载体,由骨架载体和插入其启动子p10下游的多克隆位点的一个或多个pcv2d cap蛋白编码基因构成,所述骨架载体的启动子p10和其下游的多克隆位点之间插入了如seq id.no.6所示的转录后调控序列wpre,所述骨架载体的启动子ph被替换为如seq id.no.4所示的启动子sv40,或启动子ph和启动子p10之间插入了如seq id.no.5所示的增强子hr1。
16.进一步地,所述pcv2d cap蛋白的编码基因的核苷酸序列根据杆状病毒密码子优化,优化后的核苷酸序列如seq id no.3所示。
17.进一步地,所述的骨架载体为pfast bac dual。
18.一种表达pcv2d cap蛋白的方法,将上述pcv2d cap蛋白的编码基因,与所述骨架载体连接,将所得重组杆状病毒转移质粒转化dh10bac大肠杆菌,通过转座重组,获得重组杆状病毒质粒,复苏后的菌液涂布卡那霉素、四环素、庆大霉素、iptg、x-gal平板,37℃避光培养,选白色单一菌落扩大培养,转染sf9昆虫细胞从而获得p0代重组杆状病毒,采用sf9细胞培养和重组杆状病毒增殖,结合阳离子交换树脂纯化得到pcv2d cap蛋白;
19.所述骨架载体的核苷酸序列如seq id no.1所示,具有如下组件:启动子ph被启动子sv40替换,启动子p10和其下游的多克隆位点之间插入了转录后调控序列wpre;
20.或所述骨架载体的核苷酸序列如seq id no.2所示,具有如下组件:启动子ph未被
启动子sv40替换,所述转移质粒的启动子p10和ph之间插入了增强子hrl,启动子p10和其下游的多克隆位点之间插入了转录后调控序列wpre。
21.如上所述的方法在制备pcv2d的抗原、抗体、或亚单位疫苗中的应用。
22.sv40启动子顺式作用元件的序列如seq id.no.4所示,sv40启动子序列通过合成的方式替换掉ph启动子。
23.acmnpv基因中同源重复序列(homologous region 1,简称hr1)序列如seq id.no.5所示,hr1基因通过合成的方式插入p10和ph启动子之间。
24.土拨鼠肝炎病毒转录后调控元件(woodchuck hepatitis virus post-transcriptional regulatory elemem,简称wpre)的序列如seq id.no.6所示,wpre基因通过合成的方式插入p10表达框的后面。
25.将上述的3个元件以联合作用构建到pfast bac dual载体上,得到具有sv40-wpre、或hr1-wpre的pfast bac dual载体,将2个拷贝的pcv2d cap基因构建到含有这些元件组合的pfast bac dual载体上,只含有2个拷贝的pcv2d cap的pfast bac dual载体。
26.改造后的sv40-wpre、hr1-wpre组pcv 2d cap蛋白的表达量明显增加。使用sv40启动子等顺式作用元件、增加acmnpv基因中同源重复序列(hr1)、增加转录后调控元件(wpre)来提高pcv2 cap重组蛋白的表达量。
附图说明
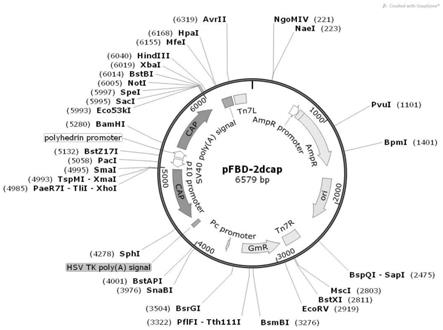
27.图1为pfast bac dual-cap的图谱;
28.图2为用引物扩增的cap蛋白编码基因的pcr鉴定结果图;
29.图3为重组的转移质粒转化后的单菌落的pcr鉴定结果图;
30.图4为转染重组杆状病毒前后sf9细胞的状态图,a为转染前的正常细胞,b为转染后的细胞;
31.图5为荧光显微镜观察7种重组杆状病毒液和空白对照在细胞孔发荧光的检测图;
32.图6为7种重组杆状病毒表达的cap蛋白的sds-page电泳结果;
33.图7为重组杆状病毒hr1-wpre组表达蛋白的sds-page电泳结果;
34.图8为tem观察重组杆状病毒hr1-wpre组表达蛋白的形貌图;
35.图9为elisa检测重组杆状病毒hr1-wpre组表达蛋白和bac-pcv2d组表达蛋白的特异性抗体水平结果对比图;
36.图10为攻毒后14天检测血清中的pcv2病毒载量结果对比图。
具体实施方式
37.下面结合具体实施例对本发明作进一步的详细说明。实施例中所述的实验方法无特别说明,即按常规分子生物学实验方法操作。
38.首先对本发明所设计的术语解释如下:
[0039]“pfastbac
tm dual载体”其特点是在单个载体上有两个启动子,用于在使用杆状病毒表达系统(baculovirus expression system)时在昆虫细胞中同时表达两个蛋白质。该载体具有两个强启动子,多角体蛋白启动子和p10启动子,因此可实现高水平表达。包含cat和β-葡萄糖醛酸酶(gus)的表达对照。
[0040]“sf9”是spodoptera frugiperda cell的缩写,即草地贪夜蛾细胞,草地贪夜蛾也称草地夜蛾。命名源于两篇文献:hegedus et al.,1998;pfeifer et al.,1997。生物实验中常用的昆虫细胞还有:ld652y、sf21、high five等。
[0041]“ii试剂”是一种阳离子脂质制剂,设计用于昆虫细胞的极佳转染。ii是原始试剂的改进版本。除提供与相同的出色性能外,ii试剂的生产旨在确保批间一致性,并经过优化,可用于更快的方案。当使用baculodirect
tm
和insectselect
tm
表达系统时,使用ii试剂进行转染可导致sf9、sf21和high five
tm
细胞的一致和高效转染。ii试剂也可用于在含血清或无血清培养基中转染贴壁或悬浮哺乳动物细胞。
[0042]
实施例1
[0043]
1、转移载体的构建
[0044]
1)cag组载体
[0045]
cag启动子顺式作用元件的序列如seq id.no.7所示,将cag启动子序列通过合成的方式替换掉pfast bac dual载体中的ph启动子。
[0046]
2)sv40组载体
[0047]
sv40启动子顺式作用元件的序列如seq id.no.4所示,将sv40启动子序列通过合成的方式替换掉pfast bac dual载体中的ph启动子。
[0048]
3)wpre组载体
[0049]
土拨鼠肝炎病毒转录后调控元件(woodchuck hepatitis virus post-transcriptional regulatory elemem,简称wpre)的序列如seq id.no.6所示,将wpre基因通过合成的方式插入pfast bac dual载体中的p10表达框的后面。4)sv40-wpre组载体
[0050]
将wpre基因通过合成的方式插入pfast bac dual载体中的p10表达框的后面,将sv40启动子序列通过合成的方式替换掉pfast bac dual载体中的ph启动子。
[0051]
5)cag-wpre组载体
[0052]
将wpre基因通过合成的方式插入pfast bac dual载体中的p10表达框的后面,将cag启动子序列通过合成的方式替换掉pfast bac dual载体中的ph启动子。
[0053]
6)hr1-wpre组载体
[0054]
acmnpv基因中同源重复序列(homologous region 1,简称hr1)序列如seq id.no.5所示,将hr1基因通过合成的方式插入pfast bac dual载体中的p10和ph启动子之间,将wpre基因通过合成的方式插入pfast bac dual载体中的p10表达框的后面。
[0055]
7)bac-pcv2d组载体
[0056]
未改造。
[0057]
2、目的基因的扩增
[0058]
将pcv2d cap蛋白编码基因进行杆状病毒密码子优化,将优化后的核苷酸序列进行基因合成,以合成后的cap蛋白编码基因为模板,用两对引物分别进行扩增,用第一对引物使bamhi和saci的限制性内切酶酶切位点分别引入到cap蛋白编码基因的上下游,用第二对引物使sphi和xhoi的限制性内切酶酶切位点分别引入到cap蛋白编码基因的上下游。引物序列如表1所示,划下横线处为酶切位点;
[0059]
表1
[0060][0061][0062]
用于扩增上述cap蛋白编码基因的pcr反应体系(总体积50μl)如下:
[0063][0064]
pcr扩增反应后,扩增产物于1%琼脂糖凝胶电泳,并凝胶回收目的片段,pcr扩增结果如图1所示,-20℃保存备用。构建好的质粒图谱如图所1示。
[0065]
3、片段和载体的酶切
[0066]
将第一对引物扩增的cap蛋白编码基因和步骤2构建的一种pfast bac dual载体分别用bamhi和saci限制性内切酶进行双酶切,将第二对引物扩增的cap蛋白编码基因和步骤2构建的一种pfast bac dual载体分别用sphi和xhoi限制性内切酶进行双酶切。酶切体系如下表2:
[0067]
表2
[0068]
模板1μl10
×
buffer2μl限制性内切酶11μl限制性内切酶21μlddh2oupto20μl
[0069]
37℃反应3小时,酶切产物用1%琼脂糖凝胶电泳后再用琼脂糖凝胶回收试剂盒回收dna片段及pfast bac dual载体骨架。
[0070]
4、连接和转化
[0071]
将回收后的目的dna片段和pfast bac dual载体骨架用t4连接酶连接进行同源重组,配好连接体系后16℃过夜,连接后的重组转移质粒转化大肠杆菌top10感受态。连接体系如下表3:
[0072]
表3
[0073][0074][0075]
5、重组质粒的鉴定
[0076]
转化后的单菌落进行pcr检测,鉴定到正确条带后送武汉生工生物有限公司进行测序,结果正确后进行下一步实验。菌落pcr鉴定结果如图3所示,构建好的pfast bac dual-2cap质粒的图谱如图1所示。
[0077]
6、筛选重组杆粒
[0078]
将100-300ng测序正确的转移质粒转化dh10bac感受态细胞,复苏后的菌液涂布卡那霉素、四环素、庆大霉素、iptg、x-gal平板,37度避光培养48h。挑选白色单克隆菌落,使用鉴定引物m13-f/r进行菌落pcr鉴定,扩增到正确条带后送测序,测序结果正确后,菌液进行扩大培养,然后提取重组杆状病毒质粒。
[0079]
7、重组杆状病毒的拯救
[0080]
(1)按照一个孔1.2
×
106个细胞数量,将对数生长期的sf9细胞铺于6孔板,静置1小时后,细胞贴壁融合度约70%~80%;
[0081]
(2)取cellfectin ii试剂3-5μl入100μl无抗无血清细胞培养液;取5μgbacmid加入100ul无抗无血清细胞培养液中混匀;
[0082]
(3)将(2)中的两管混合液合并混匀,室温孵育15min:
[0083]
(4)孵育结束后轻轻加入6孔细胞板中,27℃培养箱避光孵育4-6h后将其更换为正常培养基,注意做不改造的转移质粒的转染对照孔以及空白对照孔;
[0084]
(5)27℃培养箱中继续培养,每24h观察一次与对照组相比直至出现细胞直径增大、细胞核增大、细胞呈圆形且边界清晰、细胞内出现小泡,收细胞上清即为p0代重组杆状病毒的病毒液。如图4所示。
[0085]
8、重组杆状病毒的扩增
[0086]
使用细胞摇瓶悬浮培养sf9细胞,待细胞生长至对数生长期,密度为2
×
106cell/ml,将p0代病毒液按l:100的体积比接毒,悬浮培养96h后,收获p1代病毒,4℃,1000rpm,离心15min将上清与细胞分离,上清4℃避光保存。用该方法继续扩增p2和p3代病毒液。
[0087]
9、重组杆状病毒滴度的测定
[0088]
使用间接性免疫荧光法测定重组杆状病毒的滴度,具体步骤如下:
[0089]
(1)细胞单层制备将处于对数生长期的sf9细胞计数后,按照每孔6.5
×
104个细胞,接种96孔细胞培养板,27℃培养1h:
[0090]
(2)将待测重组杆状病毒用昆虫细胞培养基作10倍系列稀释,混匀。取10-4
、10-5
、
10-6
共3个稀释度。
[0091]
(3)将稀释好的病毒液加入细胞孔中,每孔加入100μl病毒液,一个稀释梯度做8个重复;
[0092]
(4)置27℃温箱培养96小时。
[0093]
(5)细胞固定:用4%多聚甲醛150μl每孔,4℃放置30min;
[0094]
(6)洗涤:弃去多聚甲醛,用pbst洗涤3次,200μl每孔,每次5min;
[0095]
(7)封闭:加入封闭液50μl每孔,37℃放置30min;
[0096]
(8)加入一抗:加入cap蛋白鼠的单克隆抗体,50μl每孔,37℃放置60min;
[0097]
(9)洗涤:弃去一抗,用pbst洗涤3次,200μl每孔,每次5min;
[0098]
(10)加入二抗:加入fitc标记的羊抗鼠抗体,37℃放置60min;
[0099]
(11)洗涤:弃弃去二抗,用pbst洗涤3次,200μl每孔,每次5min;
[0100]
(12)荧光显微镜观察细胞孔发荧光情况。如图5,步骤2构建的7种载体与cap蛋白编码基因重组后病毒滴度的测定结果如下表4所示:
[0101]
表4
[0102][0103]
从结果可以看到包装的重组杆状病毒的滴度在10
7-109之间,病毒滴度都较高。10、重组cap蛋白的表达和纯化分析
[0104]
将传至p2代的重组杆状病毒按照0.3moi接种sf9细胞,接种96h左右收获细胞,离心获得的上清进行sds-page电泳。用bca法测定表达的蛋白的总浓度:
[0105]
1)配制工作液:根据标准品和样品数量,按50体积bca试剂a加1体积bca试剂b试剂(50:1)配制适量的bca工作液。
[0106]
2)稀释标准品:取10μlbsa标准品用pbs稀释至100μl,使标准品的终浓度为0.5mg/ml。将标准品按0,1,2,4,8,12,16,20μl加到96孔板的蛋白标准品孔中,加pbs补足至20μl;
[0107]
3)将样品稀释至合适的浓度,总体积为20μl;
[0108]
4)各孔加200μlbca工作液,37℃放置15-30分钟。用酶标仪测定562nm下的吸光值,根据标准曲线计算出总蛋白浓度,如表5。
[0109]
表5
[0110][0111]
参考图像分析系统分析目的蛋白占总蛋白的比例得出目的蛋白的具体表达量。
[0112]
sds-page电泳结果如图6所示,相较于没有改造的bac-pcv2d组,改造后的sv40、sv40-wpre、hr1-wpre组pcv 2d cap蛋白的表达量明显增加,而改造后的cag、wpre、cag-wpre组pcv 2d cap蛋白的表达量明显减少。将表达量最高的hr1-wpre组表达的pcv2d cap蛋白用阳离子交换树脂进行纯化,将收获的培养混合物以15,000g离心10分钟来除掉细胞和细胞碎片,上清液通过0.45μm滤器以除掉细小的杂质。用平衡缓冲液(20mm tris,50mmnacl,ph 8.0)将填好的阳离子交换柱平衡,过滤后的上清与平衡好的阳离子交换柱进行结合过柱;洗杂缓冲液(20mm tris,50mmnacl,ph 8.0)过柱洗杂;洗脱缓冲液(20mm tris,500mm nacl,ph 8.0)过柱洗脱。将重组杆状病毒hr1-wpre组表达蛋白经上述阳离子交换树脂纯化,纯化后的样品跑sds-page胶,结果如图7所示,后续的试验均使用该纯化后的蛋白。
[0113]
11、重组pcv2d cap蛋白vlp颗粒的形成
[0114]
将纯化的pcv2d cap蛋白(hr1-wpre组)用hitachi的透射电镜观察,结果如图所示,视野可见大小为17nm左右的vlp颗粒,形态稳定,大小均一。结果如图8所示。
[0115]
12、疫苗的制备
[0116]
将纯化的pcv2d cap蛋白用0.22μm滤器过滤除菌后与montanide
tm isa201vg佐剂以1:1(w/w)的比例混合,制备成每毫升体积含有终浓度40μg cap蛋白的疫苗。
[0117]
13、动物试验
[0118]
1)安全性试验:8周龄的balb/c雌鼠10只,分为a、b两组,每组5只。a组每只小鼠皮下注射0.5ml,制备好的疫苗;b组每只小鼠注射0.5mlpbs溶液;连续观察14天,两组小鼠状态相同且均无异常反应,此结果表明亚单位疫苗对小鼠是安全的。
[0119]
2)免疫原性试验:将制备好的疫苗按照每只200μl免疫剂量注射到8周龄的balb/c雌鼠的腿部肌肉中,14天后进行第二次免疫。28天后收集血样分离血清并检测特异性抗体水平,采血后用pcv2d病毒进行攻毒试验,攻毒剂量为每只老鼠0.5ml,攻毒后连续观察小鼠的状态,攻毒后14天采血,分离样品的血清并提取核酸,用荧光定量pcr检测血清中pcv2d病毒的拷贝数,引物的信息如下表表6所示:
[0120]
表6elisa检测pcv2d特异性抗体水平结果如图9所示,实验组的抗体水平在
[0121][0122]
1.4左右,明显高于对照组,说明表达的pcv2 cap蛋白能够有效激发机体的体液免疫应答。pcv2感染会引发机体产生严重的病毒血症,本试验用balb/c雌鼠免疫了pcv2 cap蛋白后再进行攻毒实验,攻毒后14天检测血清中的pcv2病毒载量,结果如图10所示,对照组血清中的pcv2病毒拷贝数较高,是免疫后再攻毒组的300倍左右,说明免疫pcv2d cap蛋白后能够大大减轻小鼠的病毒血症。
[0123]
[0124]
[0125]
[0126]
[0127]
[0128]
[0129]
[0130]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。