1.本发明属于药用植物物种鉴定,具体的说是涉及采用rbcl matk组合条形码精确鉴别草麻黄与其他相似物种的方法。
背景技术:
2.草麻黄(ephedra sinica stapf),别名麻黄草、华麻黄、无叶草等,为麻黄属草本状灌木植物,是中华人民共和国《药典》收录的中药“麻黄”及“麻黄根”指定的主要基原植物。用途广、用量大,是我国许多经典名方的主要成分。
3.全世界麻黄属下有67个相似种,我国有15个种及4个变种,主要产于辽宁、吉林、内蒙古、河北、山西、河南西北部及陕西等省区,适应性强,常组成大面积的单纯群落。近年来,草麻黄市场需求量逐年上升,存在以次充好、物种混淆使用的现象,但市场上尚没有精确鉴定并区分草麻黄与其他相似物种的良好方法,这很大程度上限制了草麻黄用药安全及其现代化应用研究。
4.截止目前,草麻黄的鉴别多采用两种方法,一种是传统的形态鉴别法,该方法主要从植物外观、颜色、理化性质等指标区分,存在误差大、不够精密等缺点;第二种方法,是通过核糖体基因its、叶绿体chlb全基因、限制性片段长度多态性等分子生物学的方法分析,仅可以区分草麻黄与个别物种,或麻黄基原物种与其他物种,但无法精密而严格地区分草麻黄与木贼麻黄、中麻黄、膜果麻黄、山岭麻黄等众多同属近缘物种!而且这些方法存在成本高、操作步骤繁琐、样本分析规模较小等缺点。
5.本发明则通过开发物种间特异的matk条形码,并联合使用rbcl通用条形码对草麻黄进行鉴定,实现了草麻黄物种的精密鉴定。具有物种区分度高,操作方便、检测高效、结果可信等很多优点,是一种很有应用前景的方法技术。
技术实现要素:
6.本发明的目的在于为鉴定草麻黄植物及药材提供matk基因特异性引物。
7.本发明的另一个目的在于提供采用通用引物扩增rbcl基因目的序列,以及采用上述引物扩增matk基因目的序列,将二者综合分析以鉴定草麻黄植物及药材的方法。
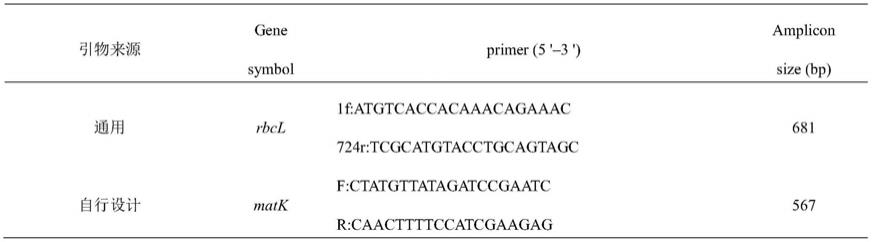
8.本发明提供及使用的引物,具体见表1
9.表1.草麻黄鉴定引物
10.11.本发明提供一种利用rbcl matk组合条形码鉴定草麻黄的方法,步骤如下:
12.1)准备草麻黄植物干或者湿的样品;
13.2)基因组dna提取:采用常规方法提取草麻黄植物的基因组dna;
14.3)采用表1中rbl通用引物,pcr扩增叶绿体基因rbl目的序列;采用25μl pcr体系:2
×
santaq pcr master mix 12.5μl,引物各1μl(10μmol/l),模板dna 2μl(100ng/μl),ddh2o 8.5μl;扩增反应程序为:94℃预变性5min;94℃变性50s,55℃退火1min,72℃延伸1min,36个循环;72℃延伸10min;
15.4)采用表1提供的matk特异性引物扩增叶绿体基因matk目的序列。采用25μl pcr体系:2
×
santaq pcr master mix 12.5μl,引物各0.5μl(10μmol/l),模板dna 2μl(100ng/μl),ddh2o 9.5μl;扩增反应程序为:94℃预变性5min;94℃变性30s,52℃退火45s,72℃延伸1min,35个循环;72℃延伸10min;
16.5)采用常规方法进行琼脂糖凝胶电泳,确认扩增产物;
17.6)采用sanger方法双向测序;
18.7)采用cexpress软件拼接序列,并利用bioedit及mega 5.1软件将麻黄属近缘物种进行多序列比对,分析得到稳定的种间遗传变异位点(见表2、表3);以k2p(kimura-2-parameter)模型计算它们的种内种间遗传距离,进行“barcoding gap”分析;随后以邻接法构建系统发育树,利用bootstrap(1000次重复)检验各分支的支持率,并选择保留50%含有空位的列,以相关类群即与麻黄目同一买麻藤亚纲(gnetidae)下买麻藤属(gnetum linn)下模式种-显轴买麻藤gnetum gnemon为outgroup,综合分析以鉴别草麻黄与同属近缘物种。
19.表2.草麻黄(ephedra sinica)与其他麻黄属近缘物种的叶绿体rbcl基因序列比较
[0020][0021][0022]
1)核苷酸位置是从测得的叶绿体基因rbcl目的序列5'端开始计数。
[0023]
表3.e.sinica与e.major的叶绿体matk基因序列比较
[0024][0025]
1)
核苷酸位置是从测得的叶绿体基因matk目的序列5'端开始计数。
[0026]
与现有技术相比本发明的有益效果:本专利采用pcr技术、cexpress、bioedit及mega5.1等软件对条形码序列(its2、psba-trnh、rbcl、rpoc1、matk)及麻黄属种间变异较大的ycf3基因基于较大规模样本数据在麻黄属多物种间的鉴别能力进行评价后,最终选择建立rbcl matk的组合条形码,基于诊断核苷酸位点、遗传距离和系统发育分析,以鉴别草麻黄与同属其他物种。该方法具有成本较低、快速、准确、实用、稳定等优点,是一种分子水平鉴定草麻黄植物和药材的良好方法。
附图说明
[0027]
图1.基于叶绿体基因rbcl目的序列的nj树
[0028]
图2.基于叶绿体基因matk目的序列的nj树
[0029]
图3.基于核糖体基因its2通用条形码的nj树
具体实施方式
[0030]
实施例中表4具体内容:
[0031]
表4.草麻黄(ephedra sinica)样本信息
[0032][0033]
实施例1
[0034]
1)准备草麻黄植物干或湿样品,具体样本见表4。
[0035]
2)基因组dna提取:采用常规方法提取表4中草麻黄植物的基因组dna。
[0036]
3)采用表1中rbl通用引物扩增叶绿体基因rbl目的序列。采用25μl pcr体系:2
×
santaq pcr master mix 12.5μl,引物各1μl(10μmol/l),模板dna 2μl(100ng/μl),ddh2o 8.5μl。扩增反应程序为:94℃预变性5min;94℃变性50s,55℃退火1min,72℃延伸1min,36个循环;72℃延伸10min。
[0037]
4)采用表1提供的matk特异性引物扩增叶绿体基因matk目的序列。采用25μl pcr体系:2
×
santaq pcr master mix 12.5μl,引物各0.5μl(10μmol/l),模板dna 2μl(100ng/μl),ddh2o 9.5μl。扩增反应程序为:94℃预变性5min;94℃变性30s,52℃退火45s,72℃延伸1min,35个循环;72℃延伸10min。
[0038]
5)采用常规方法进行琼脂糖凝胶电泳,确认扩增产物。
[0039]
6)采用sanger方法双向测序。
[0040]
7)特征分析:利用cexpress拼接得到草麻黄样本11条rbcl及4条matk目的序列,然后观察双向序列峰图质量,对每个碱基逐一人工校对。随后将高质量的rbcl序列导入到bioedit软件中与genbank中下载到的97条麻黄属rbcl目的序列(blastn比对到序列相似度较大的麻黄属相关物种序列)进行多序列比对,后将4条草麻黄样本matk目的序列与genbank中下载的20条草麻黄及4条e.major的matk目的序列进行多序列比对并观察。
[0041]
表2表明,基于叶绿体基因rbcl目的序列比对分析,发现同属物种间该序列遗传变异较小,找到1个草麻黄与同属其他物种间的稳定遗传变异位点,但未能将草麻黄与e.major鉴别开;表3表明,基于matk目的序列,则观察找到4个草麻黄与e.major两物种间的诊断核苷酸位点。这显示出rbcl matk组合条形码可基于诊断核苷酸位点分析鉴别草麻黄与同属其他物种。
[0042]
8)距离法分析:将得到的草麻黄样本11条rbcl目的序列与genbank中下载到的97条麻黄属同源序列,利用mega 5.1基于k2p(kimura-2-parameter)模型计算草麻黄种内种间遗传距离,进行“barcoding gap”分析。
[0043]
结果表明,草麻黄种内遗传距离均为0.000,同时草麻黄与ef053226.1e.major种间遗传距离也为0.000,而草麻黄与同属其他物种的种间最小遗传距离为0.002,最大遗传距离为0.006。除e.major以外,草麻黄种内遗传距离小于种间最小遗传距离,这表明基于距离法分析,可鉴别草麻黄与同属其他物种。
[0044]
9)基于叶绿体基因matk目的序列和rbcl目的序列的系统发育分析:将得到的11条草麻黄样本rbcl目的序列与与genbank中下载到的97条麻黄属rbcl目的序列,及4条草麻黄样本matk目的序列与genbank中下载的20条草麻黄及4条e.major的matk目的序列,分别用mega5.1基于k2p模型采用邻接法构建系统发育树分析,利用bootstrap(1000次重复)检验各分支的支持率,并选择保留50%含有空位的列,以相关类群即与麻黄目同一买麻藤亚纲(gnetidae)下买麻藤属(gnetum linn)下模式种-显轴买麻藤gnetum gnemon为outgroup。
[0045]
图1表明,除e.major以外,草麻黄与麻黄属其他近缘物种均能明显分开,而图2表明草麻黄均能单独聚为一支,与e.major均能分开,二者结合分析可将草麻黄与麻黄属其他近缘物种分开,这显示该方法能鉴别草麻黄与麻黄属其他物种。
[0046]
以上三种分析方法综合分析表明,rbcl matk组合条形码可提供充分的证据以鉴别草麻黄与其他相似物种。
[0047]
实施例2
[0048]
1)准备草麻黄植物干或者湿的样品,具体样本见表4。
[0049]
2)基因组dna提取:采用常规方法提取草麻黄植物的基因组dna。
[0050]
3)采用表1中rbl通用引物扩增叶绿体基因rbl目的序列。采用25μl pcr体系:2
×
santaq pcr master mix 12.5μl,引物各1μl(10μmol/l),模板dna 2μl(100ng/μl),ddh2o 8.5μl。扩增反应程序为:94℃预变性5min;94℃变性50s,55℃退火1min,72℃延伸1min,36个循环;72℃延伸10min。
[0051]
4)采用常规方法进行琼脂糖凝胶电泳,确认扩增产物。
[0052]
5)采用sanger方法双向测序。
[0053]
6)特征分析:利用cexpress软件拼接得到草麻黄样本11条rbcl,然后观察双向序列峰图质量,对每个碱基逐一人工校对。随后将高质量的rbcl序列导入到bioedit软件中与genbank中下载到的97条麻黄属rbcl目的序列(blastn比对到的麻黄属同源序列)进行多序列比对。
[0054]
表2表明,基于叶绿体基因rbcl目的序列比对分析,发现同属物种间该序列遗传变异较小,找到1个草麻黄与同属其他物种间的稳定遗传变异位点,但未能将草麻黄与e.major鉴别开。
[0055]
8)距离法分析:将得到的草麻黄样本11条rbcl目的序列与genbank中下载到的97条麻黄属rbcl序列,利用mega 5.1软件基于k2p(kimura-2-parameter)模型计算草麻黄种内种间遗传距离,进行“barcoding gap”分析。
[0056]
结果表明,草麻黄种内遗传距离均为0.000,但草麻黄与ef053226.1e.major种间遗传距离也为0.000,而草麻黄与同属其他物种的种间最小遗传距离为0.002,最大遗传距离为0.006。这表明基于距离法分析,也无法鉴别草麻黄与包括e.major在内的同属近缘物种。
[0057]
9)基于叶绿体基因rbcl目的序列的系统发育分析:将得到的11条草麻黄样本rbcl目的序列与与genbank中下载到的97条麻黄属rbcl目的序列,用mega 5.1基于k2p模型采用邻接法构建系统发育树分析,利用bootstrap(1000次重复)检验各分支的支持率,并选择保留50%含有空位的列,以相关类群即与麻黄目同一买麻藤亚纲(gnetidae)下买麻藤属(gnetum linn)下模式种-显轴买麻藤gnetum gnemon为outgroup。
[0058]
图1表明,除e.major以外,草麻黄与麻黄属其他近缘物种均能明显分开。
[0059]
以上三种分析方法综合分析表明,仅采用rbl通用条形码无法完全鉴别草麻黄与其他相似物种。
[0060]
实施例3
[0061]
1)准备草麻黄植物干或者湿的样品,具体样本见表4。
[0062]
2)基因组dna提取:采用常规方法提取草麻黄植物的基因组dna。
[0063]
3)采用表1提供的matk特异性引物扩增叶绿体基因matk目的序列。采用25μl pcr体系:2
×
santaq pcr master mix 12.5μl,引物各0.5μl(10μmol/l),模板dna 2μl(100ng/μl),ddh2o 9.5μl。扩增反应程序为:94℃预变性5min;94℃变性30s,52℃退火45s,72℃延伸1min,35个循环;72℃延伸10min。
[0064]
5)采用常规方法进行琼脂糖凝胶电泳,确认扩增产物。
[0065]
6)采用sanger方法双向测序。
[0066]
7)利用cexpress软件拼接得到草麻黄样本4条matk目的序列,然后观察双向序列峰图质量,对每个碱基逐一人工校对。随后采用blastn工具将高质量的matk目的序列在genbank库中进行比对。
[0067]
结果表明,基于matk目的序列,草麻黄样本与e.sinica、e.przewalskii、e.pseudodistachya、e.strobilacea、e.somalensis、e.pachyclada、e.lomatolepis、e.intermedia等麻黄属大多数物种序列相似性及一致性均为100%,表明采用单一matk条形码无法完全鉴别草麻黄与其他近缘物种。
[0068]
实施例4
[0069]
1)准备草麻黄植物干或者湿的样品,具体样本见表4。
[0070]
2)基因组dna提取:采用常规方法提取草麻黄植物的基因组dna。
[0071]
3)采用its2通用引物2f(5'-atgcgatacttggtgtgaat-3')和3r(5'-gacgcttctccagactacaat-3')扩增叶绿体基因its2目的序列。采用25μl pcr体系:easytaqpcr suermix 12.5μl,引物各1μl(10μmol/l),模板dna 1.5μl,ddh2o 9μl。扩增反应程序为:94℃预变性5min;94℃变性30s,56℃退火30s,72℃延伸45s,40个循环;72℃延伸10min。
[0072]
5)采用常规方法进行琼脂糖凝胶电泳,确认扩增产物。
[0073]
6)采用sanger方法测序。
[0074]
8)利用cexpress软件观察峰图质量,对每个碱基逐一人工校对。随后采用blastn工具将高质量的its2目的序列在genbank库中进行比对。
[0075]
基于its2目的序列,草麻黄样本与e.sinica、e.przewalskii、e.saxatilis、e.regeliana、e.distachya、e.monosperma、e.lomatolepis、e.intermedia等麻黄属大多数物种序列相似性及一致性均为100%,图3结果显示,草麻黄样本无法与e.sinica单独聚为一支,表明采用通用its2条形码无法鉴别草麻黄与同属近缘物种。
[0076]
上述实施例中,实施例2-4为对比实施例。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。