1.本发明是一种噻吩类导电化合物的清洁合成方法,属于电子新材料领域。
背景技术:
2.3,4-乙烯基二氧噻吩(edot)是一种性能很稳定的有机导电化合物,掺杂的edot聚合物的电导率能高达≥500s/m,目前已经是长寿命固体电子电容的关键材料,广泛应用于手机电脑主板等用途;1992年keegstra等介绍了采用3,4-二溴噻吩为起始原料制备edot(tetrahedron,1992,48(17):3633-3652);该方法价格昂贵,污染严重,反应条件苛刻,随着近几年溴素价格大幅上涨,这种方法已经被淘汰。
3.cn200810163168,cn200810044836、cn200810025428、cn201210234948、cn201110276698等都大同小异介绍了氯乙酸酯和硫化钠合成的硫代二甘酸酯为起始原料,经过和草酸酯的缩合反应,和二氯乙烷的环醚反应,水解、酸化和脱羧的5步反应合成edot,合成生产流程很长,污染极其严重,因此在工业生产中导致成本很高,生产能力很难产能放大; 。
4.cn201010522532介绍了酒石酸酯为起始原料的合成方法,经过四步合成反应,得到3,4-乙烯基二氧噻吩(edot),依旧是生产流程长,污染严重,合成操作效率依旧很低。
5.现有的edot生产工艺都存在着污染极其严重,合成工艺流程长,生产效率很低,无法有效降低成本。
技术实现要素:
6.针对以上各种edot合成工艺的问题,本发明提供一种能大规模合成3,4-乙烯基二氧噻吩(edot)的清洁方法,采用氯乙醛缩二甲醇为起始原料,经过电偶联、环硫醚和醚交换三步反应,非常清洁地低成本合成3,4-乙烯基二氧噻吩。本发明具有流程很短,成本很低,而且非常清洁环保;由于污染很少,合成工艺流程缩短为3步,生产效率能大幅提高,edot的合成成本也会大幅降低;
。
7.电偶联反应,是在带离子膜的双极电解槽中,必须在阳极室中加入自由基催化剂,并加入koh或naoh的甲醇溶液,调节物料ph保持在弱碱性。接通直流电源电解,氯乙醛缩二甲醇在阳极室自由基偶联,得到2,2,3,3-四甲氧基-1.4-二氯丁烷的式a化学结构化合物;。
8.在氯乙醛缩二甲醇分子中,只有叔碳氢具有很强的自由基活性,自由基催化剂在铂金电极上失去电子,生成的氮—氧自由基夺取氯乙醛缩二甲醇的叔碳氢,生成的叔碳自由基偶联反应,得到2,2,3,3-四甲氧基-1.4-二氯丁烷。
9.环硫醚反应,是2,2,3,3-四甲氧基-1,4-二氯丁烷和硫化钠的醇溶液回流反应,环合得到五元环的2,2,3,3-四甲氧基四氢噻吩的式b化学结构化合物。硫化钠和2,2,3,3-四甲氧基-1,4-二氯丁烷的摩尔比为1.1~1.2:1;。
10.醚交换反应,是2,2,3,3-四甲氧基四氢噻吩和乙二醇混合,加入酸性催化剂,加热裂解,醚交换合成得到3,4-乙烯基二氧噻吩,具有式c的反应式;乙二醇和2,2,3,3-四甲氧基四氢噻吩的摩尔比为1.2~1.8:1;。
11.电偶联反应的自由基催化剂,其主要特征是采用n,n,n-三羟基异氰尿酸的式d化学结构化合物;添加量为氯乙醛缩二甲醇质量的1~5%;。
12.环硫醚反应的醇溶液,采用甲醇,乙醇或异丙醇。
13.醚交换反应的酸性催化剂,采用亚磷酸,甲基膦酸,氨基磺酸,对甲苯磺酸或磷酸铝等。
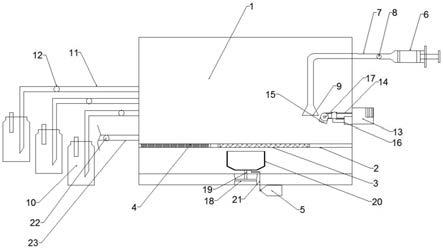
14.附图说明:图1是电偶联合成的双极电解槽:1:不锈钢阴极;2:阴极室;3:离子膜;4:阳极室;5:阳极铂金电极;采用离子膜做隔膜,阴阳极容量各为1000ml,离子膜面积170mm*240mm。
15.具体实施方式:为了更好地理解本发明,本发明介绍以下实施例,但本发明不限于以下的实施例:实施例1:在阳极储料桶中加入1240g氯乙醛缩二甲醇(10mol)、50gn,n,n-三羟基三聚氰酸、20g固体naoh和1240g无水甲醇,搅拌溶解均匀,然后用循环泵a泵送到双极电解槽阳极室不断循环;在阴极储料桶中加入3%naoh水溶液3000g,循环泵b泵送到阴极室不断循环。接通直流电源8v,阳极室不断电偶联,gc分析氯乙醛缩二甲醇含量变化,在gc分析氯乙醛缩二甲醇含量低于5%,电偶联反应结束。
16.阳极室物料减压回收甲醇和未反应氯乙醛缩二甲醇,回收套用;精馏收集130~135度/10mmhg 的式a化学结构的2,2,3,3-四甲氧基-1.4-二氯丁烷727g,gc纯度98.7%。
17.在带球型冷凝管的1000ml三口瓶中加入500g无水甲醇和41g固体氢氧化钠(1mol),磁力搅拌溶解透明后,室温下通入17~18g硫化氢气体吸收,得到新鲜的硫化钠甲醇溶液;然后加入250g精馏的2,2,3,3-四甲氧基-1.4-二氯丁烷,加热回流反应8~10h,析出大量白色nacl晶体,gc分析2,2,3,3-四甲氧基-1.4-二氯丁烷消失,环硫醚反应结束。抽滤除去白色nacl晶体,滤液减压精馏,收集112~115度/12mmhg 的馏分,得到2,2,3,3-四甲氧基四氢噻吩186g,纯度98.2%。
18.在500ml的三口瓶中加入62g乙二醇(1.0mol)和2,2,3,3-四甲氧基四氢噻吩170g(0.82mol),再加入甲基膦酸5g,85~90℃水浴加热,并减压保持在90~100mmhg进行醚交换反应,甲醇被不断分离,直到最后料温升到≥80℃,反应结束。降到室温后加入少量液碱中和到中性,然后进行减压精馏,得到87.3gedot(0.61mol),纯度99.1%。
19.实施例2:在阳极储料桶中加入1240g氯乙醛缩二甲醇(10mol)、40gn,n,n-三羟基三聚氰酸、20g固体naoh和1240g无水甲醇,搅拌溶解均匀,然后用循环泵a泵送到双极电解槽阳极室不断循环;在阴极储料桶中加入3%naoh水溶液3000g,循环泵b泵送到阴极室不断循环。接通直流电源7v,阳极室不断电偶联,gc分析氯乙醛缩二甲醇含量变化,在gc分析氯乙醛缩二甲醇含量低于5%,电偶联反应结束。
20.阳极室物料减压回收甲醇和未反应氯乙醛缩二甲醇,回收套用;精馏收集130~135度/10mmhg 的式a化学结构的2,2,3,3-四甲氧基-1.4-二氯丁烷702g,gc纯度98.4%。
21.在带球型冷凝管的2000ml三口瓶中加入900g无水乙醇和41g固体氢氧化钠(1mol),磁力搅拌溶解透明后,室温下通入17~18g硫化氢气体吸收,得到新鲜的硫化钠乙醇溶液;然后加入250g精馏的2,2,3,3-四甲氧基-1.4-二氯丁烷,加热回流反应8~10h,析出大量白色nacl晶体,gc分析2,2,3,3-四甲氧基-1.4-二氯丁烷消失,环硫醚反应结束。抽滤除去白色nacl晶体,滤液常压蒸馏乙醇回收套用,再减压精馏收集112~115度/12mmhg 的馏分,得到2,2,3,3-四甲氧基四氢噻吩189g,纯度98.6%。
22.在500ml的三口瓶中加入62g乙二醇(1.0mol)和2,2,3,3-四甲氧基四氢噻吩170g(0.82mol),再加入甲基膦酸4g,85~90℃水浴加热,并减压保持在90~100mmhg进行醚交换反应,甲醇被不断分离,直到最后料温升到≥80℃,反应结束。降到室温后加入少量液碱中和到中性,然后进行减压精馏,得到86.2gedot(0.60mol),纯度98.8%。
23.实施例3:在阳极储料桶中加入1240g氯乙醛缩二甲醇(10mol)、30gn,n,n-三羟基三聚氰酸、20g固体koh和1240g无水甲醇,搅拌溶解均匀,然后用循环泵a泵送到双极电解槽阳极室不断循环;在阴极储料桶中加入3%naoh水溶液3000g,循环泵b泵送到阴极室不断循环。接通直流电源8v,阳极室不断电偶联,gc分析氯乙醛缩二甲醇含量变化,在gc分析氯乙醛缩二甲醇含量低于5%,电偶联反应结束。
24.阳极室物料减压回收甲醇和未反应氯乙醛缩二甲醇,回收套用;精馏收集130~135度/10mmhg 的式a化学结构的2,2,3,3-四甲氧基-1.4-二氯丁烷693g,gc纯度97.8%。
25.在带球型冷凝管的2000ml三口瓶中加入1200g无水异丙醇和41g固体氢氧化钠(1mol),磁力搅拌溶解透明后,室温下通入17~18g硫化氢气体吸收,得到新鲜的硫化钠异丙醇溶液;然后加入250g精馏的2,2,3,3-四甲氧基-1.4-二氯丁烷,加热回流反应7~8h,析出大量白色nacl晶体,gc分析2,2,3,3-四甲氧基-1.4-二氯丁烷消失,环硫醚反应结束。抽滤除去白色nacl晶体,滤液常压蒸馏异丙醇回收套用,再减压精馏收集112~115度/12mmhg 的馏分,得到2,2,3,3-四甲氧基四氢噻吩178g,纯度96.5%。
26.在500ml的三口瓶中加入62g乙二醇(1.0mol)和2,2,3,3-四甲氧基四氢噻吩170g(0.82mol),再加入对甲苯磺酸4g,85~90℃水浴加热,并减压保持在90~100mmhg进行醚交换反应,甲醇被不断分离,直到最后料温升到≥80℃,反应结束。降到室温后加入少量液碱中和到中性,然后进行减压精馏,得到67.6gedot(0.48mol),纯度98.3%。
27.实施例4:在阳极储料桶中加入1240g氯乙醛缩二甲醇(10mol)、40gn,n,n-三羟基三聚氰酸、20g固体naoh和1240g无水甲醇,搅拌溶解均匀,然后用循环泵a泵送到双极电解槽阳极室不断循环;在阴极储料桶中加入3%naoh水溶液3000g,循环泵b泵送到阴极室不断循环。接通直流电源7v,阳极室不断电偶联,gc分析氯乙醛缩二甲醇含量变化,在gc分析氯乙醛缩二甲醇含量低于5%,电偶联反应结束。
28.阳极室物料减压回收甲醇和未反应氯乙醛缩二甲醇,回收套用;精馏收集130~135度/10mmhg 的式a化学结构的2,2,3,3-四甲氧基-1.4-二氯丁烷708g,gc纯度98.2%。
29.在带球型冷凝管的2000ml三口瓶中加入700g无水甲醇和41g固体氢氧化钠(1mol),磁力搅拌溶解透明后,室温下通入17~18g硫化氢气体吸收,得到新鲜的硫化钠甲醇溶液;然后加入250g精馏的2,2,3,3-四甲氧基-1.4-二氯丁烷,加热回流反应8~10h,析出大
量白色nacl晶体,gc分析2,2,3,3-四甲氧基-1.4-二氯丁烷消失,环硫醚反应结束。抽滤除去白色nacl晶体,滤液常压蒸馏甲醇回收套用,再减压精馏收集112~115度/12mmhg 的馏分,得到2,2,3,3-四甲氧基四氢噻吩183g,纯度98.3%。
30.在500ml的三口瓶中加入62g乙二醇(1.0mol)和2,2,3,3-四甲氧基四氢噻吩170g(0.82mol),再加入亚磷酸4g,85~90℃水浴加热,并减压保持在90~100mmhg进行醚交换反应,甲醇被不断分离,直到最后料温升到≥80℃,反应结束。降到室温后加入少量液碱中和到中性,然后进行减压精馏,得到77.2gedot(0.54mol),纯度98.9%。
31.实施例5:在阳极储料桶中加入1240g氯乙醛缩二甲醇(10mol)、50gn,n,n-三羟基三聚氰酸、30g固体naoh和1240g无水甲醇,搅拌溶解均匀,然后用循环泵a泵送到双极电解槽阳极室不断循环;在阴极储料桶中加入3%naoh水溶液3000g,循环泵b泵送到阴极室不断循环。接通直流电源7v,阳极室不断电偶联,gc分析氯乙醛缩二甲醇含量变化,在gc分析氯乙醛缩二甲醇含量低于5%,电偶联反应结束。
32.阳极室物料减压回收甲醇和未反应氯乙醛缩二甲醇,回收套用;精馏收集130~135度/10mmhg 的式a化学结构的2,2,3,3-四甲氧基-1.4-二氯丁烷738g,gc纯度98.5%。
33.在带球型冷凝管的2000ml三口瓶中加入800g无水甲醇和41g固体氢氧化钠(1mol),磁力搅拌溶解透明后,室温下通入17~18g硫化氢气体吸收,得到新鲜的硫化钠甲醇溶液;然后加入250g精馏的2,2,3,3-四甲氧基-1.4-二氯丁烷,加热回流反应8~10h,析出大量白色nacl晶体,gc分析2,2,3,3-四甲氧基-1.4-二氯丁烷消失,环硫醚反应结束。抽滤除去白色nacl晶体,滤液常压蒸馏甲醇回收套用,再减压精馏收集112~115度/12mmhg 的馏分,得到2,2,3,3-四甲氧基四氢噻吩181g,纯度98.6%。
34.在500ml的三口瓶中加入62g乙二醇(1.0mol)和2,2,3,3-四甲氧基四氢噻吩170g(0.82mol),再加入氨基磺酸4g,85~90℃水浴加热,并减压保持在90~100mmhg进行醚交换反应,甲醇被不断分离,直到最后料温升到≥80℃,反应结束。降到室温后加入少量液碱中和到中性,然后进行减压精馏,得到81.2gedot(0.57mol),纯度98.1%。
35.实施例6:在阳极储料桶中加入1240g氯乙醛缩二甲醇(10mol)、50gn,n,n-三羟基三聚氰酸、30g固体naoh和1240g无水甲醇,搅拌溶解均匀,然后用循环泵a泵送到双极电解槽阳极室不断循环;在阴极储料桶中加入3%naoh水溶液3000g,循环泵b泵送到阴极室不断循环。接通直流电源7v,阳极室不断电偶联,gc分析氯乙醛缩二甲醇含量变化,在gc分析氯乙醛缩二甲醇含量低于5%,电偶联反应结束。
36.阳极室物料减压回收甲醇和未反应氯乙醛缩二甲醇,回收套用;精馏收集130~135度/10mmhg 的式a化学结构的2,2,3,3-四甲氧基-1.4-二氯丁烷733g,gc纯度98.8%。
37.在带球型冷凝管的2000ml三口瓶中加入800g无水甲醇和48g固体氢氧化钠(1.2mol),磁力搅拌溶解透明后,室温下通入19~20g硫化氢气体吸收,得到新鲜的硫化钠甲醇溶液;然后加入250g精馏的2,2,3,3-四甲氧基-1.4-二氯丁烷,加热回流反应8~10h,析出大量白色nacl晶体,gc分析2,2,3,3-四甲氧基-1.4-二氯丁烷消失,环硫醚反应结束。抽滤除去白色nacl晶体,滤液常压蒸馏甲醇回收套用,再减压精馏收集112~115度/12mmhg 的馏分,得到2,2,3,3-四甲氧基四氢噻吩184g,纯度98.2%。
38.在500ml的三口瓶中加入75g乙二醇(1.2mol)和2,2,3,3-四甲氧基四氢噻吩170g(0.82mol),再加入磷酸铝4g,85~90℃水浴加热,并减压保持在90~100mmhg进行醚交换反应,甲醇被不断减压抽出分离,直到最后料温升到≥80℃,反应结束。降到室温后加入少量液碱中和到中性,然后进行减压精馏,得到51.6gedot(0.36mol),纯度94.1%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。