用于抑制e
‑
选择蛋白和cxcr4趋化因子受体的双靶向化合物
技术领域
1.本文公开了用于治疗和/或预防包括炎性疾病和癌症在内的至少一种疾病、病症和/或疾病状态的化合物、组合物和方法,具体而言,本发明涉及抑制e
‑
选择蛋白和cxcr4趋化因子受体的双靶向化合物及其用途。
背景技术:
2.当组织被感染或受损时,炎症过程将白细胞和其它免疫系统组分引导至感染或损伤部位。在此过程中,白细胞在微生物的吞噬和消化中起重要作用。白细胞向受感染或受损组织的募集对于建立有效的免疫防御至关重要。
3.选择蛋白是一类结构类似的血管内皮细胞表面受体,其主要作用之一是介导白细胞与血管内皮细胞结合,将白细胞征集至受感染或受损的组织,是免疫防御的关键步骤。
4.有三种已知的选择蛋白:e
‑
选择蛋白,p
‑
选择蛋白和l
‑
选择蛋白。e
‑
选择蛋白存在于活化的内皮细胞的表面,其排列在毛细血管的内壁上。e
‑
选择蛋白结合碳水化合物唾液酸化的
‑
lewis
x
(sle
x
),其在某些白细胞(单核细胞和嗜中性粒细胞)的表面上以糖蛋白或糖脂形式存在,并帮助这些细胞粘附在周围组织被感染或受损的区域中的毛细血管壁上;e
‑
选择蛋白也与在许多肿瘤细胞上表达的唾液酸化的
‑
lewis
a
(sle
a
)结合。p
‑
选择蛋白在发炎的内皮和血小板上表达,并且还识别sle
x
和sle
a
,但也包含与硫酸化的酪氨酸相互作用的第二位点。当毛细血管附近的组织被感染或受损时,通常e
‑
选择蛋白和p
‑
选择蛋白的表达增加。l
‑
选择蛋白在白细胞上表达。选择蛋白
‑
介导的细胞间粘附是选择蛋白
‑
介导的功能的一个实例。
5.血管生成对癌症的进展至关重要。越来越多的证据表明,选择蛋白在血管生成中起着重要作用。从活化的血管内皮细胞释放的e
‑
选择蛋白是一种粘附分子,它通过募集和激活各种白细胞至炎症部位而在炎症反应中起主要作用。肿瘤血管系统处于发炎状态,表达包括e
‑
选择蛋白在内的多个重要因子,研究也表明,e
‑
选择蛋白与肿瘤免疫抑制相关的肿瘤核心血管系统中,e
‑
选择蛋白在肿瘤周围血管系统中的表达更为广泛。
6.某些癌症全部或部分起源于骨中,如果能在癌症离开原发部位之前进行治疗,很多癌症的可治疗性都很高。然而,一旦癌症从原发部位散播,通常治疗选择就很有限了,并且存活的统计数据显著下降。因此,需要防止癌细胞离开原发部位或防止癌细胞从血流外渗并浸润其它组织。将癌细胞保留在血流中使细胞对诸如化疗的治疗的敏感性更强。
7.选择蛋白抑制剂主要用于阻止癌细胞通过血液转移。复发及转移性癌细胞表面大量表达唾液酸路易斯糖(sle
x/a
),sle
x/a
和e
‑
选凝蛋白的相互作用是癌细胞通过血液转移的关键因素之一。目前90%以上的癌症死亡是由于癌转移造成的事实,所以治疗过程中阻止癌细胞转移是治疗成功的关键。
8.研究表明,e
‑
/p
‑
选择蛋白抑制剂可以有效阻止血液癌细胞对骨髓的粘附。对于各类白血病、多发性骨髓瘤以及恶性淋巴瘤等,部分癌细胞粘附于骨髓及其它组织的血管内皮,是造成药物阻抗及病情复发的主要原因。选择蛋白抑制剂能有效阻止这种粘附作用,并
将粘附的癌细胞释放到血液中,有效提高化学治疗效果。
9.另外,cxcr4属于cxc趋化因子受体家族,是趋化因子基质细胞衍生因子
‑
i(sdf
‑
1,cxcl12)的特异性受体,与sdf
‑
i结合后会使细胞产生趋化作用。cxcr4趋化因子表达于多种细胞表面,在身体免疫系统和循环系统的生长发育中起着重要的作用。cxcr4趋化因子在很多癌组织中过量表达,在肿瘤细胞的增殖、侵润、血管增生以及转移中起到非常重要的作用。cxcr4在23种不同类型肿瘤中均有表达,是肿瘤细胞表达最为普遍的趋化因子受体。在乳腺癌患者中,cxcr4高表达和癌转移正比例相关,且预后较差。在结直肠癌患者中,cxcr4高表达与肿瘤的复发和转移正比例相关,并且相应患者生存率也较低。在卵巢癌、黑素瘤、前列腺癌、神经母细胞瘤等其他肿瘤的研宄中也得到了相似的结果。因此,cxcr4也是靶向治疗肿瘤及肿瘤转移的研究新热点。
10.目前已有一些用于抑制e
‑
选择蛋白和cxcr4趋化因子受体的异双功能化合物(参见,例如,61/174,5802009.05.01us)。但是现有的技术方案中,e
‑
/p
‑
选择蛋白的活性较低,只能与低活性的cxcr4趋化因子受体抑制剂配合连接,化合物的整体活性不高,导致最终药物剂量需求大,不利于对应疾病的治疗。
技术实现要素:
11.针对上述问题,本发明提供一种用于治疗疾病和用于改进e
‑
选择蛋白及cxcr4趋化因子受体发挥作用的化合物、组合物和方法。在本发明中,化合物是双靶向抑制剂,其中e
‑
选择蛋白抑制剂连接于cxcr4趋化因子受体抑制剂,在e
‑
选择蛋白抑制剂结构上引入硫酸酯基团,以提高e
‑
选择蛋白活性,增加p
‑
选择蛋白活性,通过连接高活性cxcr4抑制剂以提高药物的总体效果。所述化合物可以与药学可接受的载体或稀释剂组合而形成药物组合物。所述化合物可用于治疗癌细胞可能离开原发部位的癌症,或用于治疗疾病中发生细胞的粘附或迁移的炎性疾病,或用于将诸如干细胞(例如,骨髓祖细胞)的细胞释放到循环血液中并增强所述细胞在血液中的保留(例如,使细胞移出骨髓并将所述细胞保留在外周血流中)。
12.本发明提供用于抑制e
‑
选择蛋白和cxcr4趋化因子受体的双靶向化合物,该化合物具有下式:
[0013][0014]
其中:
[0015]
l为连接子基团,cd为cxcr4趋化因子受体抑制剂;
[0016]
r1为
‑
nme2、
‑
ona、
‑
nhoh、
‑
nhoch3或
‑
nc(=o)y1,其中y1为c1‑8烷基、c2‑8烯基、c2‑8炔
基、c1‑8卤代烷基、c2‑8卤代烯基或c2‑8卤代炔基;
[0017]
r2为
‑
oh、
‑
nh2、
‑
oc(=o)y2、
‑
nhc(=o)y3或
‑
nhc(=o)vy4,其中y2和y3可以相同或不同,且独立地为c1‑8烷基、c2‑8烯基、c2‑8炔基、c1‑8卤代烷基、c2‑8卤代烯基、c2‑8卤代炔基、c6‑
18
芳基或c1‑
13
杂芳基;v为化学键、(ch2)
n
基团、基团、n为1至20的整数,m为0至20的整数;y4为硫酸酯盐;
[0018]
r3为
‑
oh、
‑
nh2、
‑
oc(=o)y2、
‑
nhc(=o)y3或
‑
nhc(=o)vy4,其中y2、y3、v和y4的定义与前文相同;
[0019]
r2和r3中至少有一个为
‑
nhc(=o)vy4;和
[0020]
r4为c1‑8烷基,c1‑8卤代烷基,c3‑6环烷基。
[0021]
在一些实施方案中,y4为为其中o为2至5的整数,p为2至4的整数,q为2至3的整数。
[0022]
在一些实施方案中,该化合物具有下式:
[0023][0024]
其中l为连接子基团,cd为cxcr4趋化因子受体抑制剂。
[0025]
在一些实施方案中,该化合物具有下式:
[0026][0027]
其中l为连接子基团,cd为cxcr4趋化因子受体抑制剂。
[0028]
在一些实施方案中,cd为四氢奎诺林基cxcr4趋化因子受体抑制剂、对二甲苯烯二胺基cxcr4趋化因子受体抑制剂、吲哚基cxcr4趋化因子受体抑制剂或环五肽基cxcr4趋化因子受体抑制剂。
[0029]
在一些实施方案中,该化合物具有下式:
[0030][0031]
其中l为连接子基团。
[0032]
在一些实施方案中,该化合物具有下式:
[0033][0034]
其中l为连接子基团。
[0035]
在一些实施方案中,该化合物具有下式:
[0036][0037]
其中l为连接子基团。
[0038]
在一些实施方案中,该化合物具有下式:
[0039][0040]
其中l为连接子基团。
[0041]
在一些实施方案中,该化合物具有下式:
[0042][0043]
其中l为连接子基团。
[0044]
在一些实施方式中,化合物的连接子基团l为
‑
(ch2ch2o)
n
‑
,其中n为1至50的整数。
[0045]
在一些实施方式中,化合物的连接子基团l为
‑
nhc(o)(ch2)
n
c(o)nh
‑
,其中n为1至12的整数。
[0046]
在一些实施方式中,化合物的连接子基团l为
‑
(ch2)
n
‑
,其中n为2至22的整数。
[0047]
在一些实施方式中,化合物的连接子基团l具有下列结构式:
[0048][0049]
在一些实施方式中,化合物的连接子基团l具有下列结构式:
[0050][0051]
本发明提供包含本发明化合物和药学可接受的载体或稀释剂的药物组合物,以上化合物可以用于制备药物,该药物可以用于治疗癌细胞可能离开原发部位的癌症、希望使癌细胞从某一位置移动到血流并将所述癌细胞保留在所述血流中的癌症、将干细胞释放到循环血液中并增强所述细胞在所述血液中的保留的药物或疾病中发生细胞粘附或迁移的炎性疾病。
[0052]
本发明的有益效果是:
[0053]
(1)在e
‑
选择蛋白上引入硫酸酯基团,大幅提升了e
‑
/p
‑
选择蛋白的活性;
[0054]
(2)由于e
‑
/p
‑
选择蛋白活性得以提高,该双靶向化合物可连接更高活性的cxcr4抑制剂,化合物的整体活性亦得到提升,从而可降低最终药物的使用剂量。
附图说明
[0055]
图1(图1a、图1b、图1c、图1d及图1e)是本发明一种实施方式中双靶向化合物c4的合成示意图;
[0056]
图2是化合物a和c4对e
‑
选择蛋白的ic
50
值的比较,化合物a是已知的e
‑
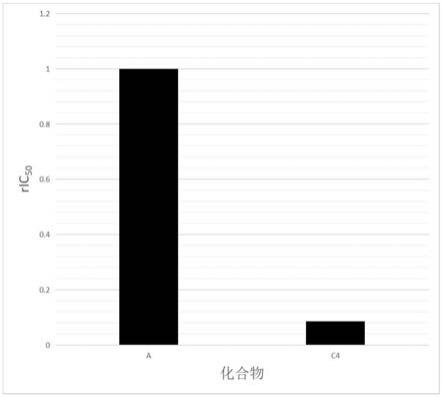
选择蛋白抑制剂,其为专利cn102421441a中的化合物#1,化合物c4为本发明提供的诸多实施例之一。
具体实施方式
[0057]
如上文所述,本发明提供了用于治疗e
‑
选择蛋白和cxcr4趋化因子受体发挥作用的基本,以及用于增强细胞在释放到循环血液中后的保留的化合物、组合物及方法。所述化合物具有多种体外和体内用途。
[0058]
本文所用的术语“e
‑
选择蛋白抑制剂”是指e
‑
选择蛋白的抑制剂,以及e
‑
选择蛋白和p
‑
选择蛋白或l
‑
选择蛋白或e
‑
选择蛋白和p
‑
选择蛋白和l
‑
选择蛋白的抑制剂。
[0059]
本发明的化合物是双靶向化合物,其中e
‑
选择蛋白抑制剂与cxcr4趋化因子受体抑制剂连接(即共价连接)。该化合物包含下式或由下式组成:e
‑
选择蛋白抑制剂——连接子基团——cxcr4趋化因子受体抑制剂。
[0060]
在一种实施方案中,化合物具有下式结构:
[0061][0062]
其中l为连接子基团,cd为cxcr4趋化因子受体抑制剂。
[0063]
在一种实施方案中,化合物具有下式结构:
[0064][0065]
其中l为连接子基团,cd为cxcr4趋化因子受体抑制剂。
[0066]
cxcr4趋化因子受体抑制剂是本领域公知的,该抑制剂通常可以阻止基质衍生因子
‑
1(sdf
‑
1)与cxcr4受体的结合。cxcr4趋化因子受体抑制剂的实施方案包括:
[0067]
四氢奎诺林基cxcr4趋化因子受体抑制剂:
[0068][0069]
对二甲苯烯二胺基cxcr4趋化因子受体抑制剂:
[0070][0071]
吲哚基cxcr4趋化因子受体抑制剂:
[0072][0073]
环五肽基cxcr4趋化因子受体抑制剂:
[0074][0075]
在本发明的化合物中,e
‑
选择蛋白抑制剂和cxcr4趋化因子受体抑制剂通过连接子基团共价连接,连接子基团可以是或包括间隔基团,例如
‑
(ch2)
n
‑
,其中n为2至22的整数。间隔基团的其它实施例包括羰基或含羰基的基团,其一种实施方案为
[0076]
其它连接子基团为本领域技术人员熟悉的或为本公开所有,例如聚乙二醇peg链
‑
(ch2ch2o)
n
‑
,其中n=1至50的整数。
[0077]
在另一种实施方案中,连接子基团为
‑
nhc(o)(ch2)
n
c(o)nh
‑
,其中n为1至12的整数。
[0078]
在另一种实施方案中,连接子基团具有下列结构式:
[0079][0080]
在本发明的一种实施方案中,该化合物具有下列结构式:
[0081][0082]
其中,l为连接子基团,cd为cxcr4趋化因子受体抑制剂。
[0083]
在本公开中,有数个化学缩写:me为甲基,bz为苯甲酰基。
[0084]
r1处取代基的选择包括
‑
nme2、
‑
ona、
‑
nhoh、
‑
nhoch3或
‑
nc(=o)y1,其中y1为c1‑8烷基、c2‑8烯基、c2‑8炔基、c1‑8卤代烷基、c2‑8卤代烯基或c2‑8卤代炔基。
[0085]
r2处取代基的选择包括
‑
oh、
‑
nh2、
‑
oc(=o)y2、
‑
nhc(=o)y3或
‑
nhc(=o)vy4,其中y2和y3可以相同或不同,且独立地为c1‑8烷基、c2‑8烯基、c2‑8炔基、c1‑8卤代烷基、c2‑8卤代烯基、c2‑8卤代炔基、c6‑
18
芳基或c1‑
13
杂芳基;v为化学键、(ch2)
n
基团、基团、n为1至20的整数,m为0至20的整数;y4为硫酸酯盐。
[0086]
r3处取代基的选择包括
‑
oh、
‑
nh2、
‑
oc(=o)y2、
‑
nhc(=o)y3或
‑
nhc(=o)vy4,其中y2、y3、v和y4的定义与前文相同;r2和r3中至少有一个为
‑
nhc(=o)vy4;和
[0087]
r4处取代基的选择包括c1‑8烷基,c1‑8卤代烷基或c3‑6环烷基。
[0088]
本文所用的“c1‑8烷基”是指具有1
‑
8个碳原子的烷取代基,并且可以是直链的、分支的或环状的(环烷基),实例是甲基、乙基、丙基、异丙基、丁基和t
‑
丁基。“c1‑8卤代烷基”是指具有至少一个卤素的“c1‑8烷基”,如果存在多个卤素,所存在的卤素可以是相同的或不同的。
[0089]
本文所用的“c2‑8烯基”是指具有2
‑
8个碳原子、至少一个碳
‑
碳双键的烯取代基,并且是可以直链的、分支的或环状的(环烯基),实例与“c1‑8烷基”的实例相似,但是具有至少一个碳
‑
碳双键。“c2‑8卤代烯基”是指具有至少一个卤素的“c2‑8烯基”,如果存在多个卤素,所存在的卤素可以是相同的或不同的。
[0090]
本文所用的“c2‑8炔基”是指具有2
‑
8个碳原子、至少一个碳
‑
碳三键的炔取代基,并且是可以直链的、分支的或环状的(环炔基),实例与“c1‑8烷基”的实例相似,但是具有至少一个碳
‑
碳三键。“c2‑8卤代炔基”是指具有至少一个卤素的“c2‑8炔基”,如果存在多个卤素,所存在的卤素可以是相同的或不同的。
[0091]
本文所用的“芳基”是指具有6
‑
18个碳原子的芳香族取代基,这些碳原子是作为一个或多个环中的环原子,所述一个或多个环可以被键隔开或可以是稠合的。“杂芳基”与“芳基”相似,但是芳族取代基具有至少一个取代环碳的杂原子(例如n、o或s),其可能具有少于6个碳环原子。“芳基”与“杂芳基”的实例包括苯基、萘基、吡啶基、嘧啶基、三唑基、呋喃基、恶唑基、苯硫基、喹啉基和联苯基。
[0092]
在一些实施方式中,硫酸酯盐y4为
其中o为2至5的整数,p为2至4的整数,q为2至3的整数。
[0093]
在一种实施方式中,连接子基团为cd为四氢奎诺林基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0094][0095]
在一种实施方式中,连接子基团为cd为环五肽基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0096]
[0097]
在一种实施方式中,连接子基团为cd为吲哚基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0098][0099]
在一种实施方式中,连接子基团为为四氢奎诺林基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0100][0101]
在一种实施方式中,连接子基团为为四氢奎诺林基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0102][0103]
在一种实施方式中,连接子基团为cd为对二甲苯烯二胺基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0104][0105]
在一种实施方式中,连接子基团为cd为四氢奎诺林基cxcr4趋化因子受体抑制剂,所述化合物具有下式:
[0106][0107]
本文所述的化合物可以存在于药物组合物中。药物组合物包含与一种或多种药学或生理上可接受的载体、稀释剂或赋形剂的组合的一种或多种化合物。所述组合物可以包含缓冲液、碳水化合物、甘露醇、蛋白、多肽或诸如甘氨酸的氨基酸、抗氧化剂、诸如edta或谷胱甘肽的螯合剂、诸例如氢氧化铝的佐剂或防腐剂。在其他实施方案中,本发明的组合物可以被配制为冻干制剂。可以根据任何合适的给药方式来配制本发明的组合物,所述给药方式包括例如,局部给药、口服给药、鼻腔给药、静脉内给药、颅内给药、腹膜内给药、皮下给药或肌肉内给药。
[0108]
在一个实施方案中,所述化合物可用于癌细胞可能离开原发部位的癌症的治疗方法。
[0109]
在另一实施方案中,包括其等同物在内的上文所述化合物,可用于希望使癌细胞从部位移动到血流中并将癌细胞保留在血流中的癌症的治疗方法。
[0110]
在另一实施方案中,包括其等同物在内的上文所述化合物,可用于将细胞(诸如造血干细胞)释放到循环血液中并增强所述细胞在血液中的保留的方法。
[0111]
在另一实施方案中,包括其等同物在内的上文所述化合物,可用于疾病中发生细胞粘附或迁移的炎性疾病的治疗方法。
[0112]
供以下实施例是为了举例说明,而并非限制。
[0113]
实施例1 双靶向化合物c4的合成
[0114]
如图1a内容所示,
[0115]
化合物2的合成:可参照文献j.med.chem.1999,42,4909
‑
4913,synthesis andbiological evaluation of a potent e
‑
selectinantagonist,g.thoma,et al的内容进行制备。
[0116]
化合物4的合成:将化合物3(0.9g)溶于甲醇(12ml)中,向该溶液中加入bu2sno(0.4g),并将混合物回流2小时。蒸发掉溶剂,将残留的溶剂与甲苯共蒸发3次。将残余物溶于二甲氧基乙烷(15ml)中,向该溶液中加入csf(0.8g)和化合物2(2.1g),将反应混合物在室温下搅拌过夜,蒸发掉溶剂。通过硅胶柱色谱法纯化残余物,得到化合物4(0.8g)。
[0117]
化合物8的合成:可参照文献j.med.chem.2007,50,1101
‑
1115,rational design of novel,potent small molecule pan
‑
selectin antagonists,r.kranich,et al.和
bioorganic&medicinal chemistry letters 11(2001)923
–
925,synthesis andbiological evaluation ofa sialyl lewis x mimicwith significantly improved e
‑
selectin inhibition,g.thoma,et al的内容进行制备。
[0118]
化合物10的合成:在0℃下,将br2(0.08ml)溶液滴加到乙基
‑
2,3,4
‑
三
‑
o
‑
苄基
‑
α
‑
l
‑
硫代吡喃岩藻糖苷(9,640mg)的dcm溶液(10ml)中,并在0℃下搅拌1小时,加入环己烷,将反应混合物再搅拌30分钟。将该混合物逐滴加入到含有分子筛的溶有化合物8(310mg)和et4nbr(280mg,在20℃下烘箱干燥)的dmf/ch2cl2(20ml,1:1)溶液中,在室温下搅拌24小时,用吡啶(2ml)终止反应,用硅藻土过滤,用dcm(20ml)洗涤,用盐水(50ml)洗涤有机溶液,用dcm(3
×
50ml)萃取水层3次,干燥合并得有机层(na2so4),过滤,浓缩至干燥,粗产物用硅胶柱色谱纯化,得到化合物10(144mg)。
[0119]
化合物11的合成:在室温和氮气下,取化合物4(3g)、化合物10(6g)和活化的粉末状分子筛4a在dcm(30ml)中混合搅拌2小时,然后在1小时时间内分四等分地加入dmtst(3g),在室温下搅拌,持续搅24小时,用硅藻土过滤,用dcm(200ml)稀释,用盐水(2x50ml)洗涤,干燥合并得有机层(na2so4),过滤,浓缩至干燥,粗产物用硅胶柱色谱纯化,得到化合物11(3g)。
[0120]
如图1b内容所示,
[0121]
化合物13的合成:向配制在dmso(10ml)中的化合物12(1g)的溶液添加4
‑
羟甲基苯甲醛(0.3g)、nabh3cn(0.5g)和acoh(0.6g),将反应混合物在60℃下搅拌2小时,蒸除溶剂,粗产物用硅胶柱色谱纯化,得到化合物13(0.95g)。
[0122]
化合物14的合成:将配制在dcm(3ml)中的(cocl)2(0.15ml)溶液冷却至
‑
78℃,向该溶液逐滴搅拌添加dmso(0.25ml),且在
‑
78℃下持续搅拌15分钟。在
‑
78℃下,将配制在dcm(3ml)中的化合物13(1g)逐滴搅拌添加至上述混合物。在
‑
78℃下持续搅拌15分钟,添加dipea(1.17ml)并搅拌15分钟。将反应混合物缓慢升温至室温,浓缩干燥,通过硅胶柱色谱纯化粗产物,从而得到化合物14(800mg)。
[0123]
如图1c内容所示,
[0124]
化合物15的合成:在化合物11(100g)的dcm(200ml)溶液中加入三苯基膦(5g)和三丁基氢化锡(2g),在室温下搅拌30分钟,再加入正三氟乙酰甘氨酸酐(22g),继续搅拌。浓缩干燥,通过硅胶柱色谱纯化粗产物,从而得到化合物15(95g)。
[0125]
化合物16的合成:向化合物15(10g)在甲醇
‑
水(10:1,50ml)溶液中加入10%pd
‑
c(1g),将反应混合物在氢气(30psi)下室温搅拌24小时,用硅藻土过滤并将溶剂蒸除,得到化合物中间体(6g),干燥后加入甲醇(80ml)和naome的甲醇溶液(25%,0.5ml),室温搅拌4小时,用乙酸中和至ph=7,将溶剂蒸除,得到化合物16(5g)。
[0126]
化合物17的合成:向化合物16(1g)在dcm溶液中加入二甲基氨,室温搅拌16小时,将溶剂蒸除,得到化合物中间体(1.1g),向该中间体(1.1g)中加入乙二胺,加热搅拌5小时,将溶剂蒸除,通过sepadeg25柱得到化合物17(0.3g)。
[0127]
化合物18的合成:向配制在dmso(20ml)中的化合物14(5g)的溶液添加化合物17(7g)和nabh3cn(8mg)和acoh(6mg),将反应混合物在60℃下搅拌2小时,蒸发掉溶剂。通过hplc(反相c18柱)纯化残留物,从而得到化合物18(6.5g)。
[0128]
如图1d内容所示,
[0129]
化合物19c的合成:将化合物19a(5g)和化合物19b(4.5g)溶解在ch2cl2(100ml)中,向该溶液中加入分子筛(4a,1g)并在室温下搅拌30分钟,然后将溶液冷却至0℃并搅拌加入bf3.et2o(0.25ml溶于5ml中),将反应混合物在0℃下搅拌2小时。加入et3n(0.5ml),蒸发溶剂,通过柱色谱(硅胶)纯化残余物,得到化合物19c(7g)。
[0130]
化合物19d的合成:向化合物19c(7g)在甲醇(100ml)溶液中加入naome的甲醇溶液,室温条件下搅拌4小时,用醋酸中和至ph=7,蒸发溶剂,得到化合物19d(4g)。
[0131]
化合物19e的合成:将化合物19d(4g)溶在n,n
‑
二甲基甲酰胺(50ml)中,加入吡啶
‑
三氧化硫溶液(5
‑
20mol/mol),保持在
‑
10℃下搅拌1小时,升温至室温后,降至
‑
20℃,反应通过加入冷水(16ml)终止,用氢氧化钠溶液将ph值调至10.0,用乙醇(3体积)稀释,在0℃下放置2小时,得到白色沉淀19e(6g)。
[0132]
化合物19f的合成:向化合物19e(6g)的甲醇
‑
水(10:1,100ml)溶液中加入10%pd
‑
c(1g),将反应混合物在氢气(30psi)下室温搅拌24小时,用硅藻土过滤并将溶剂蒸除,得到化合物19f(5g)。
[0133]
化合物19的合成:向化合物19f(5g)的dcm(50ml)溶液中加入二环己基碳二亚胺(dcc,4.5g),三乙胺(3g)和n
‑
羟基琥珀酰亚胺(2g),将反应混合物在室温搅拌24小时,蒸发溶剂,通过柱色谱(硅胶)纯化残余物,得到化合物19(4.6g)。
[0134]
如图1e内容所示,
[0135]
化合物18a的合成:向化合物18(1g)的甲醇(10ml)溶液中加入naome,将反应混物在室温搅拌4小时,蒸发溶剂,通过柱色谱(硅胶)纯化残余物,得到化合物18a(0.83g)。
[0136]
化合物20的合成:向化合物18a(0.83g)的dcm(10ml)溶液中加入化合物19(1.2g)、二环己基碳二亚胺(dcc,0.5g)和三乙胺(0.3g),将反应混合物在室温搅拌24小时,蒸发溶剂,通过柱色谱(硅胶)纯化残余物,得到化合物20(0.9g),即双靶向化合物c4。
[0137]
实施例2 评价化合物c4与cxcr4的结合的测定
[0138]
该测定评价糖拟化合物抑制藻红蛋白("pe")偶联抗cxcr4抗体与suptl细胞表面上的cxcr4结合的能力。suptl细胞是源于淋巴母细胞性白血病的t淋巴母细胞,并且在细胞表面组成型表达cxcr4。该细胞购自atcc(atcc号:crl
‑
1942)。抗人cxcr4
‑
藻红蛋白单克隆抗体(抗cxcr4
‑
pe)购自r&dsystems(目录号:fab170p、克隆12g5)。使细胞生长在补充了10%fbs的rpmi1640培养基中。通过将细胞以400g离心10分钟来洗涤约2x106个细胞,洗涤3次,并将细胞团重悬于pbs 0.05%bsa中。第三次离心后,将细胞团重悬于pbs bsa中至5x105个细胞/ml的浓度。为了封闭非特异性结合,向细胞添加人ig,至浓度为lμg/l05个细胞。然后,将200μl(l x 105个细胞)添加至5ml聚丙烯圆底管(falcon2063管)中。将化合物c4以0.5、5、10和50μm的终浓度添加至细胞。为了达到0.5μm的终浓度,将2.2μl 50μm化合物c4 19.8μl pbs/bsa添加至200μl细胞中。为了达到5μm的终浓度,将22μl 50μm化合物c4添加至200μl细胞中。为了达到10μm的终浓度,将4.4μl 500μm化合物c4 17.6μl pbs/bsa添加至200μl细胞中。为了达到50μm的终浓度,将22μl 500μm化合物c4添加至200μl细胞中。为了达到lμmamd
‑
3100的终浓度,将4.4μl 50μmamd
‑
3100 17.6μl pbs/bsa添加至200μl细胞中,且为了达到5μm的终浓度,将22μl 50μmamd
‑
3100添加至200μl细胞中。此外,用1μg/ml sdf
‑
1a(r&dsystems,目录号350
‑
ns)处理一管细胞,sdf
‑
lα是cxcr4的天然配体。将管在4℃下放置15分钟。然后,除了一管细胞加入10yl小鼠igg 2a同型对照抗体之外,其余各管加入10μl
抗cxcr4
‑
pe。将管在4℃下孵育45分钟。用pbs 0.05%bsa洗涤细胞2次,将最终的细胞团重悬于100μl pbs/bsa中。为了固定样品,向每管添加100ml2%甲醒(polysciences,inc.超纯em级,目录号04018)。利用cytomationmoflo仪器进行流式细胞术。
[0139]
结果如下表所示,化合物c4以剂量依赖方式抑制抗cxcr4
‑
pe与suptl细胞的结合,ic
50
为0.4μm。sdf
‑
1α有效抑制抗体与cxcr4的结合。
[0140][0141][0142]
实施例3 e
‑
选择蛋白活性
‑
结合测定
[0143]
筛选e
‑
选择蛋白的糖模拟拮抗剂的抑制测定是竞争性结合测定,其允许测定ic
50
值。通过在37℃下、96孔微量滴定板中孵育2小时来固定e
‑
选择蛋白/ig嵌合体。为了减少非特异性结合,向每孔添加牛血清白蛋白,并在室温孵育2小时。洗涤平板,在生物素化的sle
a
聚丙烯酰胺与链霉抗生物素/辣根过氧化物酶的偶联物的存在下,向孔中添加受试化合物的连续稀释液,并在室温孵育2小时。为了测定与洗涤后固定的e
‑
选择蛋白结合的sle
a
的量,添加过氧化物酶底物3,3',5,5'四甲基联苯胺(tmb)。3分钟后,通过添加h3po4终止酶反应,在450nm波长下测定吸光度。测定抑制50%结合所需要的受试化合物的浓度,并将其报告为每种e
‑
选择蛋白糖模拟拮抗剂的ic
50
值。
[0144]
实施例4 p
‑
选择蛋白活性
‑
结合测定
[0145]
具体步骤与实施例3大致相同,区别仅在于,筛选p
‑
选择蛋白的糖模拟拮抗剂的抑制测定是竞争性结合测定,即在微量滴定板中固定p
‑
选择蛋白/ig嵌合体。
[0146]
通过与实施例1相似的工艺可制备说明书前文提及的双靶向化合物c1、c2、c3、c5、c6和c7,各个化合物的测定结果如下表所示。
[0147][0148][0149]
通过上述测定结果可知,本发明提供的实施例中,在e
‑
选择蛋白结构的不同部位引入硫酸酯基团,以此使e
‑
选择蛋白的活性提高10倍以上,使p
‑
选择蛋白的活性提高100倍以上;同时由于e
‑
/p
‑
选择蛋白活性得以提高,本发明提供的实施例可以连接更高活性的cxcr4趋化因子受体抑制剂,提高了整个化合物的活性,降低最终药物的用药剂量。
[0150]
本领域的技术人员可以明确,在不脱离本发明的总体精神以及构思的情形下,可以做出对于以上实施例的各种变型。其均落入本发明的保护范围之内。本发明的保护方案以本发明所附的权利要求书为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。