一种n
‑
亚氨基吡啶叶立德双酰胺化合物的合成方法
技术领域
1.本发明属于有机合成领域,具体涉及一种n
‑
亚氨基吡啶叶立德双酰胺化合物的合成方法。
背景技术:
2.酰胺化合物由于其具有良好的生物活性和生理活性而被广泛应用于医药领域。因此,开发通过形成c
‑
n键来构建此类化合物的有效方法具有重要意义。传统交叉偶联反应包括ullmann
‑
戈德堡反应,buchwald
‑
hartwig胺化反应等虽然提供了有效的酰胺合成方法(a)g.j.sherborne,s.adomeit,r. menzel,j.rabeah,a.br
ü
ckner,m.r fielding,c.e.willansa,b.n.nguyen, origins of high catalyst loading in copper(i)
‑
catalysed ullmann
–
goldberg c
–
ncoupling reactions.chem.sci.2017,8,7203
‑
7210.(b)f.khan,m.fatima,m. shirzaei,y.vo,m.amarasiri,m.g.banwell,j.s.ward,m.g.gardiner, tandem ullmann
–
goldberg cross
‑
coupling/cyclopalladation
‑
reductiveelimination reactions and related sequences leading to polyfunctionalizedbenzofurans,indoles,and phthalanes.org.lett.2019,21,6342
–
6346.(c)f. ferlin,v.trombettoni,l.luciani,s.fusi,o.piermatti,s.santoroa,l.vaccaro, a waste
‑
minimized protocol for copper
‑
catalyzed ullmann
‑
type reaction in abiomass derived furfuryl alcohol/water azeotrope.green chem.2018,20, 1634
–
1639.但这些方法通常需要底物的预功能化、过量的金属氧化剂和苛刻的反应条件。相比之下,导向基团辅助的过渡金属催化的c
‑
h酰胺化反应已被证明是该领域最具吸引力的策略,因为它能够以高步骤和原子经济的方式形成c
‑
n键。y.park,y.kim,s.chang,transition metal
‑
catalyzed c
–
hamination:scope,mechanism,and applications.chem.rev.2017,117, 9247
‑
9301.然而,目前的报告主要集中在底物的单酰胺化反应方面,而双酰胺化反应仍然非常具有挑战性。所以,开发新的方法合成双酰胺化合物有着重要意义。
技术实现要素:
3.本发明的目的在于提供一种n
‑
亚氨基吡啶叶立德双酰胺化合物的合成方法,以克服上述现有技术存在的缺陷,本发明以n
‑
亚氨基吡啶叶立德、唑酮为反应原料,在溶剂的存在下,加入催化剂和碱,在温和的反应条件下高效合成n
‑
亚氨基吡啶叶立德双酰胺化合物,操作简单,底物范围广,这种转变为c
‑
n键的构建提供了高原子经济性的方法。
4.为达到上述目的,本发明采用如下技术方案:
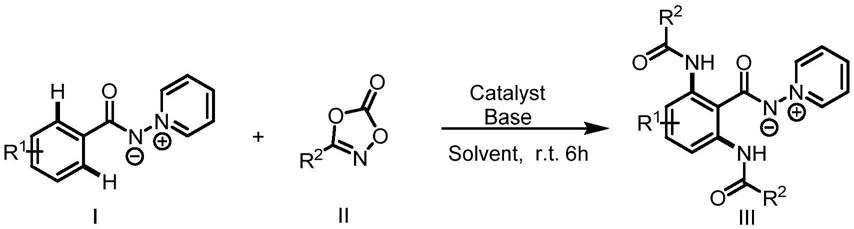
5.一种n
‑
亚氨基吡啶叶立德双酰胺化合物的合成方法,向溶剂中加入如式ⅰ所示的n
‑
亚氨基吡啶叶立德和如式ⅱ所示的唑酮,以及催化剂和碱,反应后分离提纯即得到如式ⅲ所示的n
‑
亚氨基吡啶叶立德双酰胺化合物;
[0006][0007]
其中,r1选自甲基、甲氧基或三氟甲基;r2选自甲基、甲氧基、烯基或氟。
[0008]
进一步地,所述的n
‑
亚氨基吡啶叶立德和唑酮的摩尔比为1:2。
[0009]
进一步地,所述的n
‑
亚氨基吡啶叶立德、催化剂和碱的摩尔比为20:1:40。
[0010]
进一步地,所述的反应具体为:室温下搅拌6h。
[0011]
进一步地,所述的催化剂为[cp*rhcl2]2,[cp*ircl2]2,cp*(co)i2,pd(oac)2和cui中的任意一种。
[0012]
进一步地,所述的碱为koac、kh2po4、csoac和naoac中的任意一种。
[0013]
进一步地,所述的溶剂为dce、dmf、tfe、thf、toluene和hfip中的任意一种。
[0014]
进一步地,向溶剂中加入如式ⅰ所示的n
‑
亚氨基吡啶叶立德和如式ⅱ所示的唑酮以及催化剂和碱后,n
‑
亚氨基吡啶叶立德在溶剂中的浓度为0.1摩尔/升。
[0015]
与现有技术相比,本发明具有以下有益的技术效果:
[0016]
本发明以n
‑
亚氨基吡啶叶立德、唑酮为反应原料,直接由碳氢键官能化合成目标产物,避免了底物的预功能化;以唑酮为偶联试剂,反应过程中经过氮卡宾插入方式,无需外加氧化剂;由于n
‑
亚氨基吡啶叶立德结构中酰胺氮原子上具有负电荷,使得其与金属催化剂的配位能力增加,更加容易活化邻位碳氢键,形成五元环金属物种。因此,反应在温和条件下即可进行,从而达到活化双邻位碳氢键的目的,合成n
‑
亚氨基吡啶叶立德双酰胺化合物。该方法操作简单,底物适用范围广,为双酰胺化合物的构建提供了高原子经济性的方法。
附图说明
[0017]
图1为实施例1所制备的产物的1h nmr谱图;
[0018]
图2为实施例1所制备的产物的
13
c nmr谱图;
[0019]
图3为实施例2所制备的产物的1h nmr谱图;
[0020]
图4为实施例2所制备的产物的
13
c nmr谱图;
[0021]
图5为实施例3所制备的产物的1h nmr谱图;
[0022]
图6为实施例3所制备的产物的
13
c nmr谱图;
[0023]
图7为实施例4所制备的产物的1h nmr谱图;
[0024]
图8为实施例4所制备的产物的
13
c nmr谱图;
[0025]
图9为实施例5所制备的产物的1h nmr谱图;
[0026]
图10为实施例5所制备的产物的
13
c nmr谱图;
[0027]
图11为实施例6所制备的产物的1h nmr谱图;
[0028]
图12为实施例6所制备的产物的
13
c nmr谱图;
[0029]
图13为实施例7所制备的产物的1h nmr谱图;
[0030]
图14为实施例7所制备的产物的
13
c nmr谱图;
[0031]
图15为实施例8所制备的产物的1h nmr谱图;
[0032]
图16为实施例8所制备的产物的
13
c nmr谱图;
[0033]
图17为实施例9所制备的产物的1h nmr谱图;
[0034]
图18为实施例9所制备的产物的
13
c nmr谱图。
具体实施方式
[0035]
下面对本发明的实施方式做进一步详细描述:
[0036]
一种n
‑
亚氨基吡啶叶立德双酰胺化合物的合成方法,向溶剂中加入如式ⅰ所示的n
‑
亚氨基吡啶叶立德和如式ⅱ所示的唑酮以及催化剂和碱,所述的 n
‑
亚氨基吡啶叶立德和唑酮的摩尔比为1:2,且n
‑
亚氨基吡啶叶立德、催化剂和碱的摩尔比为20:1:40,n
‑
亚氨基吡啶叶立德在溶剂中的浓度为0.1摩尔 /升,然后在室温下搅拌6h,后分离提纯即得到如式ⅲ所示的双酰胺化合物;
[0037][0038]
其中,r1选自甲基、甲氧基、三氟甲基或氟;r2选自甲基、甲氧基、三氟甲基或氟。催化剂为[cp*rhcl2]2,[cp*ircl2]2,cp*(co)i2,pd(oac)2,cui中的任意一种。碱为koac、kh2po4、csoac和naoac中的任意一种。溶剂为dce、dmf、tfe、thf、toluene和hfip中的任意一种。
[0039]
下面结合实施例对本发明做进一步详细描述:
[0040]
实施例1
[0041]
(2,6
‑
bis(benzamido)benzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0042]
将0.1mmol的n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
苯基
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05mmol[cp*rhcl2]2为催化剂, 0.2mmol kh2po4为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到36mg淡黄色固体,产率为83%,所得产品结构式如下:
[0043][0044]
如图1和图2所示,产品核磁表征:1hnmr(400mhz,cdcl3)δ12.88(s, 2h),8.62(d,j=5.9hz,2h),8.52(d,j=8.3hz,2h),8.11(t,j=7.7hz,1h),7.99 (d,j=7.5hz,4h),7.79(t,j=7.1hz,2h),7.54
‑
7.40(m,7h);
13
cnmr(100mhz, cdcl3)172.4,165.3,143.4,139.9,
139.1,136.0,131.4,131.1,128.6,127.3,126.9, 116.6,112.3
[0045]
实施例2
[0046]
(2,6
‑
bis(benzamido)
‑4‑
methylbenzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备.
[0047]
将0.1mmol的4
‑
甲基n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
苯基
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05mmol[cp*ircl2]2为催化剂, 0.2mmolkoac为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到35mg白色固体,产率为77%,所得产品结构式如下:
[0048][0049]
如图3和图4所示,产品核磁表征:1hnmr(400mhz,cdcl3)δ13.00(s, 2h),8.61(d,j=5.9hz,2h),8.40(s,2h),8.11(t,j=7.7hz,1h),7.99(d, j=7.2hz,4h),7.79(t,j=7.1hz,2h),7.51
‑
7.40(m,6h),2.49(s,3h);
13
cnmr (100mhz,cdcl3)172.6,165.3,143.4,141.8,140.0,139.0,136.1,131.4,128.5, 127.3,126.8,117.1,109.3,22.2.
[0050]
实施例3
[0051]
(2,6
‑
bis(benzamido)
‑4‑
(trifluoromethyl)benzoyl)(pyridin
‑1‑
ium
‑1‑
yl)ami de的制备
[0052]
将0.1mmol的4
‑
(3
‑
三氟甲基)n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
苯基
ꢀ‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05cp*co(co)i2为催化剂,0.2mmol kh2po4为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到29mg白色固体,产率为57%,所得产品结构式如下:
[0053][0054]
如图5和图6所示,产品核磁表征:1hnmr(400mhz,dmso)δ13.58(s, 2h),9.05(d,j=5.9hz,2h),8.92(s,2h),8.45(t,j=7.7hz,1h),8.12(t,j=7.1 hz,2h),7.94(d,j=7.4hz,4h),9.05(d,j=5.9hz,2h),7.51(d,j=7.5hz,4h);
13
cnmr(100mhz,dmso)171.3,165.5,144.3,142.0,141.5,135.2,132.6, 129.4(q,j=32.5hz),128.2,127.5,(q,j=272.2hz),114.4,111.3.
[0055]
实施例4
[0056]
(2,6
‑
bis(benzamido)
‑3‑
methoxybenzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0057]
将0.1mmol的3
‑
甲氧基n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
苯基
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05pd(oac)2为催化剂, 0.2mmolcsoac为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到18mg白色固体,产率为38%,所得产
品结构式如下:
[0058][0059]
如图7和图8所示,产品核磁表征:1hnmr(400mhz,dmso)δ11.45(s, 1h),10.31(s,1h)8.72(d,j=5.0hz,2h)8.43(d,j=9.1hz,1h),8.23(d,j=7.2 hz,1h),7.98
‑
7.88(m,6h),7.58
‑
7.47(m,6h),7.24(d,j=9.1hz,1h),3.84(s, 3h);
13
cnmr(100mhz,dmso)170.5,164.9,164.3,150.9,143.6,140.3,135.6, 135.1,132.1,131.8,131.5,129.3,128.9,128.0,127.8,127.2,126.4,123.8,118.9, 113.3,56.5.
[0060]
实施例5
[0061]
(2,6
‑
bis(4
‑
methylbenzamido)benzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0062]
将0.1mmol的n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
(4
‑
甲苯基)
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05mmol cui为催化剂, 0.2mmol kh2po4为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到33mg白色固体,产率为70%,所得产品结构式如下:
[0063][0064]
如图9和图10所示,产品核磁表征:1hnmr(400mhz,dmso)δ13.29(s, 2h),8.99(d,j=5.8hz,2h),8.47
‑
8.40(m,3h),8.09(t,j=7.2hz,2h),7.81(d, j=8.1hz,4h),7.48(t,j=8.3hz,1h),7.29(d,j=8.0hz,4h),2.35(s,6h);
13
cnmr(100mhz,dmso)172.3,164.8,144.4,142.2,141.6,140.7,130.0, 130.9,129.8,128.2,127.5,115.5,111.9,21.4.
[0065]
实施例6
[0066]
(2,6
‑
bis(4
‑
fluorobenzamido)benzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0067]
将0.1mmol的n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
(4
‑
氟苯基)
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05[cp*rhcl2]2为催化剂, 0.2mmolkoac为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到33mg白色固体,产率为69%,所得产品结构式如下:
[0068][0069]
如图11和图12所示,产品核磁表征:1hnmr(400mhz,dmso)δ13.40 (s,2h),8.99(d,j=5.6hz,2h),8.48
‑
8.41(m,3h),8.10(t,j=6.6hz,2h),7.99(t, j=6.2hz,4h),7.51(t,j=8.3hz,1h),7.94(t,j=7.9hz,4h);
13
cnmr(100 mhz,dmso)172.3,165.8(d,j=249.5hz),163.9,163.3,144.4,141.6,140.6, 132.3 132.2(d,j=2.4hz),130.9,130.2 130.1(d,j=9.3hz),128.1,116.2(d,j= 22.1hz),115.7,112.0.
[0070]
实施例7
[0071]
(2,6
‑
bis(2
‑
methylbenzamido)benzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0072]
将0.1mmol的n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
(邻甲苯基)
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05[cp*ircl2]2为催化剂, 0.2mmolcsoac为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到28mg白色固体,产率为60%,所得产品结构式如下:
[0073][0074]
如图13和图14所示,产品核磁表征:1hnmr(600mhz,dmso)δ12.67 (s,2h),8.80(d,j=5.7hz,2h),8.43(d,j=8.3hz,2h),8.29(t,j=5.2hz,1h), 7.97(t,j=7.1hz,2h),7.58(d,j=7.4hz,2h),7.47(t,j=8.3hz,1h),7.35
‑
7.32 (m,2h),7.27(d,j=7.5hz,2h),7.20(t,j=7.4hz,2h),2.43(s,6h);
13
cnmr (151mhz,dmso),171.7,167.5,144.1,141.3,140.4,137.5,136.4,131.6,130.8, 130.5,127.9,127.2,126.4,115.6,112.4,20.1.
[0075]
实施例8
[0076]
(2,6
‑
bis(3
‑
methoxybenzamido)benzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0077]
将0.1mmol的n
‑
亚氨基吡啶叶立德和0.2mmol3
‑
(3
‑
甲氧基苯基)
‑
1,4,2
‑ꢀ
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05pd(oac)2为催化剂, 0.2mmolnaoac为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到43mg白色固体,产率为87%,所得产品结构式如下:
[0078]
[0079]
如图15和图16所示,产品核磁表征:1hnmr(400mhz,dmso)δ13.34 (s,2h),9.02(d,j=5.2hz,2h),8.49(d,j=8.3hz,2h),8.42(t,j=7.5hz,1h), 8.09(t,j=6.5hz,2h),7.51
‑
7.39(m,7h),7.13(d,j=7.9hz,2h),3.72(s,6h);
13
cnmr(100mhz,dmso)172.2,164.7,159.9,144.5,140.5,137.3,131.0, 130.5,128.1,119.6,118.1,115.6,112.6,112.0,55.6.
[0080]
实施例9
[0081]
(2,6
‑
dicinnamamidobenzoyl)(pyridin
‑1‑
ium
‑1‑
yl)amide的制备
[0082]
将0.1mmol的n
‑
亚氨基吡啶叶立德和0.2mmol(e)
‑3‑
苯乙烯基
‑
1,4,2
‑
二氧唑
‑5‑
酮溶于盛有10ml dce的反应器中,以0.05[cp*rhcl2]2为催化剂, 0.2mmol kh2po4为碱,在室温下搅拌6h,用tlc监测反应的进行,经柱层析分离,得到39mg白色固体,产率为81%,所得产品结构式如下:
[0083][0084]
如图17和图18所示,产品核磁表征:1hnmr(400mhz,dmso)δ12.19 (s,2h),9.07(d,j=4.9hz,2h),8.41
‑
8.35(m,3h),8.12
‑
8.09(m,2h),7.73
‑
7.63 (m,6h),7.44(s,7h),6.86(d,j=15.7hz,2h);
13
cnmr(100mhz,dmso) 171.3,163.9,144.6,141.0,140.9,139.7,135.1,130.3,130.2,129.3,128.6,127.7, 123.7,116.2,113.8。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。