一种1,4,8,11
‑
四氮杂环十四烷类化合物及其中间体的制备方法
技术领域
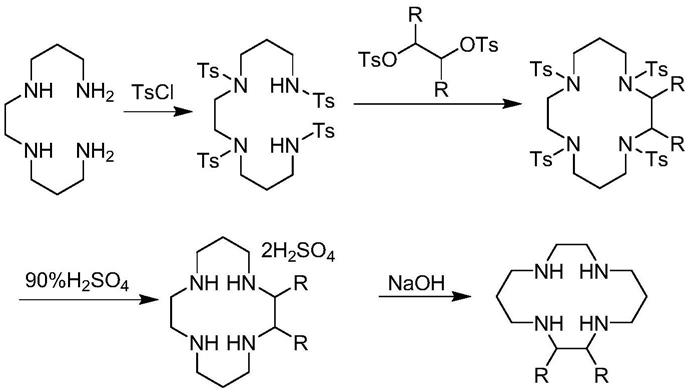
1.本发明涉及超分子化学合成技术领域,涉及一种1,4,8,11
‑
四氮杂环十四烷类化合物及其中间体的制备方法,具体涉及一种中间体1,4,8,11
‑
十氢四氮杂芘类化合物的制备方法,以及采用上述中间体制备1,4,8,11
‑
四氮杂环十四烷类化合物的方法。
背景技术:
2.大环多胺是指含有多个氨基的大环化合物,大环多胺是超分子化学中一类非常重要的主体分子,其对金属离子具有较强的配位能力,所形成的金属配合物也是一类具有独特结构和性能的化合物。由于大环多胺类的化合物及其配合物的应用十分广泛,近些年来,对它们的研究已成为新研究领域的新兴课题之一。它们不仅在过渡金属配合物方面有广泛的用途,而且在分子识别和生物医药技术方面也有着重要的应用价值。目前,其已经广泛用于化学核酸酶、生物传感器、mri照影剂、荧光探针、dna识别及酶模拟切割催化剂、金属分离与回收、放射免疫治疗药物、基础生物、医学等众多研究领域。
3.1,4,8,11
‑
四氮杂环十四烷是大环多胺中应用最广泛的一类化合物,它也是趋化因子受体拮抗剂普乐沙福的关键原料。由于1,4,8,11
‑
四氮杂环十四烷及其衍生物对过渡金属阳离子、重金属阳离子,铜系和铜系离子,甚至对有机或无机阴离子都表现出选择性配位性质,其准确行为依赖千其上取代基的性质,这种多样性使它们在众多领域具有广泛的应用价值。1,4,8,11
‑
四氮杂环十四烷分子结构如下:
[0004][0005]
现有技术中陈贝贝等人在《普乐沙福的合成工艺改进》(合成化学,2015,23,774
‑
777)中详细阐述了1,4,8,11
‑
四氮杂环十四烷及其烷烃链上衍生物的制备方法,以1,2
‑
双(3
‑
氨丙基氨基)乙烷为起始原料,经对甲苯磺酰氯保护,再与相应的二对甲苯磺酸酯关环,然后在强酸性条件下脱保护,再经碱化得到产品1,4,8,11
‑
四氮杂环十四烷或其烷烃链上衍生物。
[0006][0007]
该反应过程中存在五个比较严重的问题:第一,对甲苯磺酰基保护基团位阻非常大,导致关环效率不高,文献中报道的关环收率仅为73%;第二,对甲苯磺酰基保护基团用量大,需要使用4个当量,导致整条路线的原子经济性很低;第三,对甲苯磺酰基保护的胺基需要经过碱活化才具备亲核进攻能力,导致关环步骤必须加入大量碱,导致操作工艺比较复杂,三废量也比较多;第四,脱去对甲苯磺酰基保护基团的反应条件很苛刻,需要使用高度危险的90%硫酸,于100℃进行长时间反应,该操作危险性大,对操作人员的不友好,不符合绿色生产的需求;第五,整条路线收率较低,仅为54%,经济性较差。
[0008]
上述问题都严重限制了该工艺的进一步应用,也使得该产品的公斤级和百公斤级放大变得比较困难。因此,开发一种路线设计合理,保护剂用量少,关环效率高,原子经济性高,总收率高,操作简便,安全性好,三废少,经济性良好的1,4,8,11
‑
四氮杂环十四烷及其烷烃链上衍生物的工业化生产方法具有非常重要的意义。
技术实现要素:
[0009]
针对现有技术中的上述技术问题,本发明提供了一种1,4,8,11
‑
四氮杂环十四烷类化合物及其中间体的制备方法,解决了现有技术1,4,8,11
‑
四氮杂环十四烷类化合物制备过程中工艺复杂,产率不高,成本高等技术问题。
[0010]
本发明目的在于提供一种1,4,8,11
‑
四氮杂环十四烷类化合物中间体,具有如式3化合物所示结构:
[0011][0012]
式中,x独立选自氯、溴、碘、对甲苯磺酰氧基或甲磺酰氧基,r1独立选自h、c1
‑
c6烷基、苯基或羟甲基,r2独立选自h、c1
‑
c6烷基、苯基或羟甲基。
[0013]
本发明另一目的在于提供的上述式3化合物1,4,8,11
‑
四氮杂环十四烷类化合物中间体的制备方法,包括以下步骤,
[0014][0015]
(1)以式1化合物为原料,在惰性气体环境下,与保护剂在第一反应溶剂中,反应温度为20~200℃反应,反应完全后得式2化合物;
[0016]
(2)将式2化合物与关环试剂在第二反应溶剂中,反应温度为20~200℃反应,反应完全后得式3化合物,式中,x独立选自氯、溴、碘、对甲苯磺酰氧基或甲磺酰氧基,r1独立选自h、c1
‑
c6烷基、苯基或羟甲基,r2独立选自h、c1
‑
c6烷基、苯基或羟甲基。
[0017]
在本发明的一较佳实施例中,步骤(1)中所述的保护剂是草酸二甲酯、草酸二乙酯、草酸二异丙酯、草酸二正丙酯、草酸二叔丁酯、草酸二正丁酯、草酸二异丁酯、草酰氯、草酰氯单乙酯或草酰氯单甲酯中的一种或多种:所述第一反应溶剂是二氯甲烷、氯仿、乙腈、甲醇、乙醇、甲苯、二甲苯、四氢呋喃、2
‑
甲基四氢呋喃、乙二醇二甲醚、乙二醇二乙醚、二乙二醇二甲醚、叔丁基甲醚、氯苯、邻二氯苯或硝基苯中的一种或多种。
[0018]
在本发明的一较佳实施例中,步骤(1)中,上述第一反应溶剂与所述式1化合物的体积比为10~100:1。
[0019]
在本发明的一较佳实施例中,步骤(1)中,上述保护剂与上述式1化合物的物质的量比为0.5~5:1。
[0020]
在本发明的一较佳实施例中,步骤(2)中,上述关环试剂的结构如下:
[0021][0022]
其中,y选自氯、溴、碘、对甲苯磺酰氧基、甲磺酰氧基,r1独立选自h、c1
‑
c6烷基、苯基或羟甲基,r2独立选自h、c1
‑
c6烷基、苯基或羟甲基;上述第二关环试剂与式2化合物的物质的量比为0.5~5:1。
[0023]
在本发明的一较佳实施例中,上述第二反应溶剂是乙腈、甲醇、乙醇、异丙醇、叔丁醇、甲苯、二甲苯、四氢呋喃、2
‑
甲基四氢呋喃、乙二醇二甲醚、乙二醇二乙醚、二乙二醇二甲醚、叔丁基甲醚、丙酮、丁酮、nmp、dmf或dmso中的一种或多种。
[0024]
本发明又一目的在于提供的1,4,8,11
‑
四氮杂环十四烷类化合物的制备方法,包括上述的式3化合物1,4,8,11
‑
四氮杂环十四烷类化合物中间体的制备方法,还进一步包括:
[0025][0026]
步骤(3)中,将式3化合物与碱性试剂在第三反应溶剂中,反应温度为20~200℃反
应,得式4化合物,式中,x独立选自氯、溴、碘、对甲苯磺酰氧基或甲磺酰氧基,r1独立选自h、c1
‑
c6烷基、苯基或羟甲基,r2独立选自h、c1
‑
c6烷基、苯基或羟甲基,所述碱性试剂是氢氧化钠、氢氧化钾、或氢氧化锂中的一种或多种。
[0027]
在本发明的一较佳实施例中,步骤(3)中第三反应溶剂是水、甲醇、乙醇、异丙醇、叔丁醇中的一种或多种。
[0028]
在本发明的一较佳实施例中,步骤(3)中碱性试剂与上述式3化合物的重量比为:0.1~10:1。
[0029]
与现有技术相比,本发明生产工艺简单,原料价格较低,总收率高,产品纯度高,经济性良好,能充分满足产品工业化生产的需求,另外本发明的实施会对改善环境生产积极影响。本发明的制备工艺特别是适于制备1,4,8,11
‑
四氮杂环十四烷类化合物及其中间体,其他四氮杂环,特别是13
‑
16四氮杂元环的衍生物可以类似地制备。本发明的范围扩大包括这些类似的方法。
具体实施方式
[0030]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述地实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0031]
实施例1
[0032][0033]
步骤(1):式2化合物2,3,4,6,7,9,10,11
‑
八氢吡嗪[1,2
‑
a:4,3
‑
a']联嘧啶的制备
[0034]
保持氮气微正压,向10l反应瓶中依次加入4l 2
‑
甲基四氢呋喃,174.29g(1.0mol)式1化合物1,2
‑
双(3
‑
氨丙基氨基)乙烷和153.66g(1.05mol)草酸二乙酯,加完后,搅匀;反应液升温回流反应。保温回流反应7小时,反应完成。
[0035]
反应液旋蒸回收溶剂,剩余物用适量甲苯重结晶得到184.48g(0.959mol)黄色固体产品。
[0036]
收率95.9%,hplc纯度99.4%,1h nmr(400mhz,cdcl3):δ3.55(t,4h),3.23(s,4h),3.21(t,4h),1.86(m,4h)。
[0037]
步骤(2):式3化合物1,2,3,4,5,6,7,8,9,10
‑
十氢
‑
3a,5a,8a,10a
‑
四氮杂芘
‑
8a,10a
‑
二鎓二溴盐的制备
[0038]
保持氮气微正压,向2l反应瓶中依次加入600ml叔丁基甲醚,184.48g(0.959mol)式2化合物和180.16g(0.959mol)1,2
‑
二溴乙烷,加完后,搅匀;反应液升温至回流反应,保温反应6小时,反应完成。
[0039]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保
温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到357.61g(0.941mol)浅黄色固体状产品。
[0040]
收率98.1%,hplc纯度99.5%,1h nmr(400mhz,d6
‑
dmso):δ4.38(s,4h),4.22(t,4h),3.61(s,4h),3.28(t,4h),1.85(m,4h)。
[0041]
步骤(3):式4化合物1,4,8,11
‑
四氮杂环十四烷的制备
[0042]
保持氮气微正压,向5l反应瓶中加入2000ml水和200g氢氧化钠,搅拌下再向其中分批加入357.61g(0.941mol)式3化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钠固体。保温80℃反应6小时,反应结束。
[0043]
反应液冷却至室温,过滤,向滤液加入适量二氯甲烷进行萃取,将滤液中产品萃取干净。合并萃取液,常压蒸馏回收合适体积的二氯甲烷。向剩余溶液中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量白色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到177.69g(0.887mol)白色晶体产品。
[0044]
收率94.3%,三步反应总收率88.7%,
[0045]
产品滴定纯度:99.3%(by hclo4),
[0046]
产品核磁数据:1h nmr(400mhz,cdcl3):δ2.74(t,8h),2.68(s,8h),2.24(s,4h),1.72(m,4h),
[0047]
实施例2
[0048][0049]
步骤(1):式2化合物2,3,4,6,7,9,10,11
‑
八氢吡嗪[1,2
‑
a:4,3
‑
a']联嘧啶的制备
[0050]
保持氮气微正压,向10l反应瓶中依次加入6l四氢呋喃,174.29g(1.0mol)式1化合物1,2
‑
双(3
‑
氨丙基氨基)乙烷和236.18g(2.0mol)草酸二甲酯,加完后,搅匀;反应液升温回流反应。保温回流反应7小时,反应完成。
[0051]
反应液旋蒸回收溶剂,剩余物用适量甲苯重结晶得到181.70g(0.945mol)黄色固体产品。
[0052]
收率94.5%,hplc纯度99.1%,1h nmr(400mhz,cdcl3):δ3.55(t,4h),3.23(s,4h),3.21(t,4h),1.86(m,4h)。
[0053]
步骤(2):式3化合物1,2,3,4,5,6,7,8,9,10
‑
十氢
‑
3a,5a,8a,10a
‑
四氮杂芘
‑
8a,10a
‑
二鎓二氯盐的制备
[0054]
保持氮气微正压,向2l反应瓶中依次加入600ml四氢呋喃,181.70g(0.945mol)式2化合物和187.03g(1.89mol)1,2
‑
二氯乙烷,加完后,搅匀;反应液升温至回流反应,保温反应6小时,反应完成。
[0055]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到
265.66g(0.912mol)浅黄色固体状产品。
[0056]
收率96.5%,hplc纯度99.7%,1h nmr(400mhz,d6
‑
dmso):δ4.39(s,4h),4.22(t,4h),3.63(s,4h),3.28(t,4h),1.85(m,4h)。
[0057]
步骤(3):式4化合物1,4,8,11
‑
四氮杂环十四烷的制备
[0058]
保持氮气微正压,向5l反应瓶中加入500ml甲醇、1000ml水和265.66g氢氧化锂,搅拌下再向其中分批加入265.66g(0.912mol)式3化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化锂固体。保温80℃反应6小时,反应结束。
[0059]
反应液冷却至室温,过滤,向滤液加入适量二氯甲烷进行萃取,将滤液中产品萃取干净。合并萃取液,常压蒸馏回收合适体积的二氯甲烷。向剩余溶液中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量白色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到173.20g(0.865mol)白色晶体产品。
[0060]
收率94.8%,三步反应总收率86.5%,
[0061]
产品滴定纯度:99.5%(by hclo4),
[0062]
产品核磁数据:1h nmr(400mhz,cdcl3):δ2.72(t,8h),2.68(s,8h),2.24(s,4h),1.73(m,4h)。
[0063]
实施例3
[0064][0065]
步骤(1):式2化合物2,3,4,6,7,9,10,11
‑
八氢吡嗪[1,2
‑
a:4,3
‑
a']联嘧啶的制备
[0066]
保持氮气微正压,向20l反应瓶中依次加入13l甲苯,174.29g(1.0mol)式1化合物1,2
‑
双(3
‑
氨丙基氨基)乙烷和380.79g(3.0mol)草酰氯,加完后,搅匀;再向其中加入303.57g(3.0mol)三乙胺,搅匀,反应液升温回流反应。保温回流反应7小时,反应完成。
[0067]
反应液趁热过滤,滤液旋蒸回收溶剂,剩余物用适量甲苯重结晶得到179.77g(0.935mol)黄色固体产品。
[0068]
收率93.5%,hplc纯度99.3%,1h nmr(400mhz,cdcl3):δ3.55(t,4h),3.23(s,4h),3.21(t,4h),1.86(m,4h)。
[0069]
步骤(2):式3化合物1,2,3,4,5,6,7,8,9,10
‑
十氢
‑
3a,5a,8a,10a
‑
四氮杂芘
‑
8a,10a
‑
二鎓二碘盐的制备
[0070]
保持氮气微正压,向2l反应瓶中依次加入600ml甲苯,179.77g(0.935mol)式2化合物和790.62g(2.805mol)1,2
‑
二碘乙烷,加完后,搅匀;反应液升温至回流反应,保温反应6小时,反应完成。
[0071]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到430.89g(0.909mol)浅黄色固体状产品。
[0072]
收率97.2%,hplc纯度99.4%,1h nmr(400mhz,d6
‑
dmso):δ4.38(s,4h),4.26(t,4h),3.61(s,4h),3.29(t,4h),1.85(m,4h)。
[0073]
步骤(3):式4化合物1,4,8,11
‑
四氮杂环十四烷的制备
[0074]
保持氮气微正压,向5l反应瓶中加入500ml乙醇、1000ml水和200g氢氧化钾,搅拌下再向其中分批加入430.89g(0.909mol)式3化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钾固体。保温80℃反应6小时,反应结束。
[0075]
反应液冷却至室温,过滤,向滤液加入适量二氯甲烷进行萃取,将滤液中产品萃取干净。合并萃取液,常压蒸馏回收合适体积的二氯甲烷。向剩余溶液中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量白色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到173.36g(0.865mol)白色晶体产品。
[0076]
收率95.2%,三步反应总收率86.5%,
[0077]
产品滴定纯度:99.2%(by hclo4),
[0078]
产品核磁数据:1h nmr(400mhz,cdcl3):δ2.73(t,8h),2.70(s,8h),2.20(s,4h),1.72(m,4h)。
[0079]
实施例4
[0080][0081]
步骤(1):式2化合物2,3,4,6,7,9,10,11
‑
八氢吡嗪[1,2
‑
a:4,3
‑
a']联嘧啶的制备
[0082]
保持氮气微正压,向10l反应瓶中依次加入3l乙二醇二甲醚,174.29g(1.0mol)式1化合物1,2
‑
双(3
‑
氨丙基氨基)乙烷和546.13g(4mol)草酰氯单乙酯,加完后,搅匀;再向其中加入303.57g(3.0mol)三乙胺,搅匀,反应液升温回流反应。保温回流反应7小时,反应完成。
[0083]
反应液趁热过滤,滤液旋蒸回收溶剂,剩余物用适量甲苯重结晶得到181.12g(0.942mol)黄色固体产品。
[0084]
收率94.2%,hplc纯度99.3%,1h nmr(400mhz,cdcl3):δ3.55(t,4h),3.23(s,4h),3.21(t,4h),1.86(m,4h)。
[0085]
步骤(2):式3化合物1,2,3,4,5,6,7,8,9,10
‑
十氢
‑
3a,5a,8a,10a
‑
四氮杂芘
‑
8a,10a
‑
二鎓二甲磺酸盐的制备
[0086]
保持氮气微正压,向2l反应瓶中依次加入600ml异丙醇,181.12g(0.942mol)式2化合物和822.10g(3.768mol)1,2
‑
二甲磺酰氧基乙烷,加完后,搅匀;反应液升温至回流反应,保温反应6小时,反应完成。
[0087]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到369.68g(0.901mol)浅黄色固体状产品。
[0088]
收率95.6%,hplc纯度99.6%,1h nmr(400mhz,d6
‑
dmso):δ4.37(s,4h),4.22(t,4h),3.66(s,4h),3.28(t,4h),2.86(s,6h),1.84(m,4h)。
[0089]
步骤(3):式4化合物1,4,8,11
‑
四氮杂环十四烷的制备
[0090]
保持氮气微正压,向5l反应瓶中加入500ml异丙醇,1000ml水和200g氢氧化钾,搅拌下再向其中分批加入369.68g(0.901mol)式3化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钾固体。保温80℃反应6小时,反应结束。
[0091]
反应液冷却至室温,过滤,向滤液加入适量二氯甲烷进行萃取,将滤液中产品萃取干净。合并萃取液,常压蒸馏回收合适体积的二氯甲烷。向剩余溶液中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量白色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到172.74g(0.862mol)白色晶体产品。
[0092]
收率95.7%,三步反应总收率86.2%,
[0093]
产品滴定纯度:99.4%(by hclo4),
[0094]
产品核磁数据:1h nmr(400mhz,cdcl3):δ2.71(t,8h),2.68(s,8h),2.27(s,4h),1.70(m,4h)。
[0095]
实施例5
[0096][0097]
步骤(1):式2化合物2,3,4,6,7,9,10,11
‑
八氢吡嗪[1,2
‑
a:4,3
‑
a']联嘧啶的制备
[0098]
保持氮气微正压,向10l反应瓶中依次加入2l乙二醇二甲醚,174.29g(1.0mol)式1化合物1,2
‑
双(3
‑
氨丙基氨基)乙烷和612.54g(5mol)草酰氯单甲酯,加完后,搅匀;再向其中加入303.57g(3.0mol)三乙胺,搅匀,反应液升温回流反应。保温回流反应7小时,反应完成。
[0099]
反应液旋蒸回收溶剂,剩余物用适量甲苯重结晶得到180.16g(0.937mol)黄色固体产品。
[0100]
收率93.7%,hplc纯度99.5%,1h nmr(400mhz,cdcl3):δ3.55(t,4h),3.23(s,4h),3.21(t,4h),1.86(m,4h)。
[0101]
步骤(2):式3化合物9
‑
甲基
‑
1,2,3,4,5,6,7,8,9,10
‑
十氢
‑
3a,5a,8a,10a
‑
四氮杂芘
‑
8a,10a
‑
二鎓二溴盐的制备
[0102]
保持氮气微正压,向2l反应瓶中依次加入600ml乙醇,180.16g(0.937mol)和955.27g(4.685mol)1,2
‑
二溴丙烷,加完后,搅匀;反应液升温至回流反应,保温反应6小时,反应完成。
[0103]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到357.50g(0.907mol)浅黄色固体状产品。
[0104]
收率96.8%,hplc纯度99.3%,1h nmr(400mhz,d6
‑
dmso):δ4.38(d,2h),4.32(t,1h),4.22(t,4h),3.61(s,4h),3.31(t,4h),1.82(m,4h),1.01(d,3h)。
[0105]
步骤(3):式4化合物2
‑
甲基
‑
1,4,8,11
‑
四氮杂环十四烷的制备
[0106]
保持氮气微正压,向5l反应瓶中加入500ml叔丁醇,1000ml水和357.50g氢氧化锂,搅拌下再向其中分批加入357.50g(0.907mol)式3化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钠固体。保温80℃反应6小时,反应结束。
[0107]
反应液冷却至室温,过滤,向滤液加入适量二氯甲烷进行萃取,将滤液中产品萃取干净。合并萃取液,常压蒸馏回收合适体积的二氯甲烷。向剩余溶液中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量白色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到182.76g(0.853mol)白色晶体产品。
[0108]
收率94.0%,三步反应总收率85.3%,
[0109]
产品滴定纯度:99.1%(by hclo4),
[0110]
产品核磁数据:1h nmr(400mhz,cdcl3):δ2.75(t,8h),2.68(m,7h),2.23(s,4h),1.69(m,4h),0.97(d,3h)。
[0111]
实施例6
[0112][0113]
步骤(1):式2化合物2,3,4,6,7,9,10,11
‑
八氢吡嗪[1,2
‑
a:4,3
‑
a']联嘧啶的制备
[0114]
保持氮气微正压,向20l反应瓶中依次加入16l叔丁基甲醚,174.29g(1.0mol)式1化合物1,2
‑
双(3
‑
氨丙基氨基)乙烷和212.36g(1.05mol)草酸二叔丁酯,加完后,搅匀;反应液升温回流反应。保温回流反应7小时,反应完成。
[0115]
反应液旋蒸回收溶剂,剩余物用适量甲苯重结晶得到183.04g(0.952mol)黄色固体产品。
[0116]
收率95.2%,hplc纯度99.3%,1h nmr(400mhz,cdcl3):δ3.55(t,4h),3.23(s,4h),3.21(t,4h),1.86(m,4h)。
[0117]
步骤(2):式3化合物9
‑
苯基
‑
1,2,3,4,5,6,7,8,9,10
‑
十氢
‑
3a,5a,8a,10a
‑
四氮杂芘
‑
8a,10a
‑
二鎓二溴盐的制备
[0118]
保持氮气微正压,向2l反应瓶中依次加入600ml乙腈,183.04g(0.952mol)式2化合物和263.96g(1.0mol)1,2
‑
二溴乙基苯,加完后,搅匀;反应液升温至回流反应,保温反应6小时,反应完成。
[0119]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到414.78g(0.909mol)浅黄色固体状产品。
[0120]
收率95.5%,hplc纯度99.1%,1h nmr(400mhz,d6
‑
dmso):δ7.19
‑
7.28(m,5h),
5.32(t,1h),4.62(d,2h),4.23(m,4h),3.64(s,4h),3.28(m,4h),1.85(m,4h)。
[0121]
步骤(3):式4化合物2
‑
苯基
‑
1,4,8,11
‑
四氮杂环十四烷的制备
[0122]
保持氮气微正压,向5l反应瓶中加入500ml乙醇,1000ml水和200g氢氧化钠,搅拌下再向其中分批加入414.78g(0.909mol)式3化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钠固体。保温80℃反应6小时,反应结束。
[0123]
反应液冷却至室温,过滤,向滤液加入适量二氯甲烷进行萃取,将滤液中产品萃取干净。合并萃取液,常压蒸馏回收合适体积的二氯甲烷。向剩余溶液中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量白色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到240.47g(0.870mol)白色晶体产品。
[0124]
收率95.7%,三步反应总收率87.0%,
[0125]
产品滴定纯度:99.5%(by hclo4),
[0126]
产品核磁数据:1h nmr(400mhz,cdcl3):δ7.10
‑
7.22(m,5h),3.92(t,1h),3.11(d,2h),2.74(m,8h),2.69(s,4h),2.25(s,4h),1.72(m,4h)。
[0127]
对比例1
[0128]
根据常规技术中公开的方案(合成化学,2015,23,774
‑
777中化合物5的合成方案)以1,2
‑
双(3
‑
氨丙基氨基)乙烷为起始原料来制备1,4,8,11
‑
四氮杂环十四烷。
[0129]
总收率:62.1%,产品滴定纯度:97.3%(by hclo4)。产品ir,ms,元素分析均与文献(合成化学,2015,23,774
‑
777)报道数据一致。
[0130]
综合实施例1
‑
6和对比例1
‑
2的总收率和产品纯度结果对比分析
[0131]
类型实施例1实施例2实施例3实施例4实施例5实施例6对比例1总收率88.7%86.5%86.5%86.2%85.3%87.0%62.1%纯度99.3%99.5%99.2%99.4%99.1%99.4%97.3%
[0132]
由此可见,本专利的制备方法具有以下优点:
[0133]
1、保护剂用量少,理论上只需要使用1当量,比现有方法的4当量大为减少,保护剂利用率大为提高;
[0134]
2、保护剂分子量较小,使整条路线的原子经济比较高;
[0135]
3、保护基脱去比较方便,不需要使用现有方法中的危险反应条件,反应安全性大为提高,操作也更为便捷;
[0136]
4、化合物不需要额外的碱来进行活化,并且反应位点的位阻较小,从而使关环步骤的效率很高,关环步骤的收率达到了98.1%,比现有方法的73%高了25.1%;
[0137]
5、收率高,总收率达到了88.7%,比原工艺的54%高了34.7%,路线经济性大为提升;
[0138]
6、避免了使用大量硫酸,使反应过程中生成的酸性废水大为减少,更加绿色环保。
[0139]
尽管以上对本发明的实施方案进行了描述,但本发明并不局限于上述的具体实施方案和应用领域,上述的具体实施方案仅仅是示意性的、指导性的,而不是限制性的。本领域的普通技术人员在本说明书的启示下和在不脱离本发明权利要求所保护的范围的情况下,还可以做出很多种的形式,这些均属于本发明保护之列。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。