1.本发明涉及药物技术领域,具体涉及二萜类化合物及制备方法和应用、二萜类化合物药学上可接受的衍生物、药物组合物及应用。
背景技术:
2.银屑病是一种常见的受多基因遗传控制以及环境因素刺激诱导影响的系统性、慢性、炎症性自身免疫性疾病,目前尚不能完全治愈,是当今世界皮肤病领域重点防治对象之一。效应记忆淋巴t
em
细胞活化增殖后分泌的大量炎症因子是介导银屑病引起皮肤免疫损伤的重要原因。当银屑病发病时,t
em
细胞被活化,kv1.3离子通道在其细胞膜上的数量急剧上升(从约250个/细胞上升到1500个/细胞)并激活开放,促使t
em
细胞进一步增殖并分泌大量相关炎症因子引起后续皮肤病变,抑制该通道可从上游阻止银屑病的病程发展,这一特性使kv1.3离子通道成为开发银屑病治疗药物的重要靶点。
3.泽漆(euphorbia helioscopial)是大戟科大戟属植物,广泛分布于我国除新疆、西藏以外的各个地区。该植物全草均可入药,有行水消肿、化痰止咳、解毒杀虫的功效,主治水气肿满、痰饮喘咳疟疾、菌痢、瘰疬、结核性瘘管、银屑病等症。已有泽漆提取物具有较好的抗银屑病作用的报道,例如,中国专利cn201610016841.0公开了了一种泽漆提取物,“泽漆部分化学成分研究”(参见赵杰,吴繁荣,李宁,等.泽漆部分化学成分研究[j].安徽医药,2019,23(11):2142
‑
2145.),然而,上述泽漆提取物均是通过抑制角质形成细胞及降低炎症因子(il
‑
6)实现抗银屑病的作用。目前,尚未有泽漆中二萜类化合物的抗银屑病的相关靶点进行研究,开发抑制kv1.3离子通道从而从上游阻止银屑病的病程发展的新药物具有重要的意义。
技术实现要素:
[0004]
有鉴于此,本发明的目的在于提供二萜类化合物及制备方法和应用、二萜类化合物药学上可接受的衍生物、药物组合物及应用,本发明提供的二萜类化合物及其药学上可接受的对映体和/或消旋体以及药物组合物具有优异的抑制kv1.3离子通道作用,抗银屑病性能优异。
[0005]
为了实现上述发明目的,本发明提供以下技术方案:
[0006]
本发明提供了一种二萜类化合物,具有式i~v所示的结构中的任意一种:
[0007][0008]
所述式ii所示的结构中:
[0009]
当r1为β
‑
ch3时,r2为oac,r3为oac,r4为α
‑
oh,记为式ii
‑
1;
[0010]
记为式ii
‑
2当r1为α
‑
ch3时,r2为oh,r3为=o,r4为α
‑
oh,记为式ii
‑
2;
[0011]
当r1为β
‑
ch3时,r2为oh,r3为=o,r4为h,记为式ii
‑
3;
[0012]
当r1为β
‑
ch3时,r2为oh,r3为oac,r4为α
‑
oh,记为式ii
‑
4;
[0013]
当r1为α
‑
ch3时,r2为oh,r3为oac,r4为α
‑
oh,记为式ii
‑
5;
[0014]
所述式iii所示的结构中:
[0015]
当r6为α
‑
ch3时,r7为oac,r8为oac,r9为β
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
1;
[0016]
当r6为β
‑
ch3时,r7为oac,r8为=o,r9为α
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
2;
[0017]
当r6为α
‑
ch3时,r7为oac,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为oh,记为式iii
‑
3;
[0018]
当r6为α
‑
ch3时,r7为oac,r8为=o,r9为β
‑
ch3,r
10
为oac,r
11
为oh,记为式iii
‑
4。
[0019]
本发明提供了上述技术方案所述二萜类化合物的制备方法,包括以下步骤:
[0020]
(1)将泽漆全草进行醇提,将得到的粗醇提物与萃取剂混合进行萃取,得到醇提物浸膏;
[0021]
(2)将所述醇提物浸膏进行第一硅胶柱色谱分离,得到组分frab、组分frad和组分frae,所述第一硅胶柱色谱分离的洗脱剂包括石油醚
‑
乙酸乙酯溶剂,所述石油醚
‑
乙酸乙酯溶剂中石油醚和乙酸乙酯的体积比为50:1~0:1;
[0022]
(3)将所述组分frab进行第一反相硅胶柱分离,得到组分fb2、组分fb3和组分fb4;
[0023]
将所述组分fb2进行第一lh20凝胶柱分离,得到组分fb2a和组分fb2b;将所述组分fb2a进行第一薄层色谱硅胶板分离,得到具有式iii
‑
3和式v所示结构的二萜类化合物;将所述组分fb2b进行第二薄层色谱硅胶板分离,得具有式iii
‑
1所示结构的二萜类化合物;
[0024]
将所述组分fb3进行第二硅胶柱色谱分离,得到组分fb3b和组分fb3c;将所述组分fb3b进行重结晶,得到具有式ii
‑
1所示结构的二萜类化合物;将所述组分fb3c进行第三薄层色谱硅胶板分离,得具有式iii
‑
2所示结构的二萜类化合物;所述第二硅胶柱色谱分离采用的洗脱剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为4:1~1:5;所述重结晶采用的溶剂为甲醇;
[0025]
将所述组分fb4进行第三硅胶柱色谱分离,得到组分fb4a、组分fb4b和组分fb4c;将所述组分fb4a进行第一半制备高效液相色谱分离,得到具有式ii
‑
2所示结构的二萜类化合物;将所述组分fb4b进行第二半制备高效液相色谱分离,得到具有式ii
‑
4所示结构的二萜类化合物;将所述组分fb4c经进行第三半制备高效液相色谱分离,得到具有式ii
‑
5所示结构的二萜类化合物;所述第三硅胶柱色谱分离采用的洗脱剂为氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比为250:1~10:1;
[0026]
(4)将所述组分frad进行第二反相硅胶柱分离,得到组分fd1、组分fd2和组分fd3;
[0027]
将所述组分fd1进行第二lh20凝胶柱分离,得到组分fd1b;将所述组分fd1b进行第四半制备高效液相色谱分离,得到具有式iv所示结构的二萜类化合物;
[0028]
将所述组分fd2进行第三lh20凝胶柱分离,得到组分fd2c;将所述组分fd2c进行第五半制备高效液相色谱分离,得到具有式iii
‑
4所示结构的二萜类化合物;
[0029]
将所述组分fd3进行第四lh20凝胶柱分离,得到组分fd3b;将所述组分fd3b进行第六半制备高效液相色谱分离,得到具有式i所示结构的二萜类化合物;
[0030]
(5)将所述组分frae进行第三反相硅胶柱分离,得到组分fe1;将所述组分fe1进行第五lh20凝胶柱分离,得到组分fe1a;将所述组分fe1a进行第七半制备高效液相色谱分离,得到具有式ii
‑
3所示结构的二萜类化合物;
[0031]
所述第一反相硅胶柱分离、第二反相硅胶柱分离和第三反相硅胶柱分离采用的洗脱剂为甲醇
‑
水溶剂,所述甲醇
‑
水溶剂中甲醇和水的体积比为40:60~100:0;
[0032]
所述第一lh20凝胶柱分离、第二lh20凝胶柱分离、第三lh20凝胶柱分离、第四lh20凝胶柱分离和第五lh20凝胶柱分离采用的洗脱剂为甲醇;
[0033]
所述第一半制备高效液相色谱分离、第二半制备高效液、第五半制备高效液相色谱分离、第六半制备高效液相色谱分离和第七半制备高效液相色谱分离采用的溶剂为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%;
[0034]
所述步骤(3)~步骤(5)没有时间上的先后顺序。
[0035]
优选的,所述醇提的温度为50~65℃,时间为2~3h。
[0036]
优选的,所述萃取剂优选包括小极性溶剂
‑
大极性溶剂溶剂,所述小极性溶剂包括石油醚和/或氯仿;所述大极性溶剂包括水、乙酸乙酯、异丙醇、丙酮和甲醇中得一种或几种。
[0037]
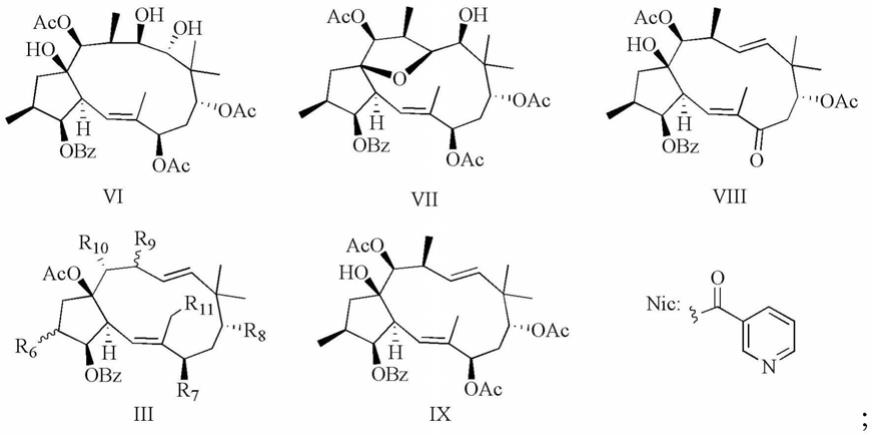
本发明提供了一种二萜类化合物的药学上可接受的衍生物,包括上述技术方案所述二萜类化合物的衍生物、具有式iii和式vi~ix所示结构中的任意一种的二萜类化合物的衍生物;所述衍生物包括对映体和/或消旋体;
[0038][0039]
所述式iii所示的结构中:
[0040]
当r6为α
‑
ch3时,r7为oh,r8为=o,r9为α
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
5;
[0041]
当r6为β
‑
ch3时,r7为oac,r8为oac,r9为β
‑
ch3,r
10
为oac,r
11
为h,记为式iii
‑
6;
[0042]
当r6为α
‑
ch3时,r7为oh,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为h,记为式iii
‑
7;
[0043]
当r6为α
‑
ch3时,r7为onic,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为h,记为式iii
‑
8。
[0044]
本发明提供了一种药物组合物,包括上述技术方案所述的二萜类化合物和/或上述技术方案所述的二萜类化合物的药学上可接受的衍生物。
[0045]
优选的,所述药物组合物的制剂剂型包括液体制剂、固体制剂、喷剂或雾剂。
[0046]
优选的,所述药物组合物中二萜类化合物和/或二萜类化合物的药学上可接受的衍生物的总含量≥0.9wt%。
[0047]
本发明还提供了上述技术方案所述的二萜类化合物、上述技术方案所述的二萜类化合物的药学上可接受的衍生物和上述技术方案所述的药物组合物中的一种或几种在制备kv1.3离子通道介导的疾病的药物中的应用。
[0048]
优选的,所述kv1.3离子通道介导的疾病包括银屑病。
[0049]
本发明提供了一种二萜类化合物,具有式i~v所示的结构中的任意一种;所述式ii所示的结构中:当r1为β
‑
ch3时,r2为oac,r3为oac,r4为α
‑
oh,r5为=o,记为式ii
‑
1;当r1为α
‑
ch3时,r2为oh,r3为=o,r4为α
‑
oh,r5为=o,记为式ii
‑
2;当r1为β
‑
ch3时,r2为oh,r3为=o,r4为h,r5为=o,记为式ii
‑
3;当r1为β
‑
ch3时,r2为oh,r3为oac,r4为α
‑
oh,r5为=o,记为式ii
‑
4;当r1为α
‑
ch3时,r2为oh,r3为oac,r4为α
‑
oh,r5为=o,记为式ii
‑
5;所述式iii所示的结构中:当r6为α
‑
ch3时,r7为oac,r8为oac,r9为β
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
1;当r6为β
‑
ch3时,r7为oac,r8为=o,r9为α
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
2;当r6为α
‑
ch3时,r7为oac,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为oh,记为式iii
‑
3;当r6为α
‑
ch3时,r7为oac,r8为=o,r9为β
‑
ch3,r
10
为oac,r
11
为oh,记为式iii
‑
4。本发明提供的二萜类化合物可关闭钾离子转运通道,具有优异的抑制kv1.3离子通道作用,从而从上游阻止银屑病的病程发展,在制备抗银屑药物方面具有良好的应用前景。
[0050]
本发明提供了上述技术方案所述二萜类化合物的制备方法。本发明以泽漆为原材料制备得到上述二萜类化合物,源于中草药,安全性和有效性高,极大的拓展了泽漆的药学
经济价值;制备方法操作简单,原料廉价易得,成本低,适宜工业化生产;而且,本发明提供得制备方法能够避免使用大量的有机原料,绿色环保。
[0051]
本发明提供的二萜类化合物的药学上可接受的衍生物以及药物组合物具有优异的抑制kv1.3离子通道功能,抗银屑病性能优异。
具体实施方式
[0052]
本发明提供了一种二萜类化合物,具有式i~v所示的结构中的任意一种:
[0053][0054]
在本发明中,所述式ii所示的结构中:
[0055]
当r1为β
‑
ch3时,r2为oac,r3为oac,r4为α
‑
oh,记为式ii
‑
1;
[0056]
当r1为α
‑
ch3时,r2为oh,r3为=o,r4为α
‑
oh,记为式ii
‑
2;
[0057]
当r1为β
‑
ch3时,r2为oh,r3为=o,r4为h,记为式ii
‑
3;
[0058]
当r1为β
‑
ch3时,r2为oh,r3为oac,r4为α
‑
oh,记为式ii
‑
4;
[0059]
当r1为α
‑
ch3时,r2为oh,r3为oac,r4为α
‑
oh,记为式ii
‑
5。
[0060]
在本发明中,所述式iii所示的结构中:
[0061]
当r6为α
‑
ch3时,r7为oac,r8为oac,r9为β
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
1;
[0062]
当r6为β
‑
ch3时,r7为oac,r8为=o,r9为α
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
2;
[0063]
当r6为α
‑
ch3时,r7为oac,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为oh,记为式iii
‑
3;
[0064]
当r6为α
‑
ch3时,r7为oac,r8为=o,r9为β
‑
ch3,r
10
为oac,r
11
为oh,记为式iii
‑
4。
[0065]
在本发明中,所述式ii和式iii所示的结构中,波浪线表示取代基为α型或β型。
[0066]
本发明提供了上述技术方案所述二萜类化合物的制备方法,包括以下步骤:
[0067]
(1)将泽漆全草进行醇提,将得到的粗醇提物与萃取剂混合进行萃取,得到醇提物浸膏;
[0068]
(2)将所述醇提取物浸膏进行第一硅胶柱色谱分离,得到组分frab、组分frad和组分frae,所述第一硅胶柱色谱分离的洗脱剂包括石油醚
‑
乙酸乙酯溶剂,所述石油醚
‑
乙酸乙酯溶剂中石油醚和乙酸乙酯的体积比为50:1~0:1;
[0069]
(3)将所述组分frab进行第一反相硅胶柱分离,得到组分fb2、组分fb3和组分fb4;
[0070]
将所述组分fb2进行第一lh20凝胶柱分离,得到组分fb2a和组分fb2b;将所述组分
fb2a进行第一薄层色谱硅胶板分离,得到具有式iii
‑
3和式v所示结构的二萜类化合物;将所述组分fb2b进行第二薄层色谱硅胶板分离,得具有式iii
‑
1所示结构的二萜类化合物;
[0071]
将所述组分fb3进行第二硅胶柱色谱分离,得到组分fb3b和组分fb3c;将所述组分fb3b进行重结晶,得到具有式ii
‑
1所示结构的二萜类化合物;将所述组分fb3c进行第三薄层色谱硅胶板分离,得具有式iii
‑
2所示结构的二萜类化合物;所述第二硅胶柱色谱分离采用的洗脱剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为4:1~1:5;所述重结晶采用的溶剂为甲醇;
[0072]
将所述组分fb4进行第三硅胶柱色谱分离,得到组分fb4a、组分fb4b和组分fb4c;将所述组分fb4a进行第一半制备高效液相色谱分离,得到具有式ii
‑
2所示结构的二萜类化合物;将所述组分fb4b进行第二半制备高效液相色谱分离,得到具有式ii
‑
4所示结构的二萜类化合物;将所述组分fb4c经进行第三半制备高效液相色谱分离,得到具有式ii
‑
5所示结构的二萜类化合物;所述第三硅胶柱色谱分离采用的洗脱剂为氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比为250:1~10:1;
[0073]
(4)将所述组分frad进行第二反相硅胶柱分离,得到组分fd1、组分fd2和组分fd3;
[0074]
将所述组分fd1进行第二lh20凝胶柱分离,得到组分fd1b;将所述组分fd1b进行第四半制备高效液相色谱分离,得到具有式iv所示结构的二萜类化合物;
[0075]
将所述组分fd2进行第三lh20凝胶柱分离,得到组分fd2c;将所述组分fd2c进行第五半制备高效液相色谱分离,得到具有式iii
‑
4所示结构的二萜类化合物;
[0076]
将所述组分fd3进行第四lh20凝胶柱分离,得到组分fd3b;将所述组分fd3b进行第六半制备高效液相色谱分离,得到具有式i所示结构的二萜类化合物;
[0077]
(5)将所述组分frae进行第三反相硅胶柱分离,得到组分fe1;将所述组分fe1进行第五lh20凝胶柱分离,得到组分fe1a;将所述组分fe1a进行第七半制备高效液相色谱分离,得到具有式ii
‑
3所示结构的二萜类化合物;
[0078]
所述第一反相硅胶柱分离、第二反相硅胶柱分离和第三反相硅胶柱分离采用的洗脱剂为甲醇
‑
水溶剂,所述甲醇
‑
水溶剂中甲醇和水的体积比为40:60~100:0;
[0079]
所述第一lh20凝胶柱分离、第二lh20凝胶柱分离、第三lh20凝胶柱分离、第四lh20凝胶柱分离和第五lh20凝胶柱分离采用的洗脱剂为甲醇;
[0080]
所述第一半制备高效液相色谱分离、第二半制备高效液、第五半制备高效液相色谱分离、第六半制备高效液相色谱分离和第七半制备高效液相色谱分离采用的溶剂为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%;
[0081]
所述步骤(3)~步骤(5)没有时间上的先后顺序。
[0082]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0083]
本发明将泽漆全草进行醇提,将得到的粗醇提物与萃取剂混合进行萃取,得到醇提物浸膏。
[0084]
在本发明中,所述泽漆全草在使用前优选还包括将所述泽漆全草进行干燥后粉碎。本发明对于所述干燥的温度和时间没有特殊限定,采用本领域熟知的干燥温度和时间即可。本发明对于所述粉碎的方式没有特殊限定,粉碎后的泽漆全草粉末的粒径为1~2cm即可。
[0085]
在本发明中,所述醇提采用的醇类溶剂优选包括甲醇和/或乙醇。在本发明中,所述醇提的温度优选为50~65℃更优选为60~65℃;所述醇提的总时间优选为1~4h,更优选为2~3h;所述醇提的次数优选为2~4次,更优选为2~3次。在本发明中,所述泽漆全草的质量和单次醇提用醇类溶剂的体积之比优选为1kg:(1~5)l,更优选为1kg:(3~4)l。所述醇提后,本发明优选还包括合并醇提取液后进行浓缩,得到粗醇提物。本发明对于所述浓缩没有特殊限定,采用本领域熟知的浓缩方式即可,具体如减压蒸馏。
[0086]
在本发明中,所述萃取剂优选为小极性溶剂
‑
大极性溶剂溶剂,所述小极性溶剂优选括石油醚和/或氯仿,更优选包括石油醚或氯仿;所述大极性溶剂优选包括水、乙酸乙酯、异丙醇、丙酮和甲醇中得一种或几种,更优选包括水、乙酸乙酯、异丙醇、丙酮或甲醇;所述萃取剂具体优选为石油醚
‑
水混合溶剂、石油醚
‑
乙酸乙酯混合溶剂、石油醚
‑
异丙醇混合溶剂、石油醚
‑
丙酮混合溶剂、氯仿
‑
甲醇混合溶剂、氯仿
‑
异丙醇混合溶剂;所述小极性溶剂
‑
大极性溶剂溶剂中小极性溶剂和大极性溶剂的体积比优选为1:0~1,更优选为1:0.01~0.2。在本发明中,所述泽漆全草的质量和萃取用萃取剂的体积之比优选为1kg:(0.5~2)l,更优选为1kg:(1~1.5)l。所述萃取后,本发明优选还包括合并萃取液后进行浓缩,得到提取物浸膏。本发明对于所述浓缩没有特殊限定,采用本领域熟知的浓缩方式即可,具体如减压蒸馏。
[0087]
得到醇提取物浸膏后,本发明将所述醇提物浸膏进行第一硅胶柱色谱分离,得到组分frab、组分frad和组分frae。在本发明中,所述第一硅胶柱色谱分离的洗脱剂包括石油醚
‑
乙酸乙酯溶剂,所述石油醚
‑
乙酸乙酯溶剂中石油醚和乙酸乙酯的体积比为50:1~0:1;所述第一硅胶柱色谱分离的洗脱方式优选为梯度洗脱;具体的,所述梯度洗脱优选为依次采用体积比为50:1、20:1、10:1、8:1、4:1、1:1和0:1的石油醚
‑
乙酸乙酯溶剂进行,通过薄层色谱跟踪检识,得到7个组分,依次编号为组分fraa、组分frab、组分frac、组分frad、组分frae、组分fraf和组分frag。
[0088]
得到组分frab后,本发明将所述组分frab进行第一反相硅胶柱分离、得到组分fb2、组分fb3和组分fb4。在本发明中,所述第一反相硅胶柱分离采用的洗脱剂为甲醇
‑
水溶剂,所述甲醇
‑
水溶剂中甲醇和水的体积比为40:60~100:0;所述第一反相硅胶柱分离的洗脱方式优选为梯度洗脱,所述梯度洗脱优选为依次采用体积比为40:60、50:50、70:30、90:10和100:0的甲醇
‑
水溶剂进行,根据薄层色谱跟踪检识,得到6个组分,依次编号为组分fb1、组分fb2、组分fb3、组分fb4、组分fb5和组分fb6。
[0089]
得到组分fb2后,本发明将所述组分fb2进行第一lh20凝胶柱分离,得到组分fb2a和组分fb2b;将所述组分fb2a进行第一薄层色谱硅胶板分离,得到具有式iii
‑
3和式v所示结构的二萜类化合物;将所述组分fb2b进行第二薄层色谱硅胶板分离,得具有式iii
‑
1所示结构的二萜类化合物。
[0090]
得到组分fb2后,本发明将所述组分fb2进行第一lh20凝胶柱分离,得到组分fb2a和组分fb2b。在本发明中,所述第一lh20凝胶柱分离采用的洗脱剂为甲醇;所述第一lh20凝胶柱分离过程中,根据薄层色谱跟踪检识,得到4个组分,依次编号为组分fb2a、组分fb2b、组分fb2c和组分fb2d。
[0091]
得到组分fb2a后,本发明将所述组分fb2a进行第一薄层色谱硅胶板分离,得到具有式iii
‑
3和式v所示结构的二萜类化合物。在本发明中,所述第一薄层色谱硅胶板分离和
第二薄层色谱硅胶板分离采用的展开剂为氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比为20:1。
[0092]
得到组分fb3后,本发明将所述组分fb3进行第二硅胶柱色谱分离,得到组分fb3b和组分fb3c;将所述组分fb3b进行重结晶,得到具有式ii
‑
1所示结构的二萜类化合物;将所述组分fb3c进行第三薄层色谱硅胶板分离,得具有式iii
‑
2所示结构的二萜类化合物;所述第二硅胶柱色谱分离采用的洗脱剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为4:1~1:5;所述重结晶采用的溶剂为甲醇。
[0093]
得到组分fb3后,本发明将所述组分fb3进行第二硅胶柱色谱分离,得到组分fb3b和组分fb3c。在本发明中,所述第二硅胶柱色谱分离采用的洗脱剂为石油醚
‑
乙酸乙酯混合溶剂,所述石油醚
‑
乙酸乙酯混合溶剂中石油醚和乙酸乙酯的体积比为4:1~1:5;所述硅胶柱色谱分离的洗脱方式优选为梯度洗脱,所述梯度洗脱优选为依次采用体积比为4:1、3:1、2:1、1:1、1:2和1:5的石油醚
‑
乙酸乙酯混合溶剂进行,根据薄层色谱跟踪检识,得到4个组分,依次编号为组分fb3a、组分fb3b、组分fb3c和组分fb3d。
[0094]
得到组分fb3b后,本发明将所述组分fb3b进行重结晶,得到具有式ii
‑
1所示结构的二萜类化合物。在本发明中,所述重结晶用溶剂为甲醇。
[0095]
得到组分fb3c后,本发明将所述组分fb3c进行第三薄层色谱硅胶板分离,得具有式iii
‑
2所示结构的二萜类化合物。在本发明中,所述第三薄层色谱硅胶板分离采用的展开剂为氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比为20:1。
[0096]
得到组分fb4后,本发明将所述组分fb4进行第三硅胶柱色谱分离,得到组分fb4a、组分fb4b和组分fb4c;将所述组分fb4a进行第一半制备高效液相色谱分离,得到具有式ii
‑
2所示结构的二萜类化合物;将所述组分fb4b进行第二半制备高效液相色谱分离,得到具有式ii
‑
4所示结构的二萜类化合物;将所述组分fb4c经进行第三半制备高效液相色谱分离,得到具有式ii
‑
5所示结构的二萜类化合物;所述第三硅胶柱色谱分离采用的洗脱剂为氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比为250:1~10:1。
[0097]
得到组分fb4后,本发明将所述组分fb4进行第三硅胶柱色谱分离,得到组分fb4a、组分fb4b和组分fb4c。在本发明中,所述第三硅胶柱色谱分离采用的洗脱剂为氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比为250:1~10:1;所述硅胶柱色谱分离的洗脱方式优选为梯度洗脱,所述梯度洗脱优选为依次采用体积比为250:1、100:1、80:1、50:1、30:1、20:1和10:1的氯仿
‑
甲醇混合溶剂进行,根据薄层色谱跟踪检识,得到6个组分,依次编号为组分fb4a、组分fb4b、组分fb4c、组分fb4d、组分fb4e和组分fb4f。
[0098]
得到组分fb4a后,本发明将所述组分fb4a进行第一半制备高效液相色谱分离,得到具有式ii
‑
2所示结构的二萜类化合物。在本发明中,所述第一半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%,优选为64%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0099]
得到组分fb4b后,本发明将所述组分fb4b进行第二半制备高效液相色谱分离,得到具有式ii
‑
4所示结构的二萜类化合物。在本发明中,所述第二半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
谁混合溶剂中乙腈的体积分数为63~65%,优选为64%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为
254nm。
[0100]
得到组分fb4c后,本发明将所述组分fb4c经进行第三半制备高效液相色谱分离,得到具有式ii
‑
5所示结构的二萜类化合物。在本发明中,所述第三半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%,优选为64%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0101]
得到组分frad后,本发明将所述组分frad进行第二反相硅胶柱分离,得到组分fd1、组分fd2和组分fd3;
[0102]
将所述组分fd1进行第二lh20凝胶柱分离,得到组分fd1b;将所述组分fd1b进行第四半制备高效液相色谱分离,得到具有式iv所示结构的二萜类化合物;
[0103]
将所述组分fd2进行第三lh20凝胶柱分离,得到组分fd2c;将所述组分fd2c进行第五半制备高效液相色谱分离,得到具有式iii
‑
4所示结构的二萜类化合物;
[0104]
将所述组分fd3进行第四lh20凝胶柱分离,得到组分fd3b;将所述组分fd3b进行第六半制备高效液相色谱分离,得到具有式i所示结构的二萜类化合物。
[0105]
得到组分frad后,本发明将所述组分frad进行第二反相硅胶柱分离,得到组分fd1、组分fd2和组分fd3。在本发明中,所述第二反相硅胶柱分离采用的洗脱剂为甲醇
‑
水溶剂,所述甲醇
‑
水溶剂中甲醇和水的体积比为40:60~100:0;所述反相硅胶柱分离的洗脱方式优选为梯度洗脱,所述梯度洗脱优选依次采用体积比为40:60、50:50、70:30、90:10和100:0的甲醇
‑
水溶剂进行,根据薄层色谱跟踪检识,得到6个组分,依次编号为组分fd1、组分fd2、组分fd3、组分fd4、组分fd5和组分fd6。
[0106]
得到组分fd1后,本发明将所述组分fd1进行第二lh20凝胶柱分离,得到组分fd1b;将所述组分fd1b进行第四半制备高效液相色谱分离,得到具有式iv所示结构的二萜类化合物。在本发明中,所述第二lh20凝胶柱分离采用的洗脱剂为甲醇。在本发明中,根据薄层色谱跟踪检识,所述组分fd1经第二lh20凝胶柱分离得到3个组分,依次编号为组分fd1a、组分fd1b和组分fd1c。在本发明中,所述第四半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%,更优选为63~64%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0107]
得到组分fd2后,本发明将所述组分fd2进行第三lh20凝胶柱分离,得到组分fd2c;将所述组分fd2c进行第五半制备高效液相色谱分离,得到具有式iii
‑
4所示结构的二萜类化合物。在本发明中,所述第三lh20凝胶柱分离采用的洗脱剂为甲醇。在本发明中,根据薄层色谱跟踪检识,所述组分fd2经第三lh20凝胶柱分离得到5个组分,依次编号为组分fd2a、组分fd2b、组分fd2c、组分fd2d和组分fd2e。在本发明中,所述第五半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%,更优选为63~64%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0108]
得到组分fd3后,本发明将所述组分fd3进行第四lh20凝胶柱分离,得到组分fd3b;将所述组分fd3b进行第六半制备高效液相色谱分离,得到具有式i所示结构的二萜类化合物。在本发明中,所述第四lh20凝胶柱分离采用的洗脱剂为甲醇。在本发明中,根据薄层色谱跟踪检识,所述组分fd3经第四lh20凝胶柱分离得到3个组分,依次编号为组分fd3a、组分
fd3b和组分fd3c。在本发明中,所述第六半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%,更优选为63~64%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0109]
得到组分frae后,本发明将所述组分frae进行第三反相硅胶柱分离,得到组分fe1;将所述组分fe1进行第五lh20凝胶柱分离,得到组分fe1a;将所述组分fe1a进行第七半制备高效液相色谱分离,得到具有式ii
‑
3所示结构的二萜类化合物。
[0110]
得到组分frae后,本发明将所述组分frae进行第三反相硅胶柱分离,得到组分fe1。在本发明中,所述第三反相硅胶柱分离采用的洗脱剂为甲醇
‑
水溶剂,所述甲醇
‑
水溶剂中甲醇和水的体积比为40:60~100:0;所述反相硅胶柱分离的洗脱方式优选为梯度洗脱,所述梯度洗脱优选依次采用体积比为40:60、50:50、70:30、90:10和100:0的甲醇
‑
水溶剂进行,根据薄层色谱跟踪检识,得到5个组分,依次编号为组分fe1、组分fe2、组分fe3、组分fe4和组分fe5。
[0111]
得到组分fe1后,本发明将所述组分fe1进行第五lh20凝胶柱分离,得到组分fe1a。在本发明中,所述第五lh20凝胶柱分离采用的洗脱剂为甲醇。在本发明中,根据薄层色谱跟踪检识,所述组分fe1经第五lh20凝胶柱分离得到4个组分,依次编号为组分依次编号为组分fe1a、组分fe1b、组分fe1c和组分fe1d。在本发明中,根据薄层色谱跟踪检识,所述组分fe2经第六lh20凝胶柱分离得到4个组分,依次编号为组分fe2a、组分fe2b、组分fe2c和组分fe2d。
[0112]
得到组分fe1a后,本发明将所述组分fe1a进行第七半制备高效液相色谱分离,得到具有式ii
‑
3所示结构的二萜类化合物。在本发明中,所述第七半制备高效液相色谱分离采用的流动相为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数为63~65%,更优选为64~65%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0113]
本发明提供了一种二萜类化合物的药学上可接受的衍生物,包括上述技术方案所述二萜类化合物的衍生物和具有式iii和式vi~ix所示结构中的任意一种或几种的二萜类化合物的衍生物;所述衍生物包括对映体和/或消旋体;
[0114][0115]
在本发明中,所述式iii所示的结构中:
[0116]
当r6为α
‑
ch3时,r7为oh,r8为=o,r9为α
‑
ch3,r
10
为oh,r
11
为h,记为式iii
‑
5;
[0117]
当r6为β
‑
ch3时,r7为oac,r8为oac,r9为β
‑
ch3,r
10
为oac,r
11
为h,记为式iii
‑
6;
[0118]
当r6为α
‑
ch3时,r7为oh,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为h,记为式iii
‑
7;
[0119]
当r6为α
‑
ch3时,r7为
‑
onic,r8为=o,r9为α
‑
ch3,r
10
为oac,r
11
为h,记为式iii
‑
8。
[0120]
在本发明中,所述具有式iii和式vi~ix所示结构中的任意一种或几种的二萜类化合物的制备方法优选与前述具有式i~v所示的结构的二萜类化合物的制备方法相同,在此不再赘述。
[0121]
在本发明中,具有式viii所示的结构的二萜类化合物优选由所述组分fb2c经第四薄层色谱硅胶板分离得到,所述第四薄层色谱硅胶板分离的展开剂优选氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比优选为20:1。
[0122]
在本发明中,具有式iii
‑
5所示的结构的二萜类化合物优选由所述组分fb3b经第五薄层色谱硅胶板分离得到,所述第五薄层色谱硅胶板分离的展开剂优选为氯仿
‑
甲醇20:1。
[0123]
在本发明中,具有式ix所示的结构的二萜类化合物优选由所述组分fb5经第二重结晶得到,所述第二重结晶用溶剂优选为甲醇。
[0124]
在本发明中,具有式vi所示的结构的二萜类化合物优选由所述组分fd1b经第四半制备高效液相色谱分离得到,所述第四半制备高效液相色谱分离的流动相优选为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数优选为63%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0125]
在本发明中,具有式vii所示的结构的二萜类化合物优选由所述组分fd2a经第八半制备高效液相色谱分离得到,所述第八半制备高效液相色谱分离的流动相优选为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数优选为63%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0126]
在本发明中,具有式iii
‑
8所示的结构的二萜类化合物优选由所述组分fd3c经第六薄层色谱硅胶板分离得到,所述第六薄层色谱硅胶板分离的展开剂优选氯仿
‑
甲醇混合溶剂,所述氯仿
‑
甲醇混合溶剂中氯仿和甲醇的体积比优选为20:1。
[0127]
在本发明中,具有式iii
‑
7所示的结构的二萜类化合物优选由所述组分fe1a经第七半制备高效液相色谱分离得到,所述第七半制备高效液相色谱分离的流动相优选为乙腈
‑
水混合溶剂,所述乙腈
‑
水混合溶剂中乙腈的体积分数优选为65%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0128]
在本发明中,具有式iii
‑
6所示的结构的二萜类化合物优选由所述组分fe2b经第九半制备高效液相色谱分离得到,所述第九半制备高效液相色谱分离的流动相优选为甲醇
‑
水混合溶剂,所述甲醇
‑
水混合溶剂中甲醇的体积分数优选为75%;所述流动相的流速优选为2~3ml/min,更优选为3ml/min;检测波长优选为254nm。
[0129]
本发明提供了一种药物组合物,包括上述技术方案所述的二萜类化合物和/或上述技术方案所述的二萜类化合物的药学上可接受的衍生物;所述衍生物包括对映体和/或外消旋体。
[0130]
在本发明中,所述药物组合物还包括可药用载体和/或赋形剂。在本发明中,所述药物组合物优选包括二萜类化合物和/或其药学上可接受的衍生物、、药用载体和/或赋形
剂。在本发明中,所述药物组合物中二萜类化合物和/或二萜类化合物的药学上可接受的衍生物的总含量优选≥0.9wt%。
[0131]
本发明对于所述药用载体或所述赋形剂没有特殊限定,采用本领域熟知的药用载体或赋形剂即可,具体如固体、半固体或液体稀释剂,填料或药物制品辅剂中的一种或多种。在本发明中,所述药物组合物的制剂剂型优选包括液体制剂、固体制剂、酊剂、喷剂或雾剂;所述液体制剂优选包括注射剂、混悬剂、乳剂或溶液剂;所述固体制剂优选包括片剂、粉剂、胶囊剂、膏剂、霜剂、颗粒剂或冲剂。本发明中对于所述药物组合物的制备方法没有特殊限定,采用本领域熟知的制备方法即可。
[0132]
在本发明中,所述药物组合物的给药方式优选为注射、口服、舌下给药或粘膜透析;所述注射优选包括静脉注射、静脉滴注、肌肉注射、腹腔注射或皮下注射。
[0133]
在本发明中,所述二萜类化合物药物优选以单位体重服用量的形式使用。在本发明中,所述单位体重服用量优选为1~10mg/kg。
[0134]
本发明还提供了上述技术方案所述二萜类化合物、上述技术方案所述二萜类化合物的药学上可接受的衍生物和上述技术方案所述药物组合物中的一种或几种在制备kv1.3离子通道介导的疾病的药物中的应用。在本发明中,所述kv1.3离子通道介导的疾病优选包括银屑病。
[0135]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0136]
实施例1
[0137]
(1)取泽漆全草干燥后粉碎,在得到的泽漆全草粉末中加入甲醇在65℃条件下醇提3次,醇提总时间为3h,合并醇提液后减压浓缩,得到粗醇提物;将所述粗醇提物用水
‑
石油醚混合溶剂(石油醚和水的体积比为1:1)萃取5次,合并萃取液后减压浓缩,得到醇提物浸膏;其中,泽漆全草粉末质量和单次提取用甲醇的体积比为1kg:5l;泽漆全草粉末质量和单次萃取用水
‑
石油醚混合溶剂的体积比为1kg:0.5l。
[0138]
(2)将所述醇提物浸膏进行第一硅胶柱色谱分离,依次采用体积比为50:1、20:1、10:1、8:1、4:1、1:1和0:1的石油醚
‑
乙酸乙酯溶剂进行梯度洗脱,根据薄层色谱跟踪检识,得到得到7个组分,依次编号为组分fraa、组分frab、组分frac、组分frad、组分frae、组分fraf和组分frag。
[0139]
(3)具有式ii
‑
1、式ii
‑
2、式ii
‑
4、式ii
‑
5、式iii
‑
1、式iii
‑
2、式iii
‑
3、式iii
‑
5、式v、式viii和式ix所示结构的二萜类化合物的制备
[0140]
(3.1)将所述组分frab进行第一反相硅胶柱分离,依次采用体积比为40:60、50:50、70:30、90:10和100:0的甲醇
‑
水溶剂进行梯度洗脱,根据薄层色谱跟踪检识,得到6个组分,依次编号为组分fb1、组分fb2、组分fb3、组分fb4、组分fb5和组分fb6。
[0141]
(3.2)将所述组分fb2进行第一lh20凝胶柱分离,甲醇作为洗脱剂,根据薄层色谱跟踪检识,得到4个组分,依次编号为组分fb2a、组分fb2b、组分fb2c和组分fb2d。
[0142]
(3.2.1)将所述组分fb2a进行第一制备薄层色谱硅胶板分离,得到具有式iii
‑
3和式v所示结构的二萜类化合物,展开剂为体积比为20:1的氯仿
‑
甲醇混合溶剂。
[0143]
(3.2.2)将所述组分fb2b进行制备第二薄层色谱硅胶板分离,得具有式iii
‑
1所示结构的二萜类化合物,展开剂为体积比为20:1的氯仿
‑
甲醇混合溶剂。
[0144]
(3.2.3)将所述组分fb2c进行第四制备薄层色谱硅胶板分离,得具有式viii所示结构的二萜类化合物,展开剂为体积比为20:1的氯仿
‑
甲醇混合溶剂。
[0145]
(3.3)将所述组分fb3进行第二硅胶柱色谱分离,依次采用体积比为4:1、3:1、2:1、1:1、1:2和1:5的石油醚
‑
乙酸乙酯混合溶剂进行梯度洗脱,根据薄层色谱跟踪检识,得到4个组分,依次编号为组分fb3a、组分fb3b、组分fb3c和组分fb3d。
[0146]
(3.3.1)所述组分fb3b进行第一重结晶,得到具有式ii
‑
1所示结构的二萜类化合物,重结晶用溶剂为甲醇。
[0147]
(3.3.2)将所述组分fb3b进行第五制备薄层色谱硅胶板分离,得具有式iii
‑
5所示结构的二萜类化合物,展开剂为体积比为20:1的氯仿
‑
甲醇混合溶剂。
[0148]
(3.3.3)将所述组分fb3c进行第二制备薄层色谱硅胶板分离,得具有式iii
‑
2所示结构的二萜类化合物,展开剂为体积比为20:1的氯仿
‑
甲醇混合溶剂。
[0149]
(3.4)将所述组分fb4进行第三硅胶柱色谱分离,依次用体积比为250:1、100:1、80:1、50:1、30:1、20:1和10:1的氯仿
‑
甲醇混合溶剂进行梯度洗脱,根据薄层色谱跟踪检识,得到6个组分,依次编号为组分fb4a、组分fb4b、组分fb4c、组分fb4d、组分fb4e和组分fb4f。
[0150]
(3.4.1)将所述组分fb4a第一经半制备高效液相色谱分离,流动相为64%乙腈,流动相流速为3ml/min,检测波长为254nm,保留时间为12.5min,得到具有式ii
‑
2所示结构的二萜类化合物。
[0151]
(3.4.2)将所述组分fb4b进行第二半制备高效液相色谱分离,流动相为64%乙腈
‑
水混合溶剂,流动相流速为3ml/min,检测波长为254nm,保留时间为13.9min,得到具有式ii
‑
4所示结构的二萜类化合物。
[0152]
(3.4.3)将所述组分fb4c进行第三半制备高效液相色谱分离,流动相为64%乙腈
‑
水混合溶剂,流动相流速为3ml/min,检测波长为254nm,保留时间为11.7min,得到具有式ii
‑
5所示结构的二萜类化合物。
[0153]
(3.5)将所述组分fb5用甲醇作为溶剂进行第二重结晶,得到具有式ix所示结构的二萜类化合物。
[0154]
(4)具有式i、式iii
‑
4、式iii
‑
8、式iv、式vi和式vii所示结构的二萜类化合物的制备
[0155]
(4.1)将所述组分frad进行第二反相硅胶柱分离,采用体积比为40:60、50:50、70:30、90:10和100:0的甲醇
‑
水溶剂进行梯度洗脱,根据薄层色谱跟踪检识,得到6个组分,依次编号为组分fd1、组分fd2、组分fd3、组分fd4、组分fd5和组分fd6。
[0156]
(4.2)将所述组分fd1进行第二lh20凝胶柱分离,以甲醇作为洗脱剂,根据薄层色谱跟踪检识,得到3个组分,依次编号为组分fd1a、组分fd1b和组分fd1c;
[0157]
将所述组分fd1b进行第四半制备高效液相色谱分离,流动相为63%乙腈
‑
水混合溶剂,流动相流速为3ml/min,检测波长为254nm,得到具有式iv和式vi所示结构的二萜类化合物,保留时间分别为14.1min和12.7min。
[0158]
(4.3)将所述组分fd2进行第三lh20凝胶柱分离,甲醇作为洗脱剂,根据薄层色谱
跟踪检识,得到5个组分,依次编号为组分fd2a、组分fd2b、组分fd2c、组分fd2d和组分fd2e。
[0159]
(4.3.1)将所述组分fd2a进行第八半制备高效液相色谱分离,流动相为63%乙腈
‑
水混合溶剂,流速为3ml/min,检测波长为254nm,保留时间为13.5min,得到具有式vii所示结构的二萜类化合物。
[0160]
(4.3.2)将所述组分fd2c进行第五半制备高效液相色谱分离,流动相为63%乙腈
‑
水混合溶剂,流速为3ml/min,检测波长为254nm,保留时间为11.9min,得到具有式iii
‑
4所示结构的二萜类化合物。
[0161]
(4.4)将所述组分fd3进行第四lh20凝胶柱分离,甲醇为洗脱剂,流速为3ml/min,检测波长为254nm,保留时间为11.9min,根据薄层色谱跟踪检识,得到3个组分,依次编号为组分fd3a、组分fd3b和组分fd3c。
[0162]
(4.4.1)将所述组分fd3b进行第六半制备高效液相色谱分离,流动相为63%乙腈
‑
水混合溶剂,流速33ml/min,检测波长为254nm,保留时间为13.2min,得到具有式i所示结构的二萜类化合物。
[0163]
(4.4.2)将所述组分fd3c进行第六薄层色谱硅胶板分离,以体积比为20:1的氯仿
‑
甲醇混合溶剂为展开剂,得到具有式iii
‑
8所示结构的二萜类化合物。
[0164]
(5)具有式ii
‑
3、式iii
‑
6和式iii
‑
7所示结构的二萜类化合物的制备
[0165]
将所述组分frae进行第三反相硅胶柱分离,依次采用体积比为40:60、50:50、70:30、90:10和100:0的甲醇
‑
水溶剂进行梯度洗脱,根据薄层色谱跟踪检识,得到5组分,依次编号为组分fe1、组分fe2、组分fe3、组分fe4和组分fe5。
[0166]
(5.1)将所述组分fe1进行第五lh20凝胶柱分离,甲醇为洗脱剂,根据薄层色谱跟踪检识,得到4个组分,依次编号为组分依次编号为组分fe1a、组分fe1b、组分fe1c和组分fe1d;
[0167]
将所述组分fe1a进行第七半制备高效液相色谱分离,流动相为65%乙腈
‑
水混合溶剂,流速为3ml/min,检测波长为254nm,得到具有式ii
‑
3和式iii
‑
7所示结构的二萜类化合物,保留时间分别为11.7min和13.3min。
[0168]
(5.2)将所述组分fe2进行第六lh20凝胶柱分离,甲醇作为洗脱剂,根据薄层色谱跟踪检识,得到4个组分,依次编号为组分fe2a、组分fe2b、组分fe2c和组分fe2d;
[0169]
将所述组分fe2b进行第九半制备高效液相色谱分离,流动相为75%甲醇
‑
水混合溶剂,流速33ml/min,检测波长为254nm,保留时间为14.8min,得到具有式iii
‑
6所示结构的二萜类化合物。
[0170]
本实施例制备的二萜类化合物的波谱数据表1~4所示。
[0171]
表1具有式i、ii
‑
1和ii
‑
2的二萜类化合物在氯仿中核磁位移值(耦合常数j,单位hz)
[0172]
[0173][0174]
表2具有式ii
‑
3、ii
‑
4和ii
‑
5的二萜类化合物在氯仿中核磁位移值(耦合常数j,单位hz)
[0175]
[0176][0177]
表3具有式iii
‑
1、iii
‑
2和iii
‑
3的二萜类化合物在氯仿中核磁位移值(耦合常数j,单位hz)
[0178]
[0179][0180]
表4化具有iii
‑
4、iv和v的二萜类化合物在氯仿中核磁位移值(耦合常数j,单位hz)
[0181]
[0182][0183][0184]
实施例2
[0185]
(1)细胞培养
[0186]
使用稳定表达kv1.3通道的人胚胎肾293(hek293,北京爱思普益生物科技股份有限公司)细胞。将hek293细胞在添加有10%fbs(胎牛血清)的dmem培养基中,在培养箱温度为37℃和含5%二氧化碳条件下培养48h,将2.5
×
105个细胞接种在6cm培养皿(最终培养基体积为5ml)中进行常规维护、扩增或维持细胞。为了维持电生理性能,细胞密度维持在≤80%,四环素诱导电流18h。所有细胞培养程序均符合细胞培养规范化流程。
[0187]
(2)膜片钳系统测试
[0188]
在
‑
80mv的钳制电位下记录kv1.3电流,然后在1秒内将钳制电位阶跃至 40mv,每隔10秒重复该步骤,以测试实施例1制备得20个二萜类化合物对kv1.3离子通道电流峰值的影响,数据由epc
‑
10放大器进一步收集,并存储在patchmaster(heka)软件中进行数据分析。用微量移液管拉拔器制备玻璃移液管,在显微镜下使用微操作器操纵玻璃移液管。当接触到单个hek293细胞后,施加轻微吸力以获得高密封电阻(gω)。在实现高密封性后,进行快速电容(pf)补偿,同时刺穿细胞膜。在全细胞模式表征后,通过全细胞补偿电解液优化细胞电容(in
‑
pf)。
[0189]
形成全细胞记录模式后,将实施例1得到的20个二萜类化合物分别配置测试溶液(浓度分别为1μmol/l和30μmol/l,具有式i和式ii
‑
1所示结构的二萜类化合物的浓度还包括10μmol/l、3μmol/l和0.3μmol/l),作为测试组,通过重力供液输送系统泵入安装在倒置显微镜台上的记录室。实验结束后,通过真空抽吸将测试液从记录室中吸出。每一个细胞在不含二萜类化合物的液体中检测到的电流作为自己的对照组。
[0190]
(3)计算方法
[0191]
二萜类化合物对kv1.3离子通道的抑制率(%)=(1
‑
测试组电流强度值/对照组电流强度值)
×
100%,测试结果如表1所示:
[0192]
表1实施例1制备的20个二萜类化合物对kv1.3离子通道的抑制作用
[0193][0194]
nt表示未检测
[0195]
由表1可知,本发明制备的二萜类化合物在10μm(μm即为μmol/l)浓度下均可显著抑制kv1.3离子通道功能。其中具有式ii
‑
1和式iv所示结构的二萜类化合物对kv1.3离子通道的抑制效果最为显著,其c
50
值均在0.9μm,说明,本发明制备的二萜类化合物均可用于制备抗银屑病药物。
[0196]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。