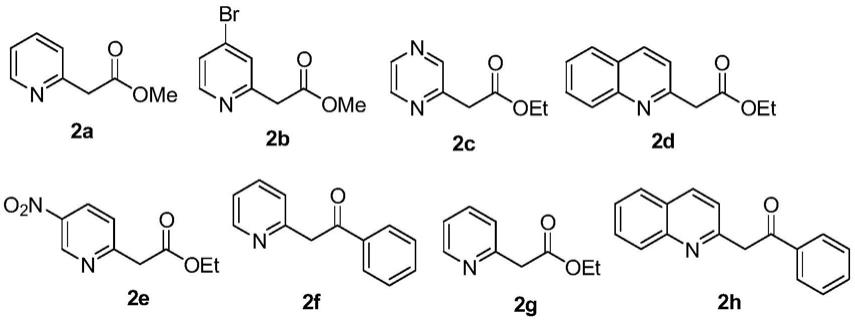
1.本发明属于有机合成领域,特别涉及一种二氢吲嗪并吡咯酮衍生物及类似物及其合成方法。
背景技术:
2.吲哚是药物化学中非常重要的化合物之一,市面上有许多基于吲哚开发的药物已被广泛用于各种疾病的治疗。而作为吲哚的同分异构体吲嗪(i),是一类含有两个稠合芳香杂环结构的吡啶并吡咯化合物,广泛存在于天然产物中。人们发现吲嗪类化合物具有独特的生物活性和药物活性[j.natural product reports,2002,19(6):742
‑
760.],因此常被用于抗结核剂[j.archiv der pharmazie,2003,336(3):191
‑
195.]、抗癌剂[j.bioorganic&medicinal chemistry,2008,16(8):4617
‑
4625.]、除草剂[j.pest management science,2005,61(1):16
‑
24.]、抗氧化剂[j.bioorganic&medicinal chemistry letters,1998,8(14):1829
‑
1832.]等领域。
[0003]
吡咯烷并吡咯酮类衍生物(ii)是以两个吡咯为骨架衍生出的杂环类化合物,具有较高的生物和药理活性[j.journal of medicinal chemistry.1998,41(21):3919
‑
3922;j.journal of organic chemistry.1999,64(14):5166
‑
5175.],可以作为人类中性粒细胞弹性蛋白酶的活性抑制剂用于治疗呼吸系统疾病[j.journal of medicinal chemistry.2002,45(18):3878
‑
3890]。
[0004][0005]
目前主流的合成吲嗪骨架的方法主要是通过吡啶衍生物或吡咯衍生物为原料进行的偶联环化反应[d.郑州大学,肖珂,2019.]。
[0006]
方法一、chen课题组[j.organic letters,2014,16(2):580
‑
583.]报道了金属钐催化c(sp3)
‑
h活化合成吲嗪衍生物的方法。该反应以三氟甲磺酸钐为催化剂,无溶剂下加热120℃得到多取代吲嗪衍生物。
[0007][0008]
方法二、vedejs课题组开发了一种吡咯衍生物在醋酐的回流条件下发生脱羧反应,随后在碱性条件下环化得到c7位取代的吲嗪衍生物的方法。
[0009][0010]
而关于制备吡咯并吡咯酮衍生物的方法则非常少,主要是依据macdonald和borthwick课题组开发的方法[j.journal of organic chemistry,1999,64(14):5166
‑
5175.j.journal of medicinal chemistry,2000,43(23):4452
‑
4464.]。macdonald课题组以蛋氨酸为原料经过9步反应以8%的总产率得到吡咯并吡咯酮。
[0011][0012]
borthwick课题以2,4
‑
二氨基丁酸为原料,经过双氨基保护、羧基还原、氧化、缩合、michael加成环化、脱boc、碱性条件环化八步反应以10%的总收率得到反式的吡咯并吡咯酮。
[0013][0014]
从易制得的原料通过尽可能简便的步骤构建高价值的骨架化合物一直是药物研发的目标。鉴于具有吲嗪骨架和吡咯烷并吡咯酮骨架的化合物存在广泛的药物活性,发明人设计将这两种杂环骨架合并于同一化合物中,合成一类新颖的的吲嗪并吡咯酮骨架的杂环化合物(iii),预测其可能具有良好的生物及药物活性。
技术实现要素:
[0015]
本发明以α
‑
丙二酸二乙酯取代的o
‑
酰基酮肟与吡啶类衍生物为原料,利用α
‑
丙二酸二乙酯取代的o
‑
酰基酮肟在铜盐催化下生成具有双亲电中心的环状中间体与吡啶类衍
生物进行双亲核加成反应,首次实现了一步高效构建各类取代的二氢吲嗪并吡咯酮衍生物及类似物的新方法。
[0016]
本发明合成方法为:丙二酸二乙酯取代的o
‑
酰基酮肟(1)与吡啶类衍生物(2)在溶剂中,铜盐催化剂和碱存在,氮气保护条件下加热至60
‑
100℃,反应1
‑
5小时,合成二氢吲嗪并吡咯酮衍生物及类似物(3),其合成路线如下:
[0017][0018]
其中,r1包括ph,4
‑
brph,4
‑
clph,4
‑
och3ph;
[0019]
r2包括br,no2;
[0020]
r3包括co2et,co2ch3,coph;
[0021]
x包括c,n原子。
[0022]
其中,丙二酸二乙酯取代的o
‑
酰基酮肟(1)为:
[0023][0024]
吡啶类化合物(2)为:
[0025][0026]
所用铜催化剂为cu(oac)2、cucl2、cucl、cui、cubr、cubr
‑
sme2、cu(otf)2、cu(clo4)2·
6h2o中的一种;
[0027]
碱为k2co3、na2co3、li2co3、naoac、nahco3中的一种;
[0028]
溶剂为甲苯、乙酸乙酯、乙腈、二氧六环、1,2
‑
二氯乙烷、二甲基亚砜、n,n
‑
二甲基
甲酰胺、乙酸正丁酯中的一种;
[0029]
α
‑
丙二酸二乙酯取代的o
‑
酰基酮肟、吡啶类化合物、铜盐催化剂、碱的摩尔比为1.0
‑
3.0:1.0
‑
2.0:0.5
‑
2.0:1.0
‑
2.0。
[0030]
该方法合成的二氢吲嗪并吡咯酮衍生物及类似物分子结构稳定,除了具有潜在的生物及结构多样性,可能还同时具有两种骨架的生物及药物活性。本发明中原料简单易得,所用催化剂及碱廉价,来源广泛,反应时间短,条件温和,不同官能团兼容性良好。
[0031]
本发明的有益效果:本发明以α
‑
丙二酸二乙酯取代的o
‑
酰基酮肟与吡啶类化合物为原料,铜盐为催化剂,加入合适的碱在有机溶剂中加热反应首次制得二氢吲嗪并吡咯酮衍生物及类似物。本发明所用的原料易制备,操作步骤简单,催化剂廉价易得,为制备多环化合物的合成方法提供了新的思路。实验结果表明,含各种取代基的丙二酸酯取代的o
‑
酰基酮肟均能以中等至优异的收率获得目标产物二氢吲嗪并吡咯酮衍生物及类似物。此外,该方法还具有条件温和,底物范围广等优点。
[0032]
本发明采用mtt法对三个典型化合物3aa、3ad、3ac进行抗癌活性测试,结果显示化合物3aa、3ad在50μm时对hepg2具有较好的抑制效果及选择性,其中3ad对a549、hepg2抑制效果更好,为之后的研究带来新的方向。
附图说明:
[0033]
图1、2为化合物3aa的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,cdcl3)谱图;
[0034]
图3、4为化合物3ab的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,d6‑
dmso)谱图;
[0035]
图5、6为化合物3ac的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,d6‑
dmso)谱图;
[0036]
图7、8为化合物3ad的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,cdcl3)谱图;
[0037]
图9、10为化合物3ad
′
的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,cdcl3)谱图;
[0038]
图11、12为化合物3ae的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,d6‑
dmso)谱图;
[0039]
图13、14为化合物3af的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,d6‑
dmso)谱图;
[0040]
图15、16为化合物3ag的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,d6‑
dmso)谱图;
[0041]
图17、18为化合物3ah的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,cdcl3)谱图;
[0042]
图19、20为化合物3ba的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,cdcl3)谱图;
[0043]
图21、22为化合物3ca的1h
‑
nmr(300mhz,d6‑
dmso)谱图和
13
c
‑
nmr(75mhz,d6‑
dmso)谱图;
[0044]
图23、24为化合物3da的1h
‑
nmr(300mhz,cdcl3)谱图和
13
c
‑
nmr(75mhz,cdcl3)谱图。
具体实施方式
[0045]
下面结合实施例对本发明做进一步描述,但不限于此。对所得产物进行测试,测试所用的仪器为:avance 300mhz型核磁共振仪(bruker公司,tms为内标);sgw x
‑
4显微熔点仪(温度计未经校正)。以下实施例测试方法与本实施例相同。
[0046]
实施例1
[0047]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3aa,产量为99.5mg,产率为85%。
[0048][0049]
3aa(yellow solid):1h nmr(300mhz,cdcl3)δ7.61(br,1h),7.44(d,j=7.4hz,2h),7.37(t,j=7.4hz,2h),7.28
‑
7.33(m,1h),7.11(t,j=6.8hz,1h),6.98(d,j=6.7hz,1h),6.70(br,1h),6.07(t,j=6.2hz,1h),4.81(d,j=3.9hz,1h),4.27(q,j=7.1hz,2h),3.63(d,j=4.0hz,1h),3.50(br,3h),1.30(t,j=7.1hz,3h);
13
c nmr(75mhz,cdcl3)δ168.6,167.5,165.5,154.6,143.5,137.3,133.0,128.8,127.9,125.5,117.9,108.9,90.2,73.7,71.1,62.7,56.3,50.2,14.1.
[0050]
实施例2
[0051]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.36mmol)、2
‑
(5
‑
溴
‑2‑
吡啶基)乙酸甲酯2b(0.3mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3ab,产量为121.8mg,产率为89%。
[0052][0053]
3ab(yellow solid,102.4mg):1h nmr(300mhz,cdcl3)δ7.70(br,1h),7.47
‑
7.29(m,5h),6.81(d,j=7.1hz,1h),6.73(br,1h),6.12(dd,j=7.1hz,1.6hz,1h),4.75(d,j=4.2hz,1h),4.28(q,j=7.1hz,2h),3.60(d,j=4.2hz,1h),3.57(br,3h),1.30(t,j=7.1hz,3h);
13
c nmr(75mhz,d6‑
dmso)δ168.9,167.6,164.4,152.5,144.7,136.9,133.7,128.2,127.0,125.5,117.4,111.1,91.3,73.2,71.1,61.4,55.4,49.6,13.8.
[0054]
实施例3
[0055]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.6mmol)、吡嗪
‑2‑
乙酸甲酯2c(0.44mmol)为原料,依次加入醋酸铜(0.088mmol)、碳酸锂(0.44mmol),溶剂甲苯(4.5ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3ac,产量为139.7mg,产率为84%。
[0056][0057]
3ac(red solid,139.7mg):1h nmr(300mhz,d6‑
cdcl3)δ8.89(br,1h),7.29
‑
7.44(m,5h),7.11(br,1h),6.80(br,1h),6.71(d,j=3.6hz,1h),4.77(d,j=3.9hz,1h),4.27(q,j=7.1hz,2h),3.58
‑
3.67(m,4h),1.30(t,j=7.1hz,3h);
13
c nmr(75mhz,d6‑
dmso)δ169.2,167.5,164.0,160.5,145.4,144.5,144.3,128.2,127.1,125.5,125.2,93.1,73.4,70.8,61.4,54.9,49.9,13.8.
[0058]
实施例4
[0059]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.6mmol)、喹啉
‑2‑
乙酸乙酯2d(0.5mmol)为原料,依次加入醋酸铜(0.1mmol)、碳酸锂(0.5mmol),溶剂甲苯(5.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(40ml)淬灭,用乙酸乙酯(3
×
15ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得到两个异构产物:3ad,产量为82.4mg,产率为37%;3ad
′
,产量为62.2mg,产率为28%。
[0060][0061]
3ad(yellow solid,82.4mg):1h nmr(300mhz,d6‑
cdcl3)δ7.65(d,j=9.3hz,1h),7.28
‑
7.48(m,8h),7.09(td,j=7.5hz,0.6hz,1h),6.84(br,1h),6.70(d,j=8.3hz,1h),5.07(d,j=3.8hz,1h),4.23
‑
4.45(m,2h),3.93
‑
4.21(m,2h),3.63(d,j=3.8hz,1h),1.32(t,j=7.1hz,3h),1.07(br,3h);
13
c nmr(75mhz,cdcl3)δ169.1,168.2,165.3,151.6,143.5,137.1,136.7,131.6,129.2,128.9,128.0,125.4,123.1,122.4,118.0,112.3,97.3,71.6,70.6,62.8,59.2,55.5,14.5,14.2.
[0062]
3ad
′
(yellow solid,62.2mg):1h nmr(300mhz,cdcl3)δ8.34(s,1h),7.66(d,j=9.7hz,1h),7.30
‑
7.45(m,6h),7.27(d,j=9.7hz,1h),7.08(ddd,j=8.5,7.3,1.3hz,1h),6.94(t,j=7.3hz,1h),6.43(d,j=8.3hz,1h),4.13
‑
4.26(m,3h),4.07(dq,j=10.7,7.1hz,1h),4.04(d,j=4.4hz,1h),3.59(d,j=4.5hz,1h),1.26(t,j=7.1hz,3h),1.22(t,j=7.1hz,3h);
13
c nmr(75mhz,cdcl3)δ174.8,168.7,165.5,151.5,140.3,136.4,136.3,130.8,129.6,129.1,129.0,124.7,123.3,122.0,117.9,113.6,92.4,86.8,62.0,59.2,57.6,56.3,14.5,14.2.
[0063]
实施例5
[0064]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.48mmol)、2
‑
(5
‑
硝
基吡啶
‑2‑
基)乙酸甲酯2e(0.4mmol)为原料,依次加入醋酸铜(0.08mmol)、碳酸锂(0.4mmol),溶剂甲苯(4.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(40ml)淬灭,用乙酸乙酯(3
×
15ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3ae,产量为101.2mg,产率为65%。
[0065][0066]
3ae(yellow solid,100.5mg):1h nmr(300mhz,d6‑
cdcl3)δ8.23(d,j=2.0hz,1h),7.67(dd,j=10.3hz,2.1hz,1h),7.44
‑
7.31(m,6h),6.80(s,1h),4.95(d,j=4.1hz,1h),4.31(q,j=7.1hz,2h),3.97
‑
4.20(m,2h),3.70(d,j=4.1hz,1h),1.34(t,j=7.2hz,3h),1.10(t,j=7.1hz,3h);
13
c nmr(75mhz,d6‑
dmso)δ169.5,167.6,164.0,150.0,144.2,141.1,131.5,130.4,128.5,127.6,125.8,115.0,101.3,73.4,71.8,61.7,59.0,55.5,14.4,14.1.
[0067]
实施例6
[0068]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.6mmol)、1
‑
苯基
‑2‑
吡啶
‑2‑
基乙酮2f(0.5mmol)为原料,依次加入醋酸铜(0.1mmol)、碳酸锂(0.5mmol),溶剂甲苯(5.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(40ml)淬灭,用乙酸乙酯(3
×
15ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3af,产量为92.1mg,产率为46%。
[0069][0070]
3af(yellow solid,92.1mg):1h nmr(300mhz,d6‑
cdcl3)δ7.54(d,j=7.3hz,2h),7.33
‑
7.50(m,8h),7.28(t,j=7.1hz,1h),7.11(d,j=6.6hz,1h),6.83
‑
6.97(m,1h),6.00
‑
6.35(m,2h),4.86(d,j=3.5hz,1h),4.32
‑
4.20(m,2h),3.63(d,j=3.5hz,1h),1.28(t,j=7.1hz,3h);
13
c nmr(75mhz,d6‑
dmso)δ184.5,168.4,167.5,152.7,144.6,143.1,138.2,136.1,129.4,128.7,128.0,126.8,126.7,125.6,115.6,110.2,105.5,73.1,71.5,61.3,55.5,13.8.
[0071]
实施例7
[0072]
取30ml的厚壁试管,以丙二酸二乙酯取代的酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸乙酯2g(0.3mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,
过滤,减压蒸馏除去有机溶剂,柱层析得3ag,产量为111.8mg,产率为87%。
[0073][0074]
3ag(yellow solid,102.8mg):1h nmr(300mhz,cdcl3)δ7.55(br,1h),7.44(d,j=7.4hz,2h),7.37(t,j=7.4hz,2h),7.27
‑
7.32(m,1h),7.10(t,j=7.7hz,2h),6.97(d,j=6.8hz,1h),6.67(br,1h),6.07(t,j=6.2hz,1h),4.81(d,j=3.8hz,1h),4.26(q,j=7.2hz,2h),4.04(br,2h),3.63(d,j=3.8hz,1h),1.29(t,j=7.2hz,3h),0.91(br,3h);
13
c nmr(75mhz,d6‑
dmso)δ169.3,168.0,164.5,153.4,145.6,138.4,135.6,128.4,127.2,126.0,116.5,108.6,91.3,73.8,71.5,61.7,57.7,56.1,14.8,14.2.
[0075]
实施例8
[0076]
取30ml的厚壁试管,以丙二酸二乙酯取代的对甲氧基苯乙酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸甲酯2h(0.3mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3ah,产量为73.4mg,产率为57%。
[0077][0078]
3ah(red solid,73.3mg):1h nmr(300mhz,d6‑
cdcl3)δ7.52(br,1h),7.44(d,j=7.2hz,2h),7.36(t,j=7.3hz,2h),7.28(t,j=7.1hz,1h),7.06(t,j=7.6hz,2h),6.95(d,j=6.6hz,1h),6.62(br,1h),6.04(br,1h),4.78(br,1h),4.24(q,j=7.1hz,2h),3.62(d,j=3.4hz,1h),1.60
‑
0.97(m,12h);
13
c nmr(75mhz,d6‑
dmso)δ168.8,167.4,165.0,153.9,144.2,136.9,132.6,128.6,127.7,125.5,117.8,108.7,92.6,78.7,73.2,71.6,62.7,56.2,28.5,14.1.
[0079]
实施例9
[0080]
取30ml的厚壁试管,以丙二酸二乙酯取代的对甲氧基苯乙酮o
‑
乙酰基肟1b(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3ba,产量为112.5mg,产率为92%。
[0081][0082]
3ba(yellow solid,112.5mg):1h nmr(300mhz,cdcl3)δ7.55(br,1h),7.33(d,j=8.8hz,2h),7.08(t,j=7.7hz,1h),6.96(d,j=7.8hz,1h),6.86(d,j=8.9hz,2h),6.78(br,1h),6.04(t,j=6.6hz,1h),4.75(d,j=4.1hz,1h),4.26(q,j=7.1hz,2h),3.78(s,3h),3.60(d,j=4.1hz,1h),3.54(br,3h),1.29(t,j=7.1hz,3h);
13
c nmr(75mhz,cdcl3)δ168.5,167.6,165.6,159.2,154.4,137.3,135.6,133.0,126.9,117.9,114.1,108.8,90.8,73.7,70.7,62.7,56.3,55.3,50.2,14.1.
[0083]
实施例10
[0084]
取30ml的厚壁试管,以丙二酸二乙酯取代的对溴苯乙酮o
‑
乙酰基肟1c(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3ca,产量为98.4mg,产率为71%。
[0085][0086]
3ca(yellow solid,98.2mg):1h nmr(300mhz,d6‑
dmso)δ9.12(s,1h),7.54(d,j=8.6hz,2h),7.51(d,j=6.9hz,1h),7.20
‑
7.36(m,4h),6.17
‑
6.27(m,1h),4.95(d,j=3.1hz,1h),4.06(q,j=7.1hz,2h),3.79(d,j=3.1hz,1h),3.36(br,3h),1.10(t,j=7.1hz,3h);
13
c nmr(75mhz,d6‑
dmso)δ168.9,167.5,164.4,144.7,138.3,135.3,130.9,128.0,120.1,116.0,108.6,90.2,73.3,70.8,61.4,55.5,49.4,13.8.
[0087]
实施例11
[0088]
取30ml的厚壁试管,以丙二酸二乙酯取代的对氯苯乙酮o
‑
乙酰基肟1d(0.3mmol)、吡啶
‑2‑
乙酸甲酯2a(0.45mmol)为原料,依次加入醋酸铜(0.06mmol)、碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测底物消失,反应结束。待反应混合物冷却至室温,加入水(30ml)淬灭,用乙酸乙酯(3
×
10ml)萃取,合并有机相,加入无水硫酸钠干燥,过滤,减压蒸馏除去有机溶剂,柱层析得3da,产量为81.7mg,产率为59%。
[0089][0090]
3da(yellow solid,82.1mg):1h nmr(300mhz,cdcl3)δ7.60(br,1h),7.39(d,j=8.7hz,1h),7.33(d,j=8.7hz,1h),7.13(t,j=7.4hz,1h),6.98(d,j=6.8hz,1h),6.69(br,1h),6.10(t,j=6.1hz,1h),4.77(d,j=3.5hz,1h),4.27(q,j=7.1hz,2h),3.62(d,j=3.8hz,1h),3.50(br,3h),1.31(t,j=7.1hz,3h);
13
c nmr(75mhz,cdcl3)δ168.6,167.4,165.5,142.2,137.5,133.8,132.9,129.0,127.2,118.0,109.2,73.6,70.9,62.9,56.2,50.3,14.2.
[0091]
实施例12
[0092]
以丙二酸二乙酯取代苯乙酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入碘化亚铜(0.06mmol),碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测反应进行,得到最终产物3aa,产量为89.0mg,产率为78%。
[0093]
实施例13
[0094]
以丙二酸二乙酯取代苯乙酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入醋酸铜(0.06mmol),碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至100℃,tlc监测反应进行,得到最终产物3aa,产量为46.8mg,产率为41%。
[0095]
实施例14
[0096]
以丙二酸二乙酯取代苯乙酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入醋酸铜(0.06mmol),碳酸钠(0.3mmol),溶剂二甲基亚砜(3.0ml),在n2保护条件下加热至80℃,tlc监测反应进行,得到最终产物3aa,产量为62.8mg,产率为55%。
[0097]
对比实施例1
[0098]
以丙二酸二乙酯取代苯乙酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙腈2i(0.3mmol)为原料,依次加入醋酸铜(0.06mmol),碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测反应进行,观察到生成痕量的产物3ai。
[0099]
对比实施例2
[0100]
以丙二酸二乙酯取代苯乙酮o
‑
乙酰基肟1a(0.36mmol)、2
‑
硝基甲基吡啶2j(0.3mmol)为原料,依次加入醋酸铜(0.06mmol),碳酸锂(0.3mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,观察到生成痕量目标产物3aj。
[0101]
对比实施例3
[0102]
以丙二酸二乙酯取代丙酮o
‑
乙酰基肟1e(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入醋酸铜(0.06mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至
60℃,tlc监测反应进行,生成了痕量的产物3ea。
[0103]
对比实施例4
[0104]
以丙二酸二乙酯取代苯乙酮o
‑
乙酰基肟1a(0.36mmol)、吡啶
‑2‑
乙酸甲酯2a(0.3mmol)为原料,依次加入氯化铁(0.06mmol),溶剂甲苯(3.0ml),在n2保护条件下加热至60℃,tlc监测反应进行,最终没有生成目标产物3aa。
[0105]
所述实施例为本发明的优选的实施方式,但本发明并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员能够做出的任何显而易见的改进、替换或变型均属于本发明的保护范围。
[0106]
应用例
[0107]
抗癌活性研究:选取h460(肺癌细胞)、hepg2(肝癌细胞)、a549(肺癌细胞)3种癌细胞为测试细胞株,对三种典型化合物3aa、3ad、3ac做体外抗癌活性测试,并以5
‑
氟尿嘧啶作阳性对照。将对数生长期的癌细胞,离心后用1640完全培养基1ml将细胞吹打均匀。根据每孔加入100μl培养基,每孔加入1.2
×
104个细胞的要求,计算所需细胞量,然后将细胞液重悬,取出所需量用含10%fbs的1640培养基稀释,接种于96孔板中,放入37℃,5%co2培养箱进行孵育16~18h。待细胞密度达到70%~80%时,进行药物干预。加入50μm的待测化合物60μl,继续孵育72h,每个干预物设置3个复孔,每块板中均设置空白对照及阳性对照。孵育72h,加入50μl 10%mtt继续孵育4h。将96孔板吸去mtt,加入dmso 100μl/孔,震荡10min,放入酶标仪,测570nm处od值。根据od值,计算化合物50μm时对癌细胞的抑制率。结果见下表。测试结果表明,三个化合物3aa、3ad、3ac于50μm时对hepg2具有明显的选择性。当原料2用吡啶
‑2‑
乙酸甲酯代替吡嗪
‑2‑
乙酸甲酯时,对hepg2癌细胞抑制效果得到明显提高。其中,与3aa、3ac相比,化合物3ad引入喹啉
‑2‑
乙酸乙酯后,对a549、hepg2的抑制效果更好。
[0108]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。