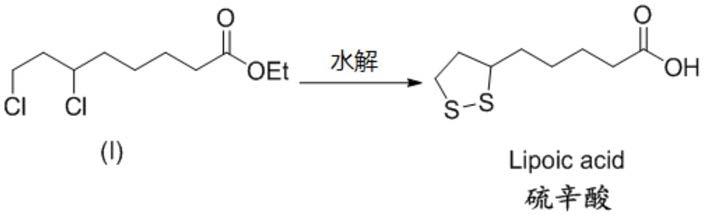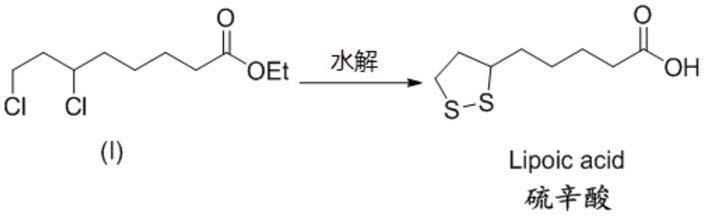
一种合成6,8
‑
二氯辛酸酯的方法
技术领域
1.本发明属于有机合成领域,具体涉及一种合成6,8
‑
二氯辛酸酯的方法。
背景技术:
2.硫辛酸(lipoic acid)化学名为1,2
‑
二硫戊环
‑3‑
戊酸,是一种存在于线粒体的辅酶,属于b族维生素中的一类化合物,在多酶系统中催化丙酮酸氧化脱羧成乙酸及α
‑
酮戊二酸氧化脱羧成琥珀酸,广泛分布于动植物等生物组织中。硫辛酸具有很强的抗氧化性能,被誉为“万能抗氧剂”,其抗氧化效果胜过维生素,能消除加速老化与致病的自由基;可改善糖尿病患者的胰岛素功能与心率变异数;治疗丙型肝炎、保护肾脏、胰脏、预防白内障等等,因而受到研究学者的广泛关注。其中6,8
‑
二氯辛酸乙酯是合成硫辛酸的关键中间体之一,通过硫化后水解乙酯即可得到产品硫辛酸:
[0003][0004]
目前6,8
‑
二氯辛酸乙酯的制备方法主要有以下几种:《浙江化工》,2010,41(5),10
‑
11,14.报道了以己二酸单乙酯为原料的合成方法。该方法先将己二酸单乙酯与三光气(btc)反应得到酰氯,然后再与乙烯发生friedel
‑
crafts烷基化反应得到氯酮中间体,氯酮中间体用硼氢化钾还原后得到氯醇中间体,最后氯醇中间体通过三光气氯代得到目标产物6,8
‑
二氯辛酸乙酯(i)。另外专利文献cn101157614a、cn105693510a分别对最后一步的氯代反应提出了改进方案。总的来说,该方法中己二酸单乙酯原料价格很高,且使用三氯化铝所产生的三废较多,还原剂硼氢化钾价格较贵,难以大规模生产。
[0005][0006]
专利文献cn107673972a报道以环己酮和乙烯基乙醚为原料,通过自由基反应得到乙基醚中间体,乙基醚中间体通过baeyer
‑
villiger氧化并开环得到开链的中间体酸,然后在三氯化铝的作用下断开醚键得到双羟基酸,最后在乙醇中与氯化亚砜反应得到6,8
‑
二氯辛酸乙酯(i)。该方法的问题在于三氯化铝的断开醚键的反应条件苛刻,三氯化铝用量大而
且收率低,很大程度上限制了该方法的应用。
[0007][0008]
专利文献cn112479878a以8
‑
羟基辛醛为原料,先将其氧化并酯化成8
‑
羟基辛酸乙酯,后经过脱水生成烯基酯中间体,烯基酯中间体再经过prins缩合反应得到甲缩醛中间体,然后经过水解加氢得到得到双羟基化合物,最后经过氯化得到6,8
‑
二氯辛酸乙酯(i)。该方法中,原料8
‑
羟基辛醛价格昂贵,而且多步用到固体催化剂,脱水反应温度达350℃,条件要求较高,不适合工业化生产。
[0009]
技术实现要素:
[0010]
为了克服了现有技术制备6,8
‑
二氯辛酸酯方法中存在的原料价格高、生产成本高、三废多、操作繁琐、不适合工业化生产的缺陷,本发明提供了一种新的合成6,8
‑
二氯辛酸酯的方法,其能够以廉价的反应原料、温和的反应条件和较高的收率制得6,8
‑
二氯辛酸酯,具有工业化生产可行性和经济性。具体而言,本发明包括以下技术方案。
[0011]
一种合成式(i)所示化合物6,8
‑
二氯辛酸酯的方法,包括以下步骤:
[0012]
a.使式(ii)所示化合物7
‑
(2
‑
羟乙基)己
‑2‑
内酯在醇溶液中发生开环氯代,得到式(i)所示化合物6,8
‑
二氯辛酸酯:
[0013][0014]
其中,r为c1
‑
c4烷基,roh选自甲醇、乙醇、正丙醇、异丙醇、正丁醇、异丁醇、叔丁醇。优选roh是乙醇或者甲醇,考虑到环保及原料经济性,更优选乙醇。
[0015]
上述步骤a中开环氯代反应所用的试剂可以为氯化亚砜socl2、光气或者三光气btc。
[0016]
在一种实施方式中,上述步骤a中式(ii)所示化合物7
‑
(2
‑
羟乙基)己
‑2‑
内酯可以通过下述步骤合成:
[0017]
b.使式(iii)所示化合物2
‑
(2
‑
羟乙基)环己
‑1‑
酮发生氧化反应,得到式(ii)所示化合物:
[0018][0019]
其中,所述氧化反应是baeyer
‑
villiger氧化反应。
[0020]
上述步骤b中氧化反应可以采用双氧水/乙酸体系、双氧水/甲酸体系、或者间氯过氧苯甲酸体系。从原料经济性角度考虑,优选双氧水/乙酸体系。
[0021]
在一种实施方式中,上述步骤b中式(iii)所示化合物2
‑
(2
‑
羟乙基)环己
‑1‑
酮可以通过下述步骤合成:
[0022]
c.使式(iv)所示化合物2
‑
氧代环己烷
‑1‑
羧酸酯与环氧乙烷加成后脱羧,得到式(iii)所示化合物:
[0023][0024]
其中,r1为c1
‑
c4烷基,选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基。
[0025]
上述步骤c中加成反应所用的碱可以选自甲醇钠、氢化钠、双三甲基硅基胺基锂、二异丙基氨基锂,从催化效率和经济性角度考虑,优选氢化钠;所用的溶剂选自甲苯、四氢呋喃、甲醇、或者它们两种以上的混合物,但不限于此。
[0026]
上述步骤c中脱羧的条件可以包括在盐酸水溶液或者硫酸水溶液中加热。
[0027]
在一种实施方式中,上述步骤c中式(iv)所示化合物2
‑
氧代环己烷
‑1‑
羧酸酯可以通过下述步骤合成:
[0028]
d.使式(v)所示化合物环己酮与碳酸酯在碱的催化下反应,得到式(iv)所示化合物:
[0029]
[0030]
其中,r1为c1
‑
c4烷基,选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基。优选地,r1为甲基或者乙基,即所述碳酸酯为碳酸二甲酯或者碳酸二乙酯。
[0031]
上述步骤d中使用的碱可以为氢化钠,所用的反应溶剂可以为甲苯。
[0032]
在一种优选的实施方式中,步骤d的产物2
‑
氧代环己烷
‑1‑
羧酸酯(iv)无需进行分离纯化,可以直接进行步骤c,且无需再加碱,得到2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii),这种连续反应操作能够降低整体工艺的生产成本,提高了经济性。
[0033]
本发明提供了一种新的6,8
‑
二氯辛酸酯的化学合成途径,该方法中,原料环己酮便宜易得,反应条件温和,产出的三废较少,操作简单,具有工业化大规模生产可行性。
具体实施方式
[0034]
本发明从降低生产成本的工业化生产的经济性构思考虑,提供了式(i)所示化合物6,8
‑
二氯辛酸酯的合成新途径。
[0035]
本文中,有时将术语“式x所示化合物”表述为“式x化合物”或“化合物x”,这是本领域技术人员能够理解的。比如式(i)所示化合物和化合物(i)都是指代相同的化合物。
[0036]
从廉价的化工原料环己酮出发、使用廉价易得的大宗化学品作为催化剂、溶剂等来制备式(i)所示化合物是比较经济的方案。
[0037][0038]
r、r1的定义如上所述。
[0039]
例如,步骤d中选用的碳酸二甲酯(r1为甲基)、步骤a中选用的乙醇(roh)都是同类化合物中价格最低廉的工业原料。此时,本发明的6,8
‑
二氯辛酸乙酯(式i中r为乙基)的合成方法包括下述步骤:
[0040]
1)以环己酮(v)为原料,在碱的作用下与碳酸二甲酯反应,得到中间体2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv);
[0041]
2)2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)与环氧乙烷加成后脱羧,得到中间体2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii);
[0042]
3)2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii)再通过baeyer
‑
villiger氧化得到中间体7
‑
(2
‑
羟乙基)己
‑2‑
内酯化合物(ii);
[0043]
4)7
‑
(2
‑
羟乙基)己
‑2‑
内酯化合物(ii)在乙醇中开环氯代,即可得到6,8
‑
二氯辛
酸乙酯(i)。
[0044][0045]
在优选的实施方式中,在上述各步骤反应完成后,可按本领域常识进行过滤、洗涤、脱色纯化、结晶、干燥等后处理操作。在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0046]
在一种优选的实施方式中,步骤1)的产物2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)无需进行分离纯化,可以直接进行步骤2),且无需再加碱,得到2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii),这种连续反应操作能够降低生产成本,提高了本发明的经济性。
[0047]
以下通过实施例进一步阐述本发明。应理解,这些实施例仅用于举例说明目的,而不是对本发明的限制。本领域技术人员根据本发明构思对其作出的各种改变或调整,均应落入本发明的保护范围内。
[0048]
本文中涉及到多种物质的添加量、含量及浓度,其中所述的百分含量,除特别说明外,皆指质量百分含量。
[0049]
本文的实施例中,如果对于反应温度或操作温度没有做出具体说明,则该温度通常指室温(15
‑
30℃)。
[0050]
实施例
[0051]
试剂:本发明实施例中使用的反应物和催化剂均为化学纯,可直接使用或根据需要经过简单纯化;有机溶剂等均为分析纯,直接使用。试剂均购自中国医药(集团)上海化学试剂公司。
[0052]
检测仪器:
[0053]
核磁共振仪型号:bruker avance iii 400mhz;
[0054]
质谱仪(液质联用(lcms)),型号:agilent 6120b。
[0055]
实施例1:2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)的制备
[0056][0057]
在1000ml四口瓶中加入400ml甲苯和50.0g碳酸二甲酯,氮气置换三次,分批加入31.0g氢化钠,加完之后缓慢升温至回流,反应30分钟,滴加27.2g环己酮(v),大约2小时加完。保持回流状态反应8小时。中控反应结束,将反应体系降至室温,滴加140ml的甲醇淬灭过量的氢化钠,得到化合物2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)的溶液。该溶液用稀盐酸洗一次,再用少量水洗涤一次,无水硫酸钠干燥后减压浓缩,得到产物粗品,减压精馏(2mmhg),收集75~80℃的馏分,得到2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)36.8g,收率:85%。
[0058]1h nmr(400mhz,cdcl3)δ3.75(s,3h),2.27(t,2h),2.22(t,2h),1.65(m,5h)。
[0059]
ms(esi)m/z=157.1(m
1)。
[0060]
实施例2:2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii)的制备
[0061][0062]
在1000ml的高压釜中,将2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)36.8g溶于500ml的甲苯,再加入甲醇钠17.7g,再加入24.4g环氧乙烷,缓慢升温至50度反应6小时。反应结束,向体系中加入100ml水和100ml浓盐酸,升温至50度反应3小时,降至室温,静置分层,甲苯相用少量水洗涤一次,无水硫酸钠干燥后减压浓缩,得到产物粗品,减压精馏(2mmhg),收集120~125℃的馏分,得到2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii)24.4g,收率73%。
[0063]1h nmr(400mhz,cdcl3)δ4.65(s,1h),3.83(t,2h),2.28(m,2h),2.12(m,1h),1.65(m,8h)。
[0064]
ms(esi)m/z=143.1(m
1)。
[0065]
重复上述还原反应操作,采用不同的溶剂和碱,分离计算收率,实验结果如下表所示:
[0066]
序号溶剂碱收率1甲苯甲醇钠73%2甲苯氢化钠77%3四氢呋喃双三甲基硅基胺基锂70%4四氢呋喃二异丙基氨基锂67%5甲醇甲醇钠52%
[0067]
由表中可知,氢化钠的催化效率最高。
[0068]
实施例3:2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii)的连续制备
[0069][0070]
在500ml四口瓶中加入200ml甲苯和25.0g碳酸二甲酯,氮气置换三次,分批加入15.5g氢化钠,加完之后缓慢升温至回流,反应30分钟,滴加13.6g环己酮(v),大约2小时加完。保持回流状态反应8小时。监控反应结束,将反应体系降至室温,滴加70ml的甲醇淬灭过量的氢化钠,得到化合物2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)的溶液。该溶液直接投入下一步反应。
[0071]
将2
‑
氧代环己烷
‑1‑
羧酸甲酯(iv)的溶液转移至高压釜中,加入12.2g环氧乙烷,缓慢升温至50℃反应6小时。反应结束,向体系中加入50ml水和25ml浓硫酸(98%),升温至50℃反应3小时,降至室温,静置分层,甲苯相用少量水洗涤一次,无水硫酸钠干燥后减压浓缩,得到产物粗品,减压精馏,收集120~125℃的馏分,得到2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii)12.2g,总收率62%。
[0072]
实施例4:7
‑
(2
‑
羟乙基)己
‑2‑
内酯化合物(ii)的制备
[0073][0074]
将2
‑
(2
‑
羟乙基)环己
‑1‑
酮(iii)10g加入乙酸20ml与双氧水(30%)20ml的混合液中,室温反应24小时。反应结束,加入二氯甲烷50ml,体系用5%碳酸钠溶液洗至中性,分离得有机层,水洗,无水硫酸钠干燥,减压浓缩溶剂。残留液减压精馏,收集130
‑
134℃馏分,得7
‑
(2
‑
羟乙基)己
‑2‑
内酯(ii)6.8g,收率:63%。
[0075]1h nmr(400mhz,cdcl3)δ4.52(m,1h),3.81(m,2h),3.60(s,1h),2.67(m,2h),1.95(m,4h),1.82(m,1h),1.64(m,3h)。
[0076]
ms(esi)m/z=159.2(m
1)。
[0077]
重复上述还原反应操作,采用不同的溶剂和氧化条件,分离计算收率,实验结果如下表所示:
[0078]
序号溶剂碱收率1醋酸双氧水63%2甲酸双氧水70%3二氯甲烷间氯过氧苯甲酸77%4乙腈间氯过氧苯甲酸75%
[0079]
从表中,可以看出间氯过氧苯甲酸/二氯甲烷体系氧化效率最高,虽然双氧水/醋酸体系氧化效率相对较低,但能满足反应要求,从原料经济性角度考虑,双氧水/乙酸体系可以接受。
[0080]
实施例5:6,8
‑
二氯辛酸乙酯(i)的制备
[0081][0082]
将7
‑
(2
‑
羟乙基)己
‑2‑
内酯(ii)5g加入到30ml乙醇中,降温至10℃,滴加11.2g氯化亚砜,升温至50℃反应3小时,反应结束,将体系浓缩干,加入50ml二氯甲烷,用5%的碳酸氢钠水溶液洗涤一次,无水硫酸钠干燥,减压浓缩溶剂得到粗品,柱层析纯化得到6,8
‑
二氯辛酸乙酯(i)5.9g,收率78%。
[0083]1h nmr(400mhz,cdcl3)δ4.01(q,2h),3.70(t,2h),3.21(m,1h),2.37(t,2h),1.79(q,2h),1.60(m,2h),1.45(q,2h),1.25(m,2h),1.03(t,3h)。
[0084]
ms(esi)m/z=242.1(m
1)。
[0085]
重复上述还原反应操作,采用不同的氯化试剂,反应完成后,分离计算收率,实验结果如下表所示:
[0086]
序号氯化试剂收率1氯化亚砜78%2三光气63%3三光气/dmf82%
[0087]
从上表中可以看出,三光气/dmf体系催化的开环氯化效率最高,氯化亚砜/乙醇催化效率也能满足反应要求,从生产安全以及原料经济性角度考虑,氯化亚砜/乙醇可以接受。
[0088]
实验表明,本发明的方法开辟了一条新的6,8
‑
二氯辛酸酯合成路线,制备的产物纯度高,而且各步骤的后处理简单,能够显著降低6,8
‑
二氯辛酸酯生产成本,因而更适合工业化大规模生产。
[0089]
虽然以上仅以6,8
‑
二氯辛酸乙酯(即式i中r为乙基)为例,对本发明的技术方案进行了验证,但是根据本发明的公开,本发明设计的工艺路线构思还可以适用于其他6,8
‑
二氯辛酸酯的合成。这对于本领域的技术人员来说是显而易见的。在不违背本发明的思想下,本领域的技术人员在此基础上,所做的各种变形、修饰与应用,均应包括在本发明的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。