1.本发明属于有机化学合成技术领域,具体涉及手性色氨酸衍生物及其合成方法。
背景技术:
2.氨基酸是一类具有生物活性的手性分子,氨基酸及其衍生物在医药、食品领域有非常广泛的应用,具有很大市场需求,因此手性氨基酸衍生物的合成已成为合成领域研究的热点。
3.l
‑
色氨酸是hopkins和kole于1901年发现和分离出来的氨基酸,它是人体所必须的氨基酸之一,其在调节人体蛋白质的合成和增强机体免疫功能等方面占据了独一无二的位置。许多研究表明,l
‑
甲基色氨酸作为色氨酸衍生物以及ido抑制剂,具有抗肿瘤活性和肿瘤化疗作用。因此,寻找其他色氨酸类似物来作为抗肿瘤制剂成为了化学家们研究的热点。此外,某些l
‑
色氨酸衍生物,可以通过pictet
‑
spengler反应,与一些具有醛基或半缩醛羟基的单萜类化合物反应而生成单萜吲哚生物碱类的化合物。由于该类化合物具有很强的抗肿瘤生物活性,引起了药学、化学、生物学、医学等众多领域科学工作者的极大关注。
4.对l
‑
色氨酸衍生物的合成研究少有文献报道,因此,对l
‑
色氨酸进行结构修饰得到结构不同的色氨酸衍生物具有一定的意义。现有技术中对于l
‑
色氨酸衍生物的制备主要是生物法,通过微生物制备得到l
‑
色氨酸衍生物,例如:us5275940a、jp1985034192a等。
5.然而,目前对于l
‑
色氨酸衍生物的化学合成,鲜有报道。
技术实现要素:
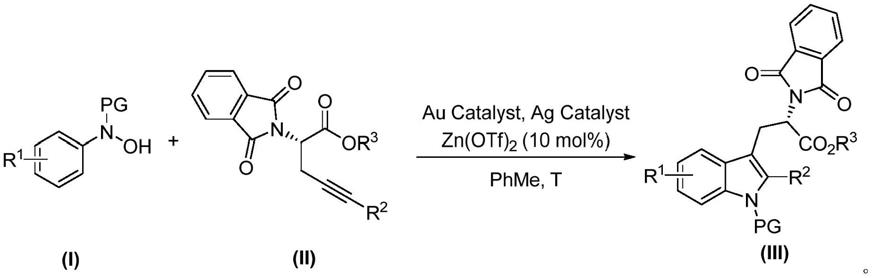
6.针对现有技术的不足,本发明提供了一种手性色氨酸衍生物的合成新方法。通过金和锌协同催化下芳基羟胺与手性炔烃的反应,高区域选择性地合成了一系列手性色氨酸的衍生物。
7.本发明的技术方案如下:
8.一种手性色氨酸衍生物,具有式(ⅲ)所示结构:
[0009][0010]
式(iii)中,r1为氢原子、烷基、烯基、芳基(包括各种取代的苯基、萘基、杂芳环取代基团等)、氧三氟甲基、氟、氯、溴、碘、三氟甲基、硫三氟甲基、酯基、吲哚基、咔唑基中的一种;
[0011]
r2为烷基、烯基、芳基(包括各种取代的苯基、萘基、杂芳环取代基团等)中的一种;
[0012]
r3为烷基、烯基、芳基(包括各种取代的苯基、萘基、杂芳环取代基团等)中的一种;
[0013]
pg为烷基、烯基、芳基(包括各种取代的苯基、萘基、杂芳环取代基团等)、苯甲酰基、乙酰基、特戊酰基、酯基、叔丁氧羰基、苄氧羰基、三氟乙酰基、9
‑
芴基甲氧基羰基中的一种。
[0014]
根据本发明,上述手性色氨酸衍生物的合成方法,包括步骤如下:
[0015]
将化合物(i)、化合物(ii)、zn(otf)2、金催化剂、银催化剂混合,在氮气氛围下加入溶剂,于氮气氛围下反应,待反应完成后,提纯,即得到目标化合物(iii);
[0016][0017]
反应过程中,可通过tlc跟踪反应进程。
[0018]
根据本发明,优选的,提纯方法如下:
[0019]
反应结束后,反应混合物减压浓缩,粗产品经过柱层析,过柱层析的洗脱剂为石油醚:乙酸乙酯=7:1,即得到目标化合物(iii)。
[0020]
根据本发明,优选的,化合物(i)和化合物(ii)的摩尔比为1:(1
‑
3),进一步优选1:1.5。
[0021]
根据本发明,优选的,所述的金催化剂为ph3paucl,cy3paucl,ipraucl或(aro)3paucl(ar=2,4
‑
di
‑
tert
‑
butylphenyl),最优选cy3paucl;
[0022]
优选的,金催化剂的用量为化合物(i)摩尔量的5
‑
20%,最优选10%。
[0023]
根据本发明,优选的,所述的银催化剂为agotf或agntf2,最优选agotf;
[0024]
优选的,银催化剂的用量为化合物(i)摩尔量的5
‑
20%,最优选10%。
[0025]
根据本发明,优选的,所述的溶剂为甲苯或1,2
‑
二氯乙烷,最优选甲苯。
[0026]
根据本发明,优选的,反应时间为10
‑
20小时,进一步优选12小时。
[0027]
根据本发明,优选的,反应温度为60
‑
80℃,最优选为80℃。
[0028]
根据本发明,所述的化合物(i)具有如下结构:以pg=bz为例
[0029][0030]
化合物(i)可按现有的技术路线制备,制备路线如下:
[0031][0032]
制备步骤:在n2氛围下,将硝基化合物(1.0当量)和5%rh/c(0.30mol%rh)溶于thf(0.324m)中,随后将反应体系冷却至0℃,逐滴加入水合肼(1.2当量)。反应混合物在0℃下搅拌1小时,然后缓慢升温至45℃,并在45℃下搅拌4小时,待反应完成后,通过硅藻土过滤反应混合物,旋蒸浓缩,重结晶,得到的粗产品羟胺直接用于下一步。
[0033]
向羟胺的乙醚(0.5m)溶液中,加入饱和nahco3水溶液,然后将溶液冷却至0℃,逐滴加入酰氯(1.1当量),在0℃下搅拌10秒后,将反应用饱和nh4cl水溶液淬灭,反应混合物用二氯甲烷萃取,有机层用盐水洗涤并经硫酸钠干燥,真空除去溶剂后,粗产品经过柱层析,过柱层析的洗脱剂为二氯甲烷:乙酸乙酯=80:1,得到化合物(i)。
[0034]
根据本发明,所述的化合物(ii)具有如下结构:
[0035][0036]
化合物(ii)可按现有的技术路线制备,制备路线如下:以r3为异丙基为例
[0037][0038]
在
‑
10℃下,向甘氨酸(4.50g,60.0mmol)的异丙醇(150ml)溶液中,逐滴加入亚硫酰氯(14.0ml,192mmol),在
‑
10℃下搅拌10分钟,随后将反应加热至40℃,反应4 h后,将反应混合物冷却至室温,蒸发浓缩除去溶剂,即得粗产物甘氨酸异丙酯。
[0039]
将粗产物甘氨酸异丙酯溶解在dcm(60ml)中,然后加入二苯甲酮亚胺(10.8ml,60mmol),反应在室温下搅拌24h后,悬浮液通过硅藻土过滤,滤液依次用水和盐水洗涤,蒸发浓缩除去溶剂后,粗产物通过重结晶(pe/etoh)纯化,即得到化合物s1。
[0040][0041]
将炔烃的溴化物(2.6ml,30.0mmol)滴加到n
‑
(二苯基亚甲基)甘氨酸异丙酯s1(1.69g,6.0mmol)和催化剂(270mg,0.6mmol)的甲苯/氯仿(7:3,30ml)混合溶剂中。然后将反应体系冷却至
‑
20℃,加入50%koh水溶液(9ml),并将反应混合物在
‑
20℃下搅拌直至原料耗尽。悬浮液用乙酸乙酯稀释,用水洗涤,用无水硫酸钠干燥,蒸发浓缩除去溶剂,粗产品经过柱层析,柱层析的洗脱剂为石油醚:乙酸乙酯=20:1,得到化合物s2。
[0042][0043]
将s2(5mmol,1.67g)溶于meoh(30ml)中,随后在室温下滴加2.0ml浓盐酸。反应混合物在室温搅拌下12h后,蒸发浓缩除去溶剂,残存物用水稀释,etoa萃取3次。水层经蒸发浓缩得到粗产物s3。
[0044]
将et3n(7.5mmol,0.9ml)滴加到粗产物s3(5.0mmol,0.82g)和邻苯二甲酸酐(5.0mmol,1.1g)的甲苯(15ml)溶液中,反应体系回流搅拌2h。反应混合物用etoac萃取,有机层用盐水洗涤并经na2so4干燥,蒸发浓缩除去溶剂后,粗产物经柱层析纯化,柱层析的洗脱剂为石油醚:乙酸乙酯=10:1,得到化合物(ii)。
[0045]
本发明的技术路线如下:
[0046][0047]
本发明的有益效果:
[0048]
本发明通过金和锌协同催化下芳基羟胺与手性炔烃的反应,高区域选择性地合成了一系列手性色氨酸的衍生物。本发明具有良好的官能团兼容性,并且以良好的产率和高区域选择性制备了多种手性色氨酸的衍生物。
附图说明
[0049]
图1为实施例1制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的1h
‑
nmr谱图;
[0050]
图2为实施例1制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的
13
c
‑
nmr谱图;
[0051]
图3为实施例1制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的hplc谱图;
[0052]
图4为实施例2制得的(s)
‑3‑
(1
‑
苯甲酰基
‑
2,5
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的1h
‑
nmr谱图;
[0053]
图5为实施例2制得的(s)
‑3‑
(1
‑
苯甲酰基
‑
2,5
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的
13
c
‑
nmr谱图;
[0054]
图6为实施例2制得的(s)
‑3‑
(1
‑
苯甲酰基
‑
2,5
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的hplc谱图;
[0055]
图7为实施例3制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑5‑
(三氟甲氧基)
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的1h
‑
nmr谱图;
[0056]
图8为实施例3制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑5‑
(三氟甲氧基)
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的
13
c
‑
nmr谱图;
[0057]
图9为实施例3制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑5‑
(三氟甲氧基)
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的
19
f
‑
nmr谱图;
[0058]
图10为实施例3制得的(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑5‑
(三氟甲氧基)
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的hplc谱图;
[0059]
图11为实施例4制得的(s)
‑3‑
(1
‑
苯甲酰基
‑5‑
氯
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的1h
‑
nmr谱图;
[0060]
图12为实施例4制得的(s)
‑3‑
(1
‑
苯甲酰基
‑5‑
氯
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的
13
c
‑
nmr谱图;
[0061]
图13为实施例4制得的(s)
‑3‑
(1
‑
苯甲酰基
‑5‑
氯
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的hplc谱图;
[0062]
图14为实施例5制得的(s)
‑3‑
(1
‑
苯甲酰基
‑
2,7
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的1h
‑
nmr谱图;
[0063]
图15为实施例5制得的(s)
‑3‑
(1
‑
苯甲酰基
‑
2,7
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的
13
c
‑
nmr谱图;
[0064]
图16为实施例5制得的(s)
‑3‑
(1
‑
苯甲酰基
‑
2,7
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的hplc谱图。
具体实施方式
[0065]
下面通过具体实施例对本发明做进一步说明,但不限于此。
[0066]
实施例1、(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的合成
[0067][0068]
将n
‑
羟基
‑
n
‑
苯基苯甲酰胺(0.2mmol,43mg)、(s)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯(0.3mmol,90mg)、zn(otf)2(0.02mmol,5.6mg)、cy3paucl(0.02mmol,10mg)、agotf(0.02mmol,5.2mg)混合,在氮气氛围下加入溶剂甲苯(2ml),在80℃下反应12h,待反应完成后,蒸发浓缩除去溶剂,粗产品经过柱层析(洗脱剂为石油醚:乙酸乙酯=7:1)得到油状产物(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯,收率为92%,ee值为92%。
[0069]1h nmr(500mhz,cdcl3):δ7.81
‑
7.75(m,2h),7.71
‑
7.66(m,2h),7.56(t,j=7.6hz,3h),7.49(d,j=7.8hz,1h),7.39(t,j=7.6hz,2h),7.08(t,j=7.4hz,1h),,7.03
‑
6.94(m,2h),5.22
–
5.06(m,2h),3.62(qd,j=14.9,7.9hz,2h),2.19(s,3h),1.28(d,j=6.3hz,3h),1.24(d,j=6.3hz,3h);
[0070]
13
c nmr(126mhz,cdcl3):δ169.6,168.3,167.4,136.4,135.6,134.9,134.2,132.8,131.7,129.7,129.4,128.7,123.5,123.2,122.5,117.8,114.4,114.2,69.9,52.1,24.1,21.8,21.7,13.2;
[0071]
hplc:the ee value was determined by hplc analysis(chiralcel odh,i
‑
proh/hexane=5/95,1.0ml/min,227nm),retention time:tmajor=9.614min,tminor=13.339min,ee=92%;[α]d
25
=
‑
267.4(c=1.97,chcl3).
[0072][0073]
实施例2、(s)
‑3‑
(1
‑
苯甲酰基
‑
2,5
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的合成
[0074][0075]
将n
‑
羟基
‑
n
‑
(对甲苯基)苯甲酰胺(0.2mmol,45mg)、(s)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯(0.3mmol,90mg)、zn(otf)2(0.02mmol,5.6mg)、cy3paucl(0.02mmol,10mg)、agotf(0.02mmol,5.2mg)混合,在氮气氛围下加入溶剂甲苯(2ml),在80℃下反应12h,待反应完成后,蒸发浓缩除去溶剂,粗产品经过柱层析(洗脱剂为石油醚:乙酸乙酯=7:1)得到油状产物(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯,收率为76%,ee值为92%。
[0076]1h nmr(500mhz,cdcl3):δ7.72
‑
7.67(m,2h),7.65
‑
7..57(m,2h),7.48(t,j=6.9hz,3h),7.31(t,j=7.6hz,2h),7.13(s,1h),6.77(d,j=8.4hz,1h),,6.67(d,j=8.4hz,1h),5.15
‑
4.98(m,2h),3.51(ddd,j=25.6,14.9,7.8hz,2h),2.20(s,3h),2.10(s,3h),1.20(d,j=6.3hz,3h),1.17(d,j=6.3hz,3h).
[0077]
13
c nmr(126mhz,cdcl3):δ169.6,168.3,167.4,135.7,134.9,134.6,134.1,132.6,132.0,131.7,129.7,129.6,128.6,124.3,123.3,117.8,114.3,113.9,69.9,52.1,24.0,21.8,21.7,21.2,13.2.
[0078]
hplc:the ee value was determined by hplc analysis(chiralcel odh,i
‑
proh/hexane=5/95,1.0ml/min,227nm),retention time:t
major
=9.527min,t
minor
=13.679min,ee=92%;[α]
d25
=
‑
261.5(c=1.60,chcl3).
[0079][0080]
实施例3、(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑5‑
(三氟甲氧基)
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的合成
[0081][0082]
将n
‑
羟基
‑
n
‑
(对三氟甲氧基)苯甲酰胺(0.2mmol,59mg)、(s)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯(0.3mmol,90mg)、zn(otf)2(0.02mmol,5.6mg)、cy3paucl(0.02mmol,10mg)、agotf(0.02mmol,5.2mg)混合,在氮气氛围下加入溶剂甲苯(2ml),在80℃下反应12h,待反应完成后,蒸发浓缩除去溶剂,粗产品经过柱层析(洗脱剂为石油醚:乙
酸乙酯=7:1)得到油状产物(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯,收率为76%,ee值为92%。
[0083]1h nmr(500mhz,cdcl3):δ7..81
‑
7.75(m,2h),7.73
‑
7.65(m,2h),7.63
‑
7.53(m,3h),7.47
‑
7.37(m,2h),7.29(s,1h),7.06(d,j=8.9hz,1h),6.83(d,j=8.9hz,1h),5.21
‑
5.12(m,1h),5.11
‑
5.04(m,1h),3.60(d,j=7.9hz,2h),2.21(s,3h),1.28(d,j=6.3hz,3h),1.24(d,j=6.2hz,3h).
[0084]
13
c nmr(126mhz,cdcl3):δ169.4,168.1,167.4,144.8,136.8,135.1,134.5,134.2,133.1,131.5,130.3,129.7,128.8,123.5,121.6,119.5,116.4,114.9,114.3,110.3,70.1,51.9,23.9,21.7,21.6,13.4.
[0085]
19
f nmr(471mhz,cdcl3):δ
‑
58.0.
[0086]
hplc:the ee value was determined by hplc analysis(chiralcel adh,i
‑
proh/hexane=5/95,1.0ml/min,227nm),retention time:t
major
=6.884min,t
minor
=9.890min,ee=92%;[α]
d25
=
‑
222.5(c=1.27,chcl3).
[0087][0088]
实施例4、(s)
‑3‑
(1
‑
苯甲酰基
‑5‑
氯
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的合成
[0089][0090]
将n
‑
(4
‑
氯苯基)
‑
n
‑
羟基苯甲酰胺(0.2mmol,50mg)、(s)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯(0.3mmol,90mg)、zn(otf)2(0.02mmol,5.6mg)、cy3paucl(0.02mmol,10mg)、agotf(0.02mmol,5.2mg)混合,在氮气氛围下加入溶剂甲苯(2ml),在80℃下反应12h,待反应完成后,蒸发浓缩除去溶剂,粗产品经过柱层析(洗脱剂为石油醚:乙酸乙酯=7:1)得到油状产物(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯,收率为67%,ee值为92%。
[0091]1h nmr(500mhz,cdcl3):δ7..74
‑
7.67(m,2h),7.67
‑
7.59(m,2h),7.55
‑
7.43(m,3h),7.33(dd,j=9.2,4.7hz,3h),6.84(dt,j=8.8,5.3hz,2h),5.09(hept,j=6.3hz,1h),4.99(dd,j=10.6,5.0hz,1h),3.49(qd,j=15.0,7.8hz,2h),2.09(s,3h),1.21(d,j=6.3hz,3h),1.17(d,j=6.3hz,3h).
[0092]
13
c nmr(126mhz,cdcl3):δ169.4,168.1,167.4,136.3,135.2,134.2,133.1,131.6,130.7,129.7,128.8,128.3,123.4,123.2,117.5,115.1,113.9,70.1,51.9,24.0,21.8,21.7,13.2.
[0093]
hplc:the ee value was determined by hplc analysis(chiralcel adh,i
‑
proh/hexane=5/95,1.0ml/min,227nm),retention time:t
major
=10.533min,t
minor
=15.490min,ee=92%;[α]
d25
=
‑
223.9(c=2.07,chcl3).
[0094][0095]
实施例5、(s)
‑3‑
(1
‑
苯甲酰基
‑
2,7
‑
二甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯的合成
[0096][0097]
将n
‑
羟基
‑
n
‑
(邻甲苯基)苯甲酰胺(0.2mmol,45mg)、(s)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯(0.3mmol,90mg)、zn(otf)2(0.02mmol,5.6mg)、cy3paucl(0.02mmol,10mg)、agotf(0.02mmol,5.2mg)混合,在氮气氛围下加入溶剂甲苯(2ml),在80℃下反应12h,待反应完成后,蒸发浓缩除去溶剂,粗产品经过柱层析(洗脱剂为石油醚:乙酸乙酯=7:1)得到油状产物(s)
‑3‑
(1
‑
苯甲酰基
‑2‑
甲基
‑
1h
‑
吲哚
‑3‑
基)
‑2‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)丙酸异丙酯,收率为62%,ee值为94%。
[0098]1h nmr(500mhz,cdcl3):δ7..69
‑
7.63(m,2h),7.61
‑
7.54(m,2h),7.51
‑
7.44(m,1h),7.40(d,j=7.2hz,2h),7.32(d,j=7.8hz,1h),7.26(t,j=7.7hz,2h),,6.97(t,j=7.6hz,1h),6.77(d,j=7.3hz,1h),5.14
‑
5.01(m,2h),3.69
‑
3.44(m,2h),1.98(s,3h),1.85(s,3h),1.20(d,j=6.3hz,3h),1.16(d,j=6.3hz,3h).
[0099]
13
c nmr(126mhz,cdcl3):δ171.2,168.4,167.4,136.1,135.7,134.3,134.1,133.7,131.7,130.2,129.5,128.8,125.9,123.3,122.6,122.2,115.8,111.6,69.8,52.2,24.2,21.8,21.7,20.2,12.1.
[0100]
hplc:the ee value was determined by hplc analysis(chiralcel adh,i
‑
proh/hexane=5/95,1.0ml/min,227nm),retention time:t
major
=12.596min,t
minor
=19.444min,ee=94%;[α]
d25
=
‑
206.7(c=0.90,chcl3).
[0101][0102]
试验例1
[0103]
以n
‑
羟基
‑
n
‑
苯基苯甲酰胺和2
‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯为原料,金催化剂和银催化剂的用量为10%mol,zn(otf)2为10%mol,甲苯为溶剂,反应温度为60℃,在氮气氛围下反应12h,探究了金催化剂和银催化剂的种类对反应的影响,如表1所示
[0104]
表1金催化剂和银催化剂的种类对反应的影响
[0105][0106]
从表1实验结果可以看出,cy3paucl和agotf为该反应的最佳金催化剂和银催化剂,其他金催化剂和银催化剂可导致产率降低。
[0107]
试验例2
[0108]
以n
‑
羟基
‑
n
‑
苯基苯甲酰胺和2
‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯为原料,cy3paucl和agotf为催化剂,cy3paucl和agotf的用量为10%mol,zn(otf)2用量为10%mol,甲苯为溶剂,在氮气氛围下反应12h,探究了温度对反应的影响,当反应温度由60℃提高到80℃时,反应收率由71%提高到96%。从实验结果得出,反应温度是80℃时反应的收率最佳。
[0109]
试验例3
[0110]
以n
‑
羟基
‑
n
‑
苯基苯甲酰胺和2
‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯为原料,cy3paucl和agotf为催化剂,cy3paucl和agotf的用量为10%mol,zn(otf)2用量为10%mol,甲苯为溶剂,在氮气氛围、80℃下反应12h,探究了金催化剂和银催化剂的用量对反应的影响,当催化剂的用量由10mol%降低到5mol%时,反应收率由96%降低到79%。从实验结果得出,金催化剂和银催化剂的用量为10mol%时反应的收率最佳。
[0111]
试验例4
[0112]
以携带不同取代基团(或保护基团)的苯基羟胺类化合物和2
‑
(1,3
‑
二氧异吲哚啉
‑2‑
基)己
‑4‑
炔酸异丙酯为原料,cy3paucl和agotf为催化剂,cy3paucl和agotf的用量为10%mol,zn(otf)2用量为10%mol,甲苯为溶剂,在氮气氛围、80℃下反应12h,探究了多种携带不同取代基团(或保护集团)的苯基羟胺类化合物在该反应中的官能团兼容性和化合物的高区域选择性。n
‑
羟基
‑
n
‑
苯基苯甲酰胺、n
‑
羟基
‑
n
‑
(对甲苯基)苯甲酰胺、n
‑
羟基
‑
n
‑
(对三氟甲氧基)苯甲酰胺、n
‑
(4
‑
氯苯基)
‑
n
‑
羟基苯甲酰胺、n
‑
羟基
‑
n
‑
(邻甲苯基)苯甲酰胺分别以92%的产率与92%ee、76%的产率与92%ee、76%的产率与92%ee、67%的产率与92%ee、62%的产率与94%ee得到相应的手性色氨酸衍生物。从实验结果看,该合成方法具备很好的官能团兼容性和高区域选择性。
[0113]
试验例5
[0114][0115]
据研究报道,该类合成方法的手性色氨酸及类似物具有抑菌、抗癌等作用,在药物
合成等领域具有很大的应用前景。在基于
18
f的色氨酸示踪剂的研究中,已证明[
18
f]l
‑
fehtp具有药理学特征,其可通过lat转运在内分泌和非内分泌肿瘤模型中积累,但不会被aadc脱羧。此外,天然存在的2,5
‑
二酮哌嗪类化合物brevianamide f不仅本身可以作为新型天然植物生长抑制剂,也是很多复杂吲哚二酮哌嗪生物碱的前体,改变氨基酸结构而得到的类似物表现出抑菌和抗癌等多样的生物活性。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。