1.本发明属于药物预测与分析领域,具体涉及一种基于端到端学习的化合物和蛋白质相互作用与亲和力预测方法。
背景技术:
2.在药物的研发过程中,事先确定靶向特定疾病的靶标蛋白是药物研发的基础,而寻找能够与特定靶标蛋白产生相互作用的化合物分子是药物研发的关键。其中,靶标是指体内与某种疾病的发生密切相关,并能与药物发生特异性结合从而产生治疗效果的生物大分子,主要包括受体、核酸、基因等。药物中的化合物分子通过调控靶标的生物活性以达到治愈或缓解相应疾病的效果。药物化合物与靶标蛋白质之间的相互作用实际上是一种特异性结合的关系,而结合关系的强弱也称之为结合亲和力。鉴定化合物和蛋白质之间的相互作用以及测定二者之间的结合亲和力是药物研发过程中的关键步骤,对药物研发具有重要意义。使用传统的实验方法进行化合物和蛋白质相互作用的鉴定以及结合亲和力的测定存在实验周期漫长且耗资昂贵等问题,也无法进行大规模地应用。开发有效的计算方法来预测化合物和蛋白质之间的相互作用和结合亲和力能够加快昂贵且耗时的实验工作,减少盲目性的生化实验,专注于更少数量而更有可能的化合物分子和靶标蛋白,从而极大地缩短药物研发的周期、降低研发成本以及研发失败所带来的风险。随着基因组学、蛋白质组学以及系统生物学等技术的不断提高,化合物和蛋白质相关的数据呈井喷式增长,为数据驱动的计算方法提供了海量的数据资源。
3.传统的计算方法既能用于分析化合物和蛋白质之间相互作用的结合模式,又能计算二者之间的结合亲和力大小,主要包括基于配体、基于结构、以及分子动力学模型等方法。但这些方法都存在一定的局限性,基于配体的方法会受到靶标已知的配体数量的限制,基于结构的方法过度依赖于靶标蛋白的三维结构数据,而分子动力学模型会受到高计算成本的限制。而目前主流的计算方法大都关注于化合物和蛋白质之间相互作用的二分类预测,即预测给定的化合物和蛋白质是否存在相互作用,而忽略了重要的相互作用强度信息,即结合亲和力的大小。虽然目前已存在一些化合物和蛋白质结合亲和力的预测方法,但是这些预测方法的生物可解释性和预测精确度上还有待进一步提高。
技术实现要素:
4.本发明提出一种基于端到端学习的化合物和蛋白质相互作用与亲和力预测方法,既可以用于化合物和蛋白质相互作用的预测,又能够预测二者之间的结合亲和力,且其生物可解释性和预测准确性较好。
5.为实现上述技术目的,本发明采用如下技术方案:
6.一种基于端到端学习的化合物和蛋白质相互作用与亲和力预测方法,包括:
7.获取化合物的分子式并将其转换为原子邻接图,将原子邻接图和随机初始化的原
子表征向量作为图注意力网络模型的输入,更新和学习得到化合物中所有原子的表征向量;
8.获取蛋白质的氨基酸序列,采用滑动窗口的方法从氨基酸序列中提取固定长度的残基,再使用卷积神经网络模型对随机初始化的残基表征向量进行更新和学习;
9.根据化合物中所有原子的表征向量和蛋白质中所有残基的表征向量,通过构建的双向注意力网络模型计算每个原子对于残基和每个残基对于原子这两个方向的注意力系数;再利用得到的注意力系数对所有原子表征向量和所有残基表征向量分别进行加权融合,得到融合的化合物特征向量和蛋白质特征向量;
10.对化合物特征向量和蛋白质特征向量进行外积运算,将运算结果展开成一维的列向量,作为第一神经网络模型的输入,用于预测化合物与蛋白质之间是否存在相互作用;对于存在相互作用的样本,将外积运算结果展开的一维列向量作为第二神经网络模型的输入,用于预测化合物与蛋白质之间的亲和力大小。
11.在更优的技术方案中,使用rdkit工具将化合物分子式转换为原子邻接图g={v,e};其中,v是原子邻接图的节点集合,所有节点与化合物的所有原子一一对应,v
i
∈v表示化合化的第i个原子;e是原子邻接图的边集合,e
ij
∈e第i个原子与第j个原子之间的化学键。
12.在更优的技术方案中,所述将原子邻接图和随机初始化的原子表征向量作为图注意力网络模型的输入,更新和学习得到化合物中所有原子的表征向量,具体为:
13.a1,按公式计算每两个原子v
i
,v
j
之间的注意力系数α
ij
,其中的分别为原子v
i
,v
j
随机初始化的表征向量,为图注意力网络模型的注意力参数;
14.a2,对于化合物每个原子v
i
,根据其所有邻居节点v
j
的表征向量以及其与所有邻居节点v
j
之间的注意力系数α
ij
,采用加权求和的方式更新该原子v
i
的表征向量的表征向量其中的n
i
为原子v
i
的所有邻居节点,化合物中与原子v
i
有化学键连接的所有原子均为原子v
i
的邻居节点。
15.在更优的技术方案中,重复步骤a1
‑
a2共k次,各原子融合k次得到的表征向量,得到化合物各原子最终的表征向量。
16.在更优的技术方案中,提取的每个残基包括氨基酸序列中相邻的3个氨基酸,则蛋白质的氨基酸序列s={s1,s2,
…
s
m
}提取得到残基序列为r={r1,r2,
…
r
l
};其中,s
i
,i=1,2,,m表示蛋白质的第i个氨基酸,r
i
,i=1,2,,l表示残基序列r中的第i个残基,且有l=m
‑
2。
17.在更优的技术方案中,所述根据化化合物中所有原子的表征向量和蛋白质中所有残基的表征向量,通过构建的双向注意力网络模型计算每个原子对于残基和每个残基对于原子这两个方向的注意力系数;再利用得到的注意力系数对所有原子表征向量和所有残基表征向量分别进行加权融合,得到融合的化合物特征向量和蛋白质特征向量,具体为:
18.b1,将化合物的原子表征向量和蛋白质的残基表征向量都转换为统一的向量维度d,并分别表示为化合物特征矩阵和蛋白质特征矩阵
19.b2,融合化合物特征矩阵c和蛋白质特征矩阵p得到相互作用矩阵a,计算公式为:
20.a=cup
t
;
21.式中,u是用于融合化合物与蛋白质特征的参数矩阵,u∈r
d
×
d
。
22.b3,分别计算残基传递到原子的化合物信息i
c
和原子传递到残基的蛋白质信息i
p
,计算公式为:
23.i
c
=apw
r2a
;
24.i
p
=apw
a2r
;
25.式中,w
r2a
和w
a2r
分别是用于计算两个不同传递方向的参数,w
r2a
∈r
d
×
d
,w
a2r
∈r
d
×
d
;
26.b4,计算原子对于残基的注意力系数α
a2r
和残基对于原子的注意力系数α
r2a
,计算公式为:
27.α
a2r
=[cw
c
||i
c
]a
a2r
;
[0028]
α
r2a
=[pw
p
||i
p
]a
r2a
;
[0029]
式中,w
c
和w
p
分别是化合物和蛋白质向量空间转换的参数,w
c
∈r
d
×
d
,w
p
∈r
d
×
d
;||表示向量拼接的操作;a
a2r
和a
r2a
分别用于计算两个不同方向的注意力系数的参数,a
a2r
∈r
d
×
d
,a
r2a
∈r
d
×
d
;
[0030]
b5,根据对应的注意力系数融合化合物原子的表征向量和蛋白质残基的表征向量,得到化合物特征向量和蛋白质特征向量计算公式为:
[0031][0032][0033]
在更优的技术方案中,重复步骤b1
‑
b5共l次,每次重复得到的和均为1个独立的双向注意力网络模型的结果,融合l个独立的双向注意力网络模型的结果,得到最终的化合物特征向量和蛋白质特征向量
[0034]
在更优的技术方案中,在步骤b5利用注意力系数进行加权融合得到化合物特征向量和蛋白质特征向量之前,先对步骤b4计算得到的注意力系数a
a2r
和a
r2a
分别进行归一化处理,再用于步骤b5加权融合计算化合物特征向量和蛋白质特征向量。
[0035]
在更优的技术方案中,所述第一神经网络采用二分类的神经网络结构,且训练样本标签只有1和0两个值,分别表示存在相互作用和不存在相互作用;所述第二神经网络采用回归分析类的神经网络结构,所有训练样本标签的值覆盖整个亲和力取值范围。
[0036]
有益效果
[0037]
本发明提出了一种基于端到端学习的化合物和蛋白质相互作用与亲和力预测方法,与现有技术相比,具有如下有益效果:该方法既可以用于化合物和蛋白质相互作用的预测,又能够预测二者之间的结合亲和力;使用双向注意力网络模型来融合化合物中所有原子的表征向量和蛋白质中所有残基的表征向量,能够增加预测方法的生物可解释性;通过大量的实验表明,该方法在相互作用预测和结合亲和力预测均能取得更好的预测准确性;该方法能够用于辅助虚拟药物筛选和药物重定位,减少盲目的实验工作,节省药物研发的
时间和成本,缓解药物研发的压力。
附图说明
[0038]
图1为本发明预测方法的流程图;
[0039]
图2为本发明与对比方法在human数据集上的auc和aupr值对比图;
[0040]
图3为发明与对比方法在c.elegans数据集上的auc和aupr值对比图;
[0041]
图4为发明与对比方法在不同结合亲和力数据集上的rmse和pcc值对比图。
具体实施方式
[0042]
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
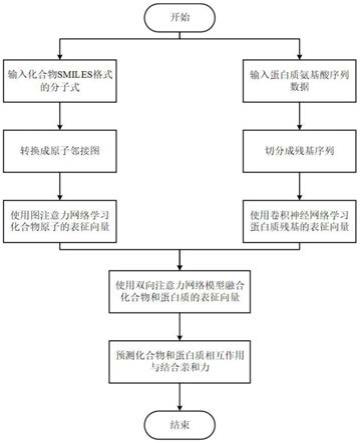
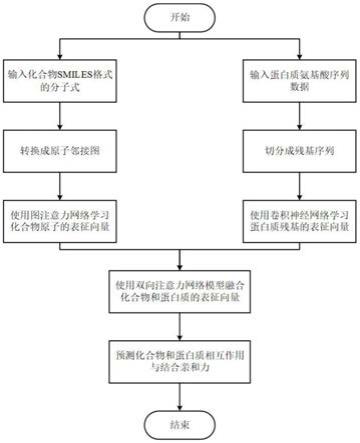
[0043]
如图1所示,本发明实施例具体公开一种基于端到端学习的化合物和蛋白质相互作用与亲和力预测方法,包括以下步骤:
[0044]
步骤1,获取化合物的分子式并将其转换为原子邻接图,将原子邻接图和随机初始化的原子表征向量作为图注意力网络模型的输入,更新和学习得到化合物中所有原子的表征向量。
[0045]
具体可以使用rdkit工具将化合物分子式转换为原子邻接图g={v,e};其中,v是原子邻接图的节点集合,所有节点与化合物的所有原子一一对应,v
i
∈v表示化合化的第i个原子;e是原子邻接图的边集合,e
ij
∈e第i个原子与第j个原子之间的化学键。
[0046]
所述图注意力网络模型,能够为原子邻接图中每个节点的邻居节点分配不同的权重,能够从化合物中每个原子的邻居节点提取特征信息,来更新和学习化合物中每个原子的表征向量。具体为:
[0047]
步骤a1,按公式计算每两个原子v
i
,v
j
之间的注意力系数α
ij
,其中的分别为原子v
i
,v
j
随机初始化的表征向量,为图注意力网络模型的注意力参数,1≤i≤n,1≤j≤n;所有注意力系数表示为:
[0048][0049]
步骤a2,对于化合物每个原子v
i
,根据其所有邻居节点v
j
的表征向量以及其与所有邻居节点v
j
之间的注意力系数α
ij
,采用加权求和的方式更新该原子v
i
的表征向量的表征向量的表征向量其中的n
i
为原子v
i
的所有邻居节点,化合物中与原子v
i
有化学键连接的所有原子均为原子v
i
的邻居节点。
[0050]
在更优的实施例中,所述图注意力网络模型采用多头注意力机制,通过融合多个独立的图注意力网络模型的计算结果,从而能够获得更准确的原子表征向量。具体体现为:重复步骤a1
‑
a2共k次,各原子融合k次得到的表征向量,得到化合物各原子最终的表征向量。
[0051]
步骤2,获取蛋白质的氨基酸序列,采用滑动窗口的方法从氨基酸序列中提取固定长度的残基,再使用卷积神经网络模型对随机初始化的残基表征向量进行更新和学习。
[0052]
采用固定长度为3、步长为1的滑动窗口,从蛋白质的氨基酸序列s={s1,s2,
…
s
m
}依次提取残基,则提取到的每个残基包括氨基酸序列中相邻的3个氨基酸,所有残基表示为残基序列r={r1,r2,
…
r
l
};其中,s
i
(i=1,2,,m)表示蛋白质的第i个氨基酸,r
i
(i=1,2,,l)表示残基序列r中的第i个残基,且有l=m
‑
2。例如,氨基酸序列为mrpsg...figa的蛋白质,可以被分割成长度为3的不同子序列:`mrp',`rps',`psg',...,`fig',`iga',其中每一个子序列都为一个残基。将每个残基都随机初始化表示为一个向量,即为残基表征向量,蛋白质的所有残基表征向量合在一起可以组成一个二维矩阵,然后输入到卷积神经网络中进行卷积和池化的操作,相当于对输入的残基表征向量进行计算和转换,深度学习里面的专业术语。
[0053]
所述的卷积神经网络的超参数主要包括卷积层数、滤波器的大小和数量,学到的残基表征向量将会输入到步骤3中的双向注意力网络模型进行预测。
[0054]
步骤3,根据化合物中所有原子的表征向量和蛋白质中所有残基的表征向量,通过构建的双向注意力网络模型计算每个原子对于残基和每个残基对于原子这两个方向的注意力系数;再利用得到的注意力系数对所有原子表征向量和所有残基表征向量分别进行加权融合,得到融合的化合物特征向量和蛋白质特征向量。具体包括:
[0055]
b1,将化合物的原子表征向量和蛋白质的残基表征向量
[0056]
都转换为统一的向量维度d,并分别表示为化合物特征矩阵和蛋白质特征矩阵
[0057]
b2,融合化合物特征矩阵c和蛋白质特征矩阵p得到相互作用矩阵a,计算公式为:
[0058]
a=cup
t
;
[0059]
式中,u是用于融合化合物与蛋白质特征的参数矩阵,u∈r
d
×
d
。
[0060]
b3,分别计算残基传递到原子的化合物信息i
c
和原子传递到残基的蛋白质信息i
p
,计算公式为:
[0061]
i
c
=apw
r2a
;
[0062]
i
p
=apw
a2r
;
[0063]
式中,w
r2a
和w
a2r
分别是用于计算两个不同传递方向的参数,w
r2a
∈r
d
×
d
,w
a2r
∈r
d
×
d
;
[0064]
b4,计算原子对于残基的注意力系数α
a2r
和残基对于原子的注意力系数α
r2a
,计算公式为:
[0065]
α
a2r
=[cw
c
||i
c
]a
a2r
;
[0066]
α
r2a
=[pw
p
||i
p
]a
r2a
;
[0067]
式中,w
c
和w
p
分别是化合物和蛋白质向量空间转换的参数,w
c
∈r
d
×
d
,w
p
∈r
d
×
d
;||表示向量拼接的操作;a
a2r
和a
r2a
分别用于计算两个不同方向的注意力系数的参数,a
a2r
∈r
d
×
d
,a
r2a
∈r
d
×
d
;
[0068]
b5,根据对应的注意力系数融合化合物原子的表征向量和蛋白质残基的表征向量,得到化合物特征向量和蛋白质特征向量计算公式为:
[0069]
regression、random forest、deepaffinity和monn)进行了对比。为了评价本发明在化合物和蛋白质结合亲和力预测上的精确度,采用均方根误差(rmse)和皮尔森相关系数(pcc)两个指标进行比较。均方根误差是衡量预测值与真实值之间误差的指标,值越小代表预测误差越小,预测模型的性能就越好。皮尔森相关系数是一种线性相关系数,用来反应预测值与真实值之间的线性相关程度,其值介于
‑
1到1之间,值大于0表示正相关,值小于0则表示负相关,值越接近于1代表预测值与真实值之间的正相关性越强,预测模型的性能就越好。rmse值和pcc值的实验结果具体如图4所示。可以看出,本发明的预测方法在ic50和ki两个较大的数据集上取得了最低的rmse值和最高的pcc值,在ec50数据集上取得了最低的rmse值以及与monn方法相同的pcc值,而在最小的kd数据集上因其样本量较少,学到的化合物和蛋白质的表征向量不够准确,取得了第二好的rmse值和pcc值(稍差于monn方法)。由此可见,本发明提出的预测方法在化合物和蛋白质结合亲和力预测上具有很好的预测效果。
[0081]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0082]
虽然结合附图描述了本发明的实施方式,但是本领域技术人员可以在不脱离本发明的精神和范围的情况下做出各种修改和变型,这样的修改和变型均落入由所附权利要求所限定的范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。