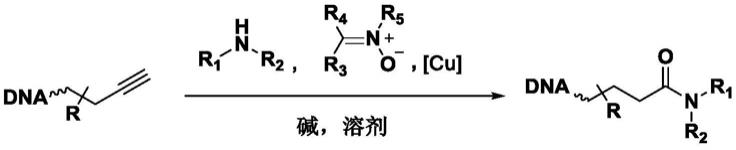
1.本发明属于生物化学领域,具体涉及一种将末端炔烃转化为酰胺的方法及其在基因编码化合物库构建中的应用。
背景技术:
2.基因编码化学库(del)最初是由brenner和lerner在1992年提出的
1.。在del中,每个化合物都与一个唯一的基因标签结合,该标签上的寡聚核酸基因序列代表其化学结构。所有库化合物混合在一起并同时针对目标蛋白进行生物筛选。与标靶蛋白结合后的结合物可以用聚合酶链反应(pcr)扩增来解码其对应的化学结构。del可以包含数十亿甚至数万亿的化合物库,而且生物筛选可以在短短几个小时内完成
[2
‑
4]
。
[0003]
如今,基因编码化合物库技术(delt)已成为生物医学研究中一个强大的活性化合物发现技术
[5]
,越来越多的制药公司采用delt发现具有生物学或药学意义的有机小分子化合物
[6]
。这些能够与标靶蛋白结合的小分子化合物的发现推动了新药研发的进程。最近几年来,在这一领域里出现的活细胞del筛选方法,是一个引人注目的成就
[7]
,它代表着通过delt平台可以筛选到具有细胞生物活性的化合物。del活细胞筛选将不再需要纯化过的靶标蛋白,也无需对蛋白进行修饰。这样不仅简化了生物筛选的过程,而且更好地保持了蛋白的原生态结构
[8]
,从而药物学家能够在此平台上找到更好的先导化合物。
[0004]
在大多数情况下,del的合成都涉及到酰胺偶联反应
[10]
。这常常是在链接寡聚核酸的游离氨基与羧酸小分子底物之间进行的。dario neri及其共同研究者发现了通过使用1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳二亚胺、1
‑
羟基
‑7‑
氮杂苯并三唑和n,n'
‑
二异丙基乙胺(edci/hoat/dipea)的偶联试剂组合,可以得到高的收率和比较好的普适性的酰胺偶联反应
[9]
。这些反应条件适用于各种伯胺和仲胺以及各种类型的含有游离氨基的寡核苷酸底物,并适用于del的合成。
[0005]
酰胺键可以通过廉价多样的羧酸与各种有机胺类化合物的酰胺偶联反应建立。但是,绝大多数酰胺反应是通过寡聚核酸
‑
游离胺类底物与小分子羧酸反应物实现的。然而,寡聚核酸
‑
游离羧酸类底物与小分子胺类的酰胺偶联反应目前仍然非常低效
[11]
。michael j.waring最近报道了一种使用表面活性剂胶束进行酰胺偶联的方法
[12]
。他们的方法必须使用很长的脂肪烷烃作为寡聚核酸与小分子文库之间的链接头。那种很长的脂肪烷烃链接头可以帮助形成使用表面活性剂胶束,因此而使得寡聚核酸上反应能够顺利进行。但是,那种很长的脂肪烷烃链接头可能对随后的生物筛选实验产生干扰。
[0006]
因此,本发明报道一种在寡聚核酸上成功应用的通过末端炔烃与游离氨类小分子反应而合成酰胺偶联的新方法。它适用于建立从羧酸到酰胺的偶联产物。而且,与传统的酰胺偶联反应不同的是,同时有游离的氨基与羧酸的小分子底物可以在不带保护基的情形下直接参与化学反应形成酰胺。在寡聚核酸上进行的每一步合成化学反应都会对核苷酸序列造成或多或少的破坏,使用尽可能少的化学反应、尽可能温和的反应条件有利于提高基因
编码库的合成效率与品质。因此,本发明具有比传统方法更高的合成效率与更好的寡聚核酸品质。
技术实现要素:
[0007]
本发明所要解决的技术问题,在于提供一种将末端炔烃转化为酰胺的方法并应用于基因编码化合物库的构建。具体地,以寡聚核酸末端炔烃化合物为底物,在铜催化剂和硝酮类试剂存在下,与含有游离氨基类小分子化合物反应将末端炔烃转化为酰胺。并以此反应为基础,建立基因编码库。
[0008]
为解决上述技术问题,本发明提供以下技术方案:
[0009]
本发明提供在基因编码化合物库构建中将末端炔烃转化为酰胺的方法,具体是以寡聚核酸末端炔烃化合物为底物,在铜催化剂和硝酮类试剂存在下,与含有游离氨基类小分子化合物反应形成酰胺化合物。具体反应方程式如下:
[0010][0011]
优选地,以寡聚核酸末端炔烃化合物为底物,在铜催化剂和硝酮类试剂存在下,与含有游离氨基类小分子化合物在0~90℃反应1~24小时反应形成酰胺化合物。
[0012]
其中,寡聚核酸末端炔烃化合物的结构式为是由寡聚核酸连接具有末端炔基的化学基团构成;所制备的酰胺化合物的结构式为是由寡聚核酸连接具有酰胺键的化学基团构成,r为氢、卤素、氨基、硝基、氰基、羟基、巯基、芳基甲酮、烷基甲酮、c1‑
c
12
烷基、c2‑
c6烯烃基、c2‑
c6炔烃基、c3‑
c8环烷基、c1‑
c6烷基氧、c4‑
c
12
芳基、c4‑
c
12
杂环芳基中的任意一种至多种或它们的任意组合;
[0013]
其中,寡聚核酸是由经人工修饰的和/或未修饰的寡核苷酸单体聚合得到的单链或双链的寡核苷酸链;
[0014]
其中,所述含有游离氨基类小分子化合物的结构式为r1‑
nh
‑
r2,可以是一级或二级胺类化合物,包括芳香类、脂肪类、碳环类化合物以及含有杂原子的环类化合物、氨基酸类化合物以及带有其他保护基团的游离氨基类化合物,r1、r2为羧酸、氢、氨基、硝基、氰基、羟基、巯基、芳基甲酮、烷基甲酮、c1‑
c
12
烷基、c2‑
c6烯烃基、c2‑
c6炔烃基、c3‑
c8环烷基、c1‑
c6烷基氧、c4‑
c
12
芳基、c4‑
c
12
杂环芳基基团中的任意一种至多种或它们的任意组合;
[0015]
其中,所述铜催化剂为醋酸铜、硫酸铜、氯化铜、硝酸铜、碳酸铜、碘化亚铜、铜
‑
β
‑
环糊精复合物、双(2,4
‑
戊二酮酸)铜、乙酰丙酮铜、四氟硼酸四(乙腈)铜、二氯(1,10
‑
菲咯啉)铜、双(8
‑
羟基喹啉)铜、三氟甲磺酸铜、双(三氟
‑
2,4
‑
戊二酮)铜、高氯酸铜、六氟磷酸四(乙腈)铜、醋酸亚铜、溴化铜、氟化铜、溴化亚铜、氯化亚铜、氯化亚铜
‑
双(氯化锂)络合物、溴化亚铜二甲硫醚;优选地,铜催化剂为碘化亚铜;
[0016]
其中,硝酮结构为结构式中r3、r4、r5为氢、卤素、氨基、硝基、氰基、羟基、巯基、芳基甲酮、烷基甲酮、c1‑
c
12
烷基、c2‑
c6烯烃基、c2‑
c6炔烃基、c3‑
c8环烷基、c1‑
c6烷基氧、c4‑
c
12
芳香基团、c4‑
c
12
杂环芳香基团中的任意一种至多种或它们的任意组合,其中r5不可为氢;优选地,硝酮结构为
[0017]
其中,所述碱为碳酸铯、碳酸钾、碳酸钠、碳酸氢钠、碳酸氢钾、碳酸锂、氢氧化锂、氢氧化钾、氢氧化钠、氢氧化铯、硼酸钠、硼酸氢钠、硼酸钾、磷酸二氢钾、磷酸二氢钠、醋酸钠、氟化钠、氟化钾、氟化铯、三甲胺、三乙胺、异丁胺、异丙胺、4
‑
二甲氨基吡啶、n,n
‑
二异丙基乙胺、1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯、n,n,n',n'
‑
四甲基乙二胺、四甲基胍、吡啶、n
‑
甲基二环己胺或二环己胺中的一种或几种的混合物;优选地,所述碱为硼酸钠;
[0018]
其中,所述反应溶剂是水、甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、叔丁醇、戊醇、环己醇、2
‑
氟乙醇、2,2
‑
二氟乙醇、2,2,2
‑
三氟乙醇、六氟异丙醇、苯甲醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、丙三醇、乙醚、环氧丙烷、异丙醚、四氢呋喃、2
‑
甲基四氢呋喃、四氢吡喃、1,4
‑
二氧六环、苯甲醚、二甲硫醚、二乙基硫醚、乙二醇二甲醚,乙二醇二乙醚、二乙二醇二甲醚、二乙二醇二乙醚、n
‑
甲基吡咯烷酮、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲基亚砜、乙腈、丙酮、环己酮、二氯甲烷、氯仿、氯苯、1,2
‑
二氯乙烷、乙酸乙酯、正己烷、环己烷、吡啶、2
‑
甲基吡啶、3
‑
甲基吡啶、4
‑
甲基吡啶、4
‑
甲氧基吡啶、甲苯、二甲苯、无机盐缓冲液、有机碱缓冲液中的任意一种或几种的混合溶剂;优选地,所述反应溶剂是无机盐缓冲液与二甲基亚砜的混合溶剂;更优选地,所述反应溶剂是ph=9.5的硼酸钠缓冲溶液与二甲基亚砜的混合溶剂。
[0019]
优选地,将5纳摩尔寡聚核酸末端炔烃化合物溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量含有游离氨基类小分子化合物(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下震荡反应3小时。
[0020]
在一个实施方案中,所述寡聚核酸末端炔烃化合物的摩尔浓度是0.1~2毫摩尔/升;优选地,寡聚核酸末端炔烃化合物的摩尔浓度为0.5~1.5毫摩尔/升;更优选地,寡聚核酸末端炔烃化合物的摩尔浓度为1.0毫摩尔/升。
[0021]
在一个实施方案中,所述铜催化剂的当量为1~100当量;优选地,铜催化剂的当量为5~35当量;更优选地,铜催化剂的当量为20当量。
[0022]
在一个实施方案中,所述硝酮类试剂的当量为5~500当量;优选地,硝酮类试剂的当量为50~150当量;更优选地,硝酮类试剂的当量为100当量。
[0023]
在一个实施方案中,所述含有游离氨基类小分子化合物的当量为5~500当量;优选地,含有游离氨基类小分子化合物的当量为100~300当量;更优选地,含有游离氨基类小分子化合物的当量为200当量。
[0024]
在一个实施方案中,所述反应的温度为0~90℃;优选地,所述反应的温度为30~70℃;更优选地,所述反应的温度为50℃。
[0025]
在一个实施方案中,所述反应的时间为0~24小时;优选地,所述反应的时间为2~5小时;更优选地,所述反应的时间为3小时。
[0026]
本发明提供了一种在基因编码化合物库构建中将末端炔烃转化为酰胺的方法,为基因编码化合物库酰胺的形成提供了新的方法。本发明提供的方法普适性好、操作方便、收率高,适用于多孔板进行的基因编码化合物库的合成。其先进性在于可以使用与常规羧酸不同的试剂来进行酰胺偶联反应,增加了反应底物的来源的多样性,特别是提供了一种直接将同时带有游离羧酸与未保护的一级或者是二级胺类小分子化合物进行酰胺偶联;扩大了反应底物的适用范围;减少了在基因编码化合物库合成中的反应步骤数目;提高了基因编码化合物库的合成效率和品质。本发明提供的方法适用于常规的脂肪及芳香类炔烃,反应适用的底物类型在图11中列出。本发明中的胺类底物适用于游离的一级胺(如图21中列出)、或者是二级胺类如图21中列出)、以及同时还有游离胺及羧酸的底物(如图21中列出)。
附图说明
[0027]
图1为本发明寡聚核酸末端炔烃化合物1
‑
2的液相色谱质谱检测结果。
[0028]
图2为本发明寡聚核酸末端炔烃化合物5
‑
5的液相色谱质谱检测结果。
[0029]
图3为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0030]
图4为本发明以寡聚核酸末端炔烃化合物1
‑
3为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0031]
图5为本发明以寡聚核酸末端炔烃化合物1
‑
6为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0032]
图6为本发明以寡聚核酸末端炔烃化合物2
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0033]
图7为本发明以寡聚核酸末端炔烃化合物3
‑
3为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0034]
图8为本发明以寡聚核酸末端炔烃化合物5
‑
4为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0035]
图9为本发明以寡聚核酸末端炔烃化合物5
‑
5为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0036]
图10为本发明以寡聚核酸末端炔烃化合物5
‑
6为底物,在铜催化剂和硝酮类试剂存在下,与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺形成酰胺的液相色谱质谱检测结果。
[0037]
图11为本发明寡聚核酸末端炔烃化合物部分代表结构式。
[0038]
图12为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与(s)
‑3‑
氨基四氢呋喃盐酸盐形成酰胺的液相色谱质谱检测结果。
[0039]
图13为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与十氢异喹啉形成酰胺的液相色谱质谱检测结果。
[0040]
图14为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂
存在下,与2
‑
氨基
‑4‑
吡啶甲胺形成酰胺的液相色谱质谱检测结果。
[0041]
图15为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与反
‑4‑
(氨基甲基)环已基羧酸形成酰胺的液相色谱质谱检测结果。
[0042]
图16为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与3
‑
氨甲基苯甲酸盐酸盐形成酰胺的液相色谱质谱检测结果。
[0043]
图17为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与1
‑
(2
‑
氨基苯基)乙胺盐形成酰胺的液相色谱质谱检测结果。
[0044]
图18为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与二环己胺形成酰胺的液相色谱质谱检测结果。
[0045]
图19为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与3
‑
氨基环己羧酸形成酰胺的液相色谱质谱检测结果。
[0046]
图20为本发明以寡聚核酸末端炔烃化合物1
‑
2为底物,在铜催化剂和硝酮类试剂存在下,与4
‑
羟基苄胺形成酰胺的液相色谱质谱检测结果。
[0047]
图21为本发明与寡聚核酸末端炔烃化合物反应的含有游离氨基类小分子化合物的部分代表结构式。
具体实施方式
[0048]
下面将对本发明的技术方案进行清楚、完整的描述。显然,所描述的实施例只是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。本发明实施例中的所有寡聚核酸原料均为寡聚核酸双链或者单链的底物。寡聚核酸末端炔烃化合物底物的合成方法十分类似。通用的合成方法可以参照实施例1或实施例2完成。
[0049]
实施例1,聚核酸末端炔烃化合物1
‑
2的合成
[0050]
将10纳摩尔寡聚核酸溶于10微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),将80当量5
‑
己炔酸(80毫摩尔/升)在室温下加入到寡聚核酸溶液中,并加入50当量2
‑
(7
‑
偶氮苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(50毫摩尔/升)及200当量n,n
‑
二异丙基乙胺(200毫摩尔/升),在室温下反应1小时。反应结束后,向反应液中加入4微升5摩尔/升的氯化钠水溶液,100微升无水乙醇,震荡混匀。放置到
‑
80℃冰箱冷冻10~30分钟后,高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到寡聚核酸末端炔烃化合物1
‑
2。通过液相色谱质谱检测产物分子量及确认收率,结果如图1。产物分子量为16284.6,产物的收率为95.0%。
[0051]
具体反应方程式如下:
[0052][0053]
实施例2,寡聚核酸末端炔烃化合物5
‑
5的合成
[0054]
将10纳摩尔寡聚核酸溶于10微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),将30当量4
‑
炔基
‑2‑
氟苯胺(30毫摩尔/升)在室温下加入到寡聚核酸溶液中,并加入20当量2
‑
(7
‑
偶氮苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(20毫摩尔/升)及20当量n,
n
‑
二异丙基乙胺(20毫摩尔/升),在室温下反应1小时。反应结束后,向反应液中加入4微升5摩尔/升的氯化钠水溶液,100微升无水乙醇,震荡混匀。放置到
‑
80℃冰箱冷冻10~30分钟后,高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到底物寡聚核酸末端炔烃化合物5
‑
5。通过液相色谱质谱检测产物分子量及确认收率,结果如图2。产物分子量为16347.9,产物的收率为93.0%。
[0055]
具体反应方程式如下:
[0056][0057]
实施例3,硝酮的合成
[0058]
0.08毫摩尔芳香硝基化合物溶于700微升乙腈,加入0.16毫摩尔芳香醛基化合物、0.096毫摩尔氯化铵、0.08毫摩尔锌粉以及100微升水,在氮气保护于40℃下搅拌反应12小时。反应结束后离心处理,将上清液直接用于下一步的反应。
[0059][0060]
以下实施例中使用的硝酮均为实施例3中制备的硝酮。
[0061]
实施例4,寡聚核酸末端炔烃化合物1
‑
2(制备方法同实施例1)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0062]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图3。产物分子量为16436.9,产物的收率为76.2%。
[0063]
实施例5,寡聚核酸末端炔烃化合物1
‑
3(制备方法同实施例1)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0064]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
3溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过
液相色谱质谱检测产物分子量及确认收率,结果如图4。产物分子量为16422.6,产物的收率为76.6%。
[0065]
实施例6,寡聚核酸末端炔烃化合物1
‑
6(制备方法同实施例1)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0066]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
6溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图5。产物分子量为16487.0,产物的收率为58.2%。
[0067]
实施例7,寡聚核酸末端炔烃化合物2
‑
2(制备方法同实施例1)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0068]
将5纳摩尔寡聚核酸末端炔烃化合物2
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图6。产物分子量为16485.3,产物的收率为60.9%。
[0069]
实施例8,寡聚核酸末端炔烃化合物3
‑
3(制备方法同实施例2)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0070]
将5纳摩尔寡聚核酸末端炔烃化合物3
‑
3溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图7。产物分子量为16434.9,产物的收率为62.1%。
[0071]
实施例9,寡聚核酸末端炔烃化合物5
‑
4(制备方法同实施例2)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0072]
将5纳摩尔寡聚核酸末端炔烃化合物5
‑
4溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,
3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图8。产物分子量为16500.5,产物的收率为58.7%。
[0073]
实施例10,寡聚核酸末端炔烃化合物5
‑
5(制备方法同实施例2)相同与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0074]
将5纳摩尔寡聚核酸末端炔烃化合物5
‑
5溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图9。产物分子量为16504.2,产物的收率为58.9%。
[0075]
实施例11,寡聚核酸末端炔烃化合物5
‑
6(制备方法同实施例2)与1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺反应转化为酰胺
[0076]
将5纳摩尔寡聚核酸末端炔烃化合物5
‑
6溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1,3,5
‑
三甲基
‑
吡唑
‑4‑
甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图10。产物分子量为16488.1,产物的收率为61.7%。
[0077]
我们验证了不同寡聚核酸末端炔烃转化为酰胺的反应,部分炔烃化合物代表结构式见图11。通过液相色谱质谱谱图,我们确认寡聚核酸末端炔烃转化为酰胺,具有反应条件温和,产率高,反应的底物普适性好的优点。
[0078]
实施例12,寡聚核酸末端炔烃化合物1
‑
2与(s)
‑3‑
氨基四氢呋喃盐酸盐反应转化为酰胺
[0079]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量(s)
‑3‑
氨基四氢呋喃盐酸盐(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),
取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图12。产物分子量为16384.1,产物的收率为72.5%。
[0080]
实施例13,寡聚核酸末端炔烃化合物1
‑
2与十氢异喹啉反应转化为酰胺
[0081]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量十氢异喹啉(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图13。产物分子量为16438.4,产物的收率为52.9%。
[0082]
实施例14,寡聚核酸末端炔烃化合物1
‑
2与2
‑
氨基
‑4‑
吡啶甲胺反应转化为酰胺
[0083]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量2
‑
氨基
‑4‑
吡啶甲胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图14。产物分子量为16420.5,产物的收率为79.5%。
[0084]
实施例15,寡聚核酸末端炔烃化合物1
‑
2与反
‑4‑
(氨基甲基)环己基羧酸反应转化为酰胺
[0085]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量反
‑4‑
(氨基甲基)环己基羧酸(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图15。产物分子量为16454.2,产物的收率为72.3%。
[0086]
实施例16,寡聚核酸末端炔烃化合物1
‑
2与3
‑
氨甲基苯甲酸盐酸盐反应转化为酰胺
[0087]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量3
‑
氨甲基苯甲酸盐酸盐(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小
时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图16。产物分子量为16449.2,产物的收率为73.8%。
[0088]
实施例17,寡聚核酸末端炔烃化合物1
‑
2与1
‑
(2
‑
氨基苯基)乙胺反应转化为酰胺
[0089]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量1
‑
(2
‑
氨基苯基)乙胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图17。产物分子量为16433.1,产物的收率为70.5%。
[0090]
实施例18,寡聚核酸末端炔烃化合物1
‑
2与二环己胺反应转化为酰胺
[0091]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量二环己胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图18。产物分子量为16476.8,产物的收率为51.8%。
[0092]
实施例19,寡聚核酸末端炔烃化合物1
‑
2与3
‑
氨基环己羧酸反应转化为酰胺
[0093]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量3
‑
氨基环己羧酸(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图19。产物分子量为16440.7,产物的收率为65.4%。
[0094]
实施例20,寡聚核酸末端炔烃化合物1
‑
2与4
‑
羟基苄胺反应转化为酰胺
[0095]
将5纳摩尔寡聚核酸末端炔烃化合物1
‑
2溶于5微升硼酸钠缓冲溶液(ph=9.5,250毫摩尔/升,1当量),并加入20微升二甲基亚砜、20当量碘化亚铜(20毫摩尔/升)、200当量4
‑
羟基苄胺(200毫摩尔/升)及100当量硝酮(200毫摩尔/升),在50℃下反应3小时。反应结束后,向反应液中加入50当量二乙基二硫代氨基甲酸钠(100毫摩尔/升)水溶液,震荡混匀,在
50℃下反应10分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),取上层溶液加入3.5微升5摩尔/升的氯化钠水溶液,87.5微升无水乙醇,震荡混匀,放置到
‑
80℃冰箱冷冻10~30分钟。高速冷冻离心分离(4℃,12000转数/分钟,5分钟),得到产物,通过液相色谱质谱检测产物分子量及确认收率,结果如图20。产物分子量为16420.0,产物的收率为77.7%。
[0096]
本发明验证了寡聚核酸末端炔烃与不同游离氨基类小分子化合物转化为酰胺的反应,部分代表化合物结构式见图21。本发明通过液相色谱质谱谱图,确认寡聚核酸末端炔烃转化为酰胺的反应具有反应条件温和,产率高,反应的底物普适性好的优点。
[0097]
综上所述,上述各实施例及附图仅为说明本发明的广适性而已,并不用以限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,皆应包含在本发明的保护范围内。
[0098]
参考文献
[0099]
[1]brenner s.;lerner r.a.encoded combinatorial chemistry[j].proc.natl.acad.sci.usa.,1992,89,5381
–
5383.
[0100]
[2]ottl j.;leder l.;schaefer j.v.;et al.encoded library technologies as integrated lead finding platforms for drug discovery[j].molecules.,2019,24,1629.
[0101]
[3]徐力昆;张东娜;窦媛媛;等.基因编码化合物库在药物筛选和发现中的研究与应用[j].国际药学研究杂志,2018,45(10),736
‑
742.
[0102]
[4]kodadek t.;paciaroni n.g.;balzarini m.;et al.beyond protein binding:recent advances in screening dna
‑
encoded libraries[j].chem.commun.,2019,55,13330
–
13341.
[0103]
[5]nielsen j.;brenner s.;janda k.d.synthetic methods for the implementation of encoded combinatorial chemistry[j].j.am.chem.soc.,1993,115,9812
–
9813.
[0104]
[6]huang y.r.;meng l.;nie q.g.;et al.selection of dna
‑
encoded chemical libraries against endogenous membrane proteins on live cells[j].nat.chem.,2021,13(1),77
‑
88.
[0105]
[7]bo c.;dongwook k.;saeed a.;et al.selection of dna
‑
encoded libraries to protein targets within and on living cells[j].j.am.chem.soc.,2019,141(43),17057
‑
17061.
[0106]
[8]wu z.n.;graybill l.t.;zeng x.;et al.cell
‑
based selection expands the utility of dna
‑
encoded small
‑
molecule library technology to cell surface drug targets:identification of novel antagonists of the nk3 tachykinin receptor[j].acs comb.sci.,2015,17(12),722
‑
731.
[0107]
[9]li y.z.;gabriele e.;samain f.;et al.optimized reaction conditions for amide bond formation in dna
‑
encoded combinatorial libraries[j].acs comb.sci.,2016,18,438
‑
443.
[0108]
[10]franzini r.m.;randolph c.chemical space of dna
‑
encoded libraries[j].j.med.chem.,2016,59(14),6629
‑
6644.
[0109]
[11]kung p.p.;bingham p.;burke b.j.;et al.characterization of specific n
‑
α
‑
acetyltransferase 50(naa50)inhibitors identified using a dna encoded library[j].acs med.chem.lett.,2020,11,1175
–
1184.
[0110]
[12]hunter j.h.;anderson m.j.;castan i.f.s.f.;et al.highly efficient on
‑
dna amide couplings promoted by micelle forming surfactants for the synthesis of dna encoded libraries[j].chem.sci.,2021,12,9475
‑
9484.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。