1.本发明属于医药领域,进一步而言,属于原料药合成工业,具体涉及到一种布美他尼的中间体的合成方法。
背景技术:
2.布美他尼为一种强效的利尿剂,临床上主要用于治疗心力衰竭、肝病、肾脏病水肿包括各种顽固性水肿及急性肺水肿,结构式如下:
[0003][0004]
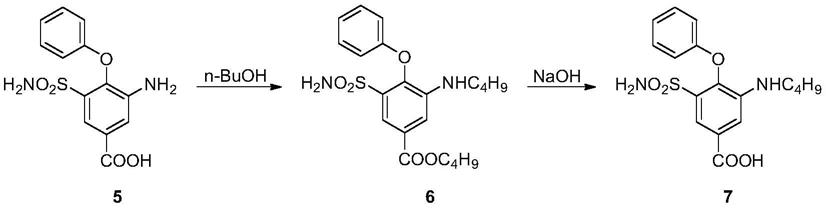
合成布美他尼的路线中,化合物5(3
‑
氨基
‑4‑
苯氧基
‑5‑
氨磺酰基苯甲酸)是一个关键中间体,其与正丁醇反应进行烷基化后,经过酯水解得到布美他尼(化合物7)。
[0005][0006]
对于化合物5的合成方法,journal of labelled compound and radiopharmaceuticals.21(2):173
‑
80,1984报道如下的合成路线。
[0007][0008]
在该合成路线中,需要使用氯磺酸,为剧毒品,购买需要特殊的许可。且反应过程中,需要使用浓硫酸和浓硝酸进行硝化反应,对安全生产提出了很大的挑战,带来较高的风险。因此,本发明提供了一种新的合成布美他尼的中间体的方法。
技术实现要素:
[0009]
发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种布美他尼的中间体的合成方法。
[0010]
为了解决上述技术问题,本发明公开了一种布美他尼的中间体的合成方法,其包括如下步骤:
[0011]
(1)采用含苄基类保护基的化合物对式1所示3
‑
氨基
‑4‑
氯苯甲酸中的氨基进行保护,得到n原子被一个苄基类保护基保护的式2所示化合物;
[0012]
(2)式2所示化合物与氯化亚砜反应后,经氨水处理,得到式3所示化合物;
[0013]
(3)式3所示化合物与苯酚反应后,得到式4所示化合物;
[0014]
(5)式4所示化合物与氧化剂反应后,得到式5所示的布美他尼的中间体,3
‑
氨基
‑4‑
苯氧基
‑5‑
氨磺酰基苯甲酸;
[0015][0016]
其中,r选自h或甲氧基。
[0017]
步骤(1)中,所述苄基类保护基为苄基或对甲氧苄基。
[0018]
步骤(1)中,所述含苄基类保护基的化合物为氯化苄、溴化苄、对甲氧基氯苄和对甲氧基溴苄中的任意一种。
[0019]
步骤(1)中,所述含苄基类保护基的化合物与式1所示3
‑
氨基
‑4‑
氯苯甲酸的摩尔比为(1
‑
1.5):1;优选地,所述含苄基类保护基的化合物与式1所示3
‑
氨基
‑4‑
氯苯甲酸的摩尔比为(1.0
‑
1.1):1;进一步优选地,所述含苄基类保护基的化合物与式1所示3
‑
氨基
‑4‑
氯苯甲酸的摩尔比为1.1:1。
[0020]
步骤(1)中,该反应的溶剂为有机溶剂,包括但不限于二氯甲烷。
[0021]
步骤(1)中,式1所示3
‑
氨基
‑4‑
氯苯甲酸与溶剂的用量比为0.4
‑
1.6mmol/ml;优选地,式1所示3
‑
氨基
‑4‑
氯苯甲酸与溶剂的用量比为0.7
‑
1.3mmol/ml;优选地,式1所示3
‑
氨基
‑4‑
氯苯甲酸与溶剂的用量比为0.9
‑
1.0mmol/ml。
[0022]
步骤(1)中,该反应还包括碱性化合物,所述碱性化合物包括但不限于三乙胺;优选地,式1所示3
‑
氨基
‑4‑
氯苯甲酸与碱性化合物的摩尔比为1:(1
‑
3);进一步优选地,式1所示3
‑
氨基
‑4‑
氯苯甲酸与碱性化合物的摩尔比为1:(1.5
‑
2.5);更进一步优选地,式1所示3
‑
氨基
‑4‑
氯苯甲酸与碱性化合物的摩尔比为1:2。
[0023]
步骤(1)中,优选地,向3
‑
氨基
‑4‑
氯苯甲酸和溶剂的溶液中加入碱性化合物,将含苄基类保护基的化合物加入,控制加入速度和温度,tlc监控反应至反应完成。
[0024]
步骤(1)中,所述反应的温度为35
‑
45℃,优选为40℃。
[0025]
步骤(1)中,所述反应时间通过tlc监控反应至反应完成;优选地,使用gf254硅胶板,展开剂为二氯甲烷:甲醇(v/v)=10:1,使用254nm紫外灯观察。
[0026]
步骤(1)中,所述反应结束后,加入氢氧化钠溶液萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,干燥,得到式2所示化合物。
[0027]
步骤(2)中,式2所示化合物与氯化亚砜的用量比为(0.2
‑
0.8)mol:300ml;优选地,式2所示化合物与氯化亚砜的用量比为(0.4
‑
0.6)mol:300ml;进一步优选地,式2所示化合物与氯化亚砜的用量比为(0.5
‑
0.55)mol:300ml。
[0028]
步骤(2)中,所述氨水与氯化亚砜的体积比为(1
‑
7):3;优选地,所述氨水与氯化亚砜的体积比为(3
‑
5):3;进一步优选地,所述氨水与氯化亚砜的体积比为4:3。
[0029]
步骤(2)中,优选地,将氯化亚砜降温至0℃以下,分批加入式2所示化合物,控制加入速度,并保持温度为10℃以下,加入完毕后,升温反应。
[0030]
步骤(2)中,所述反应的温度为35℃以下;优选地,所述反应的温度为30℃以下;进一步优选地,所述反应的温度为25℃以下。
[0031]
步骤(2)中,所述反应的时间为通过tlc监控反应至反应完成;优选地,使用gf254硅胶板,展开剂为二氯甲烷:甲醇(v/v)=50:1,使用254nm紫外灯观察。
[0032]
步骤(2)中,所述反应结束后,加入10℃以下的氨水;优选地,所述反应结束后,加入10℃以下的氨水,继续于20
±
5℃反应;进一步优选地,所述反应结束后,加入10℃以下的氨水,控制加入速度,控制反应体系温度为25℃以下,继续于20
±
5℃反应;更进一步优选地,所述反应结束后,加入10℃以下的氨水,控制加入速度,控制反应体系温度为25℃以下,继续于20
±
5℃反应0.5h以上;再更进一步优选地,所述反应结束后,加入10℃以下的氨水,控制加入速度,控制反应体系温度为25℃以下,继续于20
±
5℃反应0.5h以上,将析出的固体过滤,滤饼淋洗,干燥,得到式3所示化合物。
[0033]
步骤(3)中,式3所示化合物与苯酚的摩尔比为1:(1.5
‑
2.9);优选地,式3所示化合物与苯酚的摩尔比为1:(1.8
‑
2.6);进一步优选地,式3所示化合物与苯酚的摩尔比为1:(2.1
‑
2.3)。
[0034]
步骤(3)中,优选地,向含式3所示化合物的溶液中加入含苯酚的溶液,反应。
[0035]
其中,所述含式3所示化合物的溶液包括式3所示化合物、碳酸氢钠和水;优选地,三者的用量比为1mol:(1
‑
3)mol:(2
‑
3)l;优选地,三者的用量比为1mol:2mol:(2.5
‑
2.7)l。
[0036]
其中,所述含苯酚的溶液包括苯酚、氢氧化钠和水;优选地,三者的用量比为1mol:(0.5
‑
1.5)mol:(0.2
‑
0.9)l;优选地,三者的用量比为1mol:1mol:(0.5
‑
0.6)l。
[0037]
步骤(3)中,所述反应的温度为60
‑
100℃;优选地,所述反应的温度为70
‑
80℃。
[0038]
步骤(3)中,所述反应的时间为5h以上;优选地,所述反应的时间为5
‑
15h;进一步优选地,所述反应的时间为10h。
[0039]
步骤(3)中,所述反应结束后,将反应液冷却至0℃以下,析出的固体过滤,洗涤滤饼,干燥,即得式4所示化合物。
[0040]
步骤(4)中,所述氧化剂为二氯二氰基苯醌(ddq)和/或硝酸铈铵(can)。
[0041]
步骤(4)中,所述氧化剂与式4所示化合物的摩尔比为(2
‑
6):1。
[0042]
优选地,当所述氧化剂为二氯二氰基苯醌时,所述氧化剂与式4所示化合物的摩尔比为2.5
‑
6;进一步优选地,当所述氧化剂为二氯二氰基苯醌时,所述氧化剂与式4所示化合
物的摩尔比为3。
[0043]
优选地,当所述氧化剂为硝酸铈铵时,所述氧化剂与式4所示化合物的摩尔比为2
‑
5;进一步优选地,当所述氧化剂为硝酸铈铵时,所述氧化剂与式4所示化合物的摩尔比为2.5。
[0044]
步骤(4)中,当所述氧化剂二氯二氰基苯醌时;
[0045]
其中,所述反应的溶剂为乙腈。
[0046]
其中,所述式4所示化合物与溶剂的用量比为0.1
‑
0.5mmol/ml;优选地,式4所示化合物与溶剂的用量比为0.3
‑
0.35mmol/ml。
[0047]
其中,将式4所示化合物与溶剂加热溶解后,加入二氯二氰基苯醌反应。
[0048]
其中,所述加热的温度为30
‑
50℃。
[0049]
其中,所述反应的温度为50
‑
70℃;优选地,所述反应的温度为55
‑
65℃。
[0050]
其中,所述反应的时间为1h以上;优选地,所述反应的时间为1
‑
5h;进一步优选地,所述反应的时间为3h。
[0051]
其中,所述反应结束后,将反应液冷却,加水,用乙酸乙酯萃取,用饱和氯化钠溶液洗涤有机相,向乙酸乙酯中加入正庚烷,降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,干燥,得到式5所示化合物。
[0052]
步骤(4)中,当所述氧化剂为硝酸铈铵时;
[0053]
其中,所述反应的溶剂为乙腈和水;优选地,乙腈与水的体积比为(0.5
‑
2):1;优选地,乙腈与水的体积比为(1
‑
1.5):1;进一步优选地,乙腈与水的体积比为(1.2
‑
1.4):1。
[0054]
其中,所述式4所示化合物与溶剂的用量比为0.05
‑
0.2mmol/ml;优选地,式4所示化合物与溶剂的用量比为0.1
‑
0.15mmol/ml。
[0055]
优选地,向5℃以下的式4所示化合物和溶剂的溶液中加入硝酸铈铵,反应;进一步优选地,向0℃以下的式4所示化合物和溶剂的溶液中加入硝酸铈铵,反应。
[0056]
其中,所述反应的温度为
‑5‑
10℃;优选地所述反应温度为0
‑
5℃。
[0057]
其中,所述反应的时间为5h以上;优选地,所述反应的时间为5
‑
15h;进一步优选地,所述反应的时间为10h。
[0058]
其中,所述反应结束后,加入乙酸乙酯萃取,合并有机相,用饱和氯化钠溶液洗涤有机相次,向乙酸乙酯中滴加正庚烷,将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,干燥,得到式5所示化合物。
[0059]
有益效果:与现有技术相比,本发明具有如下优势:
[0060]
本发明提供了一种新的制备3
‑
氨基
‑4‑
苯氧基
‑5‑
氨磺酰基苯甲酸的方法,避免了剧毒品氯磺酸的使用,避免了硝化反应,便于生产组织,提高了生产的安全性。
具体实施方式
[0061]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述原料、试剂、溶剂均为常规的市售化学品,在使用前未经纯化。
[0062]
下述实施例中所述不高于40℃,不高于20℃等类似的描述指的是滴加的过程中会放热,所述温度逼近相应40℃,或20℃等,该表述为合成领域常见的表述方式。
[0063]
下述实施例中所述控制滴加速度指的是根据反应体系的温度来控制加料速度,如
果温度接近,则放慢速度或暂停加入,等温度降低些再开始滴加或提高滴加速度。
[0064]
实施例1:制备化合物2a
[0065][0066]
将3
‑
氨基
‑4‑
氯苯甲酸(100g,0.583mol)加入到反应瓶中,加入二氯甲烷(600ml)溶解,加入三乙胺(162ml,1.166mol),将氯化苄(81.18g,0.641mol)滴加进入,控制滴加速度,温度不高于40℃,滴加完毕,tlc监控反应至反应完成,加入0.5mol/l氢氧化钠溶液(500ml)萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,40
‑
50℃鼓风干燥,得到化合物2a(141.9g),收率93%。
[0067]
esi
‑
ms( ):262.2[m h]
.1h
‑
nmr(400mhz,dmso
‑
d6 d2o):7.49(d,1h,j=7.0hz),7.47(d,1h,j=7.0hz),7.31
‑
7.27(m,6h),4.31(s,2h).
[0068]
实施例2:制备化合物2a
[0069][0070]
将3
‑
氨基
‑4‑
氯苯甲酸(100g,0.583mol)加入到反应瓶中,加入二氯甲烷(600ml)溶解,加入三乙胺(162ml,1.166mol),将溴化苄(109.64g,0.641mol)滴加进入,控制滴加速度,温度不高于40℃,滴加完毕,tlc监控反应至反应完成,加入0.5mol/l氢氧化钠溶液(500ml)萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,40
‑
50℃鼓风干燥,得到化合物2a(138.8g),收率91%。
[0071]
实施例3:制备化合物2a
[0072]
将3
‑
氨基
‑4‑
氯苯甲酸(100g,0.583mol)加入到反应瓶中,加入二氯甲烷(600ml)溶解,加入三乙胺(162ml,1.166mol),将溴化苄(149.66g,0.875mol)滴加进入,控制滴加速度,温度不高于40℃,滴加完毕,tlc监控反应至反应完成,加入0.5mol/l氢氧化钠溶液(500ml)萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,40
‑
50℃鼓风干燥,得到化合物2a(140.3g),收率92%。
[0073]
实施例4:制备化合物2b
[0074][0075]
将3
‑
氨基
‑4‑
氯苯甲酸(100g,0.583mol)加入到反应瓶中,加入二氯甲烷(600ml)溶解,加入三乙胺(162ml,1.166mol),将对甲氧基氯苄(100.39g,0.641mol)滴加进入,控制滴加速度,温度不高于40℃,滴加完毕,tlc监控反应至反应完成,加入0.5mol/l氢氧化钠溶
液(500ml)萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,40
‑
50℃鼓风干燥,得到化合物2b(151.4g),收率89%。
[0076]
esi
‑
ms( ):292.1[m h]
.1h
‑
nmr(400mhz,dmso
‑
d6 d2o):7.48(d,1h,j=6.9hz),7.47(d,1h,j=7.0hz),7.31(s,1h),7.12(d,2h,j=6.6hz),7.02(d,2h,j=6.6hz),4.31(s,2h),3.77(s,3h).
[0077]
实施例5:制备化合物2b
[0078]
将3
‑
氨基
‑4‑
氯苯甲酸(100g,0.583mol)加入到反应瓶中,加入二氯甲烷(600ml)溶解,加入三乙胺(162ml,1.166mol),将对甲氧基氯苄(91.3g,0.583mol)滴加进入,控制滴加速度,温度不高于40℃,滴加完毕,tlc监控反应至反应完成,加入0.5mol/l氢氧化钠溶液(500ml)萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,40
‑
50℃鼓风干燥,得到化合物2b(141.2g),收率83%。
[0079]
实施例6:制备化合物2b
[0080][0081]
将3
‑
氨基
‑4‑
氯苯甲酸(100g,0.583mol)加入到反应瓶中,加入二氯甲烷(600ml)溶解,加入三乙胺(162ml,1.166mol),将对甲氧基溴苄(128.88g,0.641mol)滴加进入,控制滴加速度,温度不高于40℃,滴加完毕,tlc监控反应至反应完成,加入0.5mol/l氢氧化钠溶液(500ml)萃取,分出水相,用稀盐酸调节ph值至4
‑
5,在0
‑
10℃析晶,得到类白色固体,40
‑
50℃鼓风干燥,得到化合物2b(159.9g),收率94%。
[0082]
实施例7:制备化合物3a
[0083][0084]
将氯化亚砜(300ml)加入到反应瓶中,使用冷浴降温至0℃以下,将化合物2a(138.8g,0.530mol)固体分批加入,控制加速速度,保持温度在10℃以下,加入完毕,将反应体系升温至25
±
5℃,搅拌反应,tlc监控反应过程,至原料点消失。
[0085]
将氨水(400ml)降温至10℃以下,搅拌下,将反应液滴加进入,控制滴加速度,保持反应体系温度不高于25℃。滴加完毕,在20
±
5℃搅拌1小时。将析出的固体过滤,滤饼用纯化水(100ml
×
2)淋洗。在50℃以下鼓风干燥,得到化合物3a,共125.6g,收率73%。
[0086]
esi
‑
ms( ):325.1[m h]
.1h
‑
nmr(400mhz,dmso
‑
d6 d2o):7.63(s,1h),7.48(s,1h),7.32
‑
7.29(m,5h),4.31(s,2h).
[0087]
实施例8:制备化合物3a
[0088]
将氯化亚砜(300ml)加入到反应瓶中,使用冷浴降温至0℃以下,将化合物2a(52.4g,0.2mol)固体分批加入,控制加速速度,保持温度在10℃以下,加入完毕,将反应体系升温至25
±
5℃,搅拌反应,tlc监控反应过程,至原料点消失。
[0089]
将氨水(400ml)降温至10℃以下,搅拌下,将反应液滴加进入,控制滴加速度,保持反应体系温度不高于25℃。滴加完毕,在20
±
5℃搅拌1小时。将析出的固体过滤,滤饼用纯化水(100ml
×
2)淋洗。在50℃以下鼓风干燥,得到化合物3a,共43.52g,收率67%。
[0090]
实施例9:制备化合物3b
[0091][0092]
将氯化亚砜(300ml)加入到反应瓶中,使用冷浴降温至0℃以下,将化合物2b(159.0g,0.545mol)固体分批加入,控制加速速度,保持温度在10℃以下,加入完毕,将反应体系升温至25
±
5℃,搅拌反应,tlc监控反应过程,至原料点消失。
[0093]
将氨水(400ml)降温至10℃以下,搅拌下,将反应液滴加进入,控制滴加速度,保持反应体系温度不高于25℃。滴加完毕,在20
±
5℃搅拌1小时。将析出的固体过滤,滤饼用纯化水(100ml
×
2)淋洗。在50℃以下鼓风干燥,得到化合物3b,共127.6g,收率66%。
[0094]
esi
‑
ms( ):354.04[m h]
.1h
‑
nmr(400mhz,dmso
‑
d6 d2o):7.62(s,1h),7.49(s,1h),7.37(d,2h,j=6.7hz),7.29(d,2h,j=6.7hz),4.31(s,2h),3.81(s,3h).
[0095]
实施例10:制备化合物3b
[0096]
将氯化亚砜(300ml)加入到反应瓶中,使用冷浴降温至0℃以下,将化合物2b(233.4g,0.8mol)固体分批加入,控制加速速度,保持温度在10℃以下,加入完毕,将反应体系升温至25
±
5℃,搅拌反应,tlc监控反应过程,至原料点消失。
[0097]
将氨水(400ml)降温至10℃以下,搅拌下,将反应液滴加进入,控制滴加速度,保持反应体系温度不高于25℃。滴加完毕,在20
±
5℃搅拌1小时。将析出的固体过滤,滤饼用纯化水(100ml
×
2)淋洗。在50℃以下鼓风干燥,得到化合物3b,共173.1g,收率61%。
[0098]
实施例11:制备化合物4a
[0099][0100]
在反应瓶中加入纯化水(1000ml),加入化合物3a(123.0g,0.379mol),搅拌下,加入碳酸氢钠(63.7g,0.758mol),固体溶解,将反应体系升温至80
‑
85℃。将苯酚(78.5g,0.834mol)和氢氧化钠(33.4g,0.834mol)溶解在纯化水(500ml)中。将苯酚和氢氧化钠的溶液滴加到化合物3a的溶液中,控制滴加速度,保持温度在70
‑
80℃,然后在70
‑
80℃搅拌反应10小时。
[0101]
将反应液冷却至0℃以下,将析出的固体滤出,滤饼用纯化水淋洗,固体在60
±
5℃鼓风干燥12小时,得到化合物4a,共118.9g,收率82.0%。
[0102]
esi
‑
ms( ):405.2[m na]
.1h
‑
nmr(400mhz,dmso
‑
d6 d2o):7.65(s,1h),7.50(s,1h),7.42
‑
7.31(m,7h),7.18
‑
7.10(m,3h),4.32(s,2h).
[0103]
实施例12:制备化合物4a
[0104]
在反应瓶中加入纯化水(1000ml),加入化合物3a(123.0g,0.379mol),搅拌下,加入碳酸氢钠(63.7g,0.758mol),固体溶解,将反应体系升温至80
‑
85℃。将苯酚(103.52g,1.10mol)和氢氧化钠(44g,1.10mol)溶解在纯化水(500ml)中。将苯酚和氢氧化钠的溶液滴加到化合物3a的溶液中,控制滴加速度,保持温度在70
‑
80℃,然后在70
‑
80℃搅拌反应10小时。
[0105]
将反应液冷却至0℃以下,将析出的固体滤出,滤饼用纯化水淋洗,固体在60
±
5℃鼓风干燥12小时,得到化合物4a,共107.4g,收率74.1%。
[0106]
实施例13:制备化合物4b
[0107][0108]
在反应瓶中加入纯化水(1000ml),加入化合物3b(125.0g,0.352mol),搅拌下,加入碳酸氢钠(59.1g,0.704mol),固体溶解,将反应体系升温至80
‑
85℃。将苯酚(72.9g,0.774mol)和氢氧化钠(31.0g,0.774mol)溶解在纯化水(500ml)中。将苯酚和氢氧化钠的溶液滴加到化合物3b的溶液中,控制滴加速度,保持温度在90
‑
100℃,然后在90
‑
100℃搅拌反应10小时。
[0109]
将反应液冷却至0℃以下,将析出的固体滤出,滤饼用纯化水淋洗,固体在60
±
5℃鼓风干燥12小时,得到化合物4b,共116.1g,收率80.0%。
[0110]
esi
‑
ms( ):413.20[m h]
,435.11[m na]
1
h
‑
nmr(400mhz,dmso
‑
d6 d2o):7.66(s,1h),7.50(s,1h),7.42
‑
7.31(m,6h),7.18
‑
7.10(m,3h),4.32(s,2h),3.86(s,3h).
[0111]
实施例14:制备化合物4b
[0112]
在反应瓶中加入纯化水(1000ml),加入化合物3b(125.0g,0.352mol),搅拌下,加入碳酸氢钠(59.1g,0.704mol),固体溶解,将反应体系升温至80
‑
85℃。将苯酚(49.69g,0.528mol)和氢氧化钠(21.12g,0.528mol)溶解在纯化水(500ml)中。将苯酚和氢氧化钠的溶液滴加到化合物3b的溶液中,控制滴加速度,保持温度在60
‑
70℃,然后在60
‑
70℃搅拌反应10小时。
[0113]
将反应液冷却至0℃以下,将析出的固体滤出,滤饼用纯化水淋洗,固体在60
±
5℃鼓风干燥12小时,得到化合物4b,共107.4g,收率74.0%。
[0114]
实施例15:由4a制备化合物5(ddq使用2.5当量)
[0115][0116]
将化合物4a(100g,0.261mol)加入到反应瓶中,加入乙腈(800ml),加热至40℃,将
固体溶解,加入二氯二氰基苯醌(148.39g,0.65mol,2.5当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(480ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(160ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(960ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共60.46g,收率75%。
[0117]
esi
‑
ms( ):309.10[m h]
.1h
‑
nmr(400mhz,dmso
‑
d6 d2o):8.02(s,1h),7.81(s,1h),7.42(d,2h,j=6.3hz),7.18
‑
7.10(m,3h).
[0118]
实施例16:由4a制备化合物5(ddq使用3当量)
[0119][0120]
将化合物4a(200g,0.523mol)加入到反应瓶中,加入乙腈(1600ml),加热至40℃,将固体溶解,加入二氯二氰基苯醌(356.14g,1.57mol,3当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(960ml),用乙酸乙酯(640ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(320ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(1920ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共143.50g,收率89%。
[0121]
实施例17:由4a制备化合物5(ddq使用4.5当量)
[0122][0123]
将化合物4a(100g,0.261mol)加入到反应瓶中,加入乙腈(800ml),加热至40℃,将固体溶解,加入二氯二氰基苯醌(267.11g,1.18mol,4.5当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(480ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(160ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(960ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共59.66g,收率74%。
[0124]
实施例18:由4a制备化合物5(ddq使用6当量)
[0125]
[0126]
将化合物4a(150g,0.392mol)加入到反应瓶中,加入乙腈(1200ml),加热至40℃,将固体溶解,加入二氯二氰基苯醌(534.22g,2.35mol,6当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(720ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(240ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(1440ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共65.30g,收率54%。
[0127]
实施例19:由4b制备化合物5(ddq使用2.5当量)
[0128][0129]
将化合物4b(100g,0.242mol)加入到反应瓶中,加入乙腈(800ml),加热至40℃,将固体溶解,加入二氯二氰基苯醌(137.59g,0.61mol,2.5当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(480ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(160ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(960ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共47.84g,收率64%。
[0130]
实施例20:由4b制备化合物5(ddq使用3当量)
[0131][0132]
将化合物4b(100g,0.242mol)加入到反应瓶中,加入乙腈(800ml),加热至40℃,将固体溶解,加入二氯二氰基苯醌(165.11g,0.73mol,3当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(480ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(160ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(960ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共53.07g,收率71%。
[0133]
实施例21:由4b制备化合物5(ddq使用4.5当量)
[0134][0135]
将化合物4b(100g,0.242mol)加入到反应瓶中,加入乙腈(800ml),加热至40℃,将
固体溶解,加入二氯二氰基苯醌(247.66g,1.09mol,4.5当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(480ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(160ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(960ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共50.08g,收率67%。
[0136]
实施例22:由4b制备化合物5(ddq使用6当量)
[0137][0138]
将化合物4b(100g,0.242mol)加入到反应瓶中,加入乙腈(800ml),加热至40℃,将固体溶解,加入二氯二氰基苯醌(330.21g,1.45mol,6当量),将反应体系加热至60
±
5℃,反应3小时。冷却至25℃以下,加入纯化水(480ml),用乙酸乙酯(320ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(160ml
×
3)洗涤有机相。向乙酸乙酯溶液中滴加正庚烷(960ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共45.60g,收率61%。
[0139]
实施例23:由4a制备化合物5(can使用2当量)
[0140][0141]
将化合物4a(120g,0.314mol)加入到反应瓶中,加入乙腈(1200ml),加入纯化水(960ml),降温至0℃,将硝酸铈铵(344.04g,0.628mol,2当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(600ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(120ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(720ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共67.72g,收率70%。
[0142]
实施例24:由4a制备化合物5(can使用3当量)
[0143][0144]
将化合物4a(200g,0.523mol)加入到反应瓶中,加入乙腈(2000ml),加入纯化水(1600ml),降温至0℃,将硝酸铈铵(860.11g,1.569mol,3当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(1000ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(200ml
×
3)洗涤有机相,
向乙酸乙酯中滴加正庚烷(1200ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共104.80g,收率65%。
[0145]
实施例25:由4a制备化合物5(can使用2.5当量)
[0146][0147]
将化合物4a(200g,0.523mol)加入到反应瓶中,加入乙腈(2000ml),加入纯化水(1600ml),降温至0℃,将硝酸铈铵(716.76g,1.307mol,2.5当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(1000ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(200ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(1200ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共130.60g,收率81%。
[0148]
实施例26:由4a制备化合物5(can使用5当量)
[0149][0150]
将化合物4a(120g,0.314mol)加入到反应瓶中,加入乙腈(1200ml),加入纯化水(960ml),降温至0℃,将硝酸铈铵(860.11g,1.569mol,5当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(600ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(120ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(720ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共72.56g,收率75%。
[0151]
实施例27:由4b制备化合物5(can使用2当量)
[0152][0153]
将化合物4b(150g,0.364mol)加入到反应瓶中,加入乙腈(1500ml),加入纯化水(1200ml),降温至0℃,将硝酸铈铵(398.74g,0.727mol,2当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(750ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(150ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(900ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共52.70g,收率47%。
[0154]
实施例28:由4b制备化合物5(can使用3当量)
[0155][0156]
将化合物4b(150g,0.364mol)加入到反应瓶中,加入乙腈(1500ml),加入纯化水(1200ml),降温至0℃,将硝酸铈铵(598.12g,1.091mol,3当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(750ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(150ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(900ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共79.61g,收率71%。
[0157]
实施例29:由4b制备化合物5(can使用2.5当量)
[0158][0159]
将化合物4b(150g,0.364mol)加入到反应瓶中,加入乙腈(1500ml),加入纯化水(1200ml),降温至0℃,将硝酸铈铵(498.43g,0.909mol,2.5当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(750ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(150ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(900ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共87.46g,收率78%。
[0160]
实施例30:由4b制备化合物5(can使用5当量)
[0161][0162]
将化合物4b(150g,0.364mol)加入到反应瓶中,加入乙腈(1500ml),加入纯化水(1200ml),降温至0℃,将硝酸铈铵(996.86g,1.818mol,5当量)加入,在0
‑
5℃反应10小时。加入乙酸乙酯(750ml
×
3)萃取,合并有机相,用饱和氯化钠溶液(150ml
×
3)洗涤有机相,向乙酸乙酯中滴加正庚烷(900ml),将体系降温至0
‑
10℃析晶,将固体滤出,用乙酸乙酯和正庚烷1:1的混合溶剂淋洗固体,减压45
‑
55℃干燥,得到化合物5,共84.09g,收率75%。
[0163]
本发明提供了一种布美他尼的中间体的合成方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。