1.本发明涉及有机化学领域,具体涉及一种多用途的二氮杂双环类化合物、 制备方法及在合成药物中的应用。
背景技术:
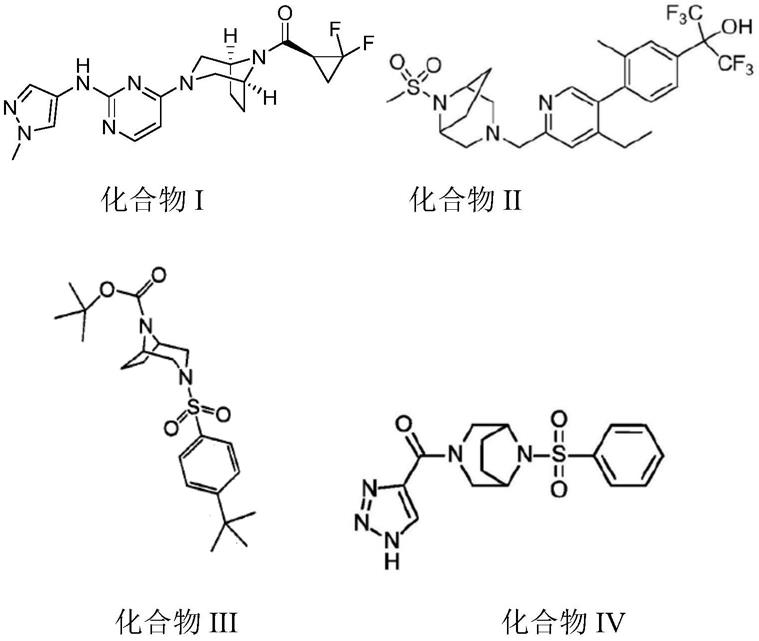
2.二氮杂桥化合物是一种非常有用的医药中间体,有大量的药物具有二氮杂 桥化合物的片段,如美国辉瑞公司开发的用于治疗多种免疫性疾病的 brepocitinib(化合物i),美国爱斯凯利尔公司开发的一种视黄酸相关的孤儿核 受体调节剂(化合物ii),美国先灵公司开发的一种用于抑制i型11β
‑
羟基类固 醇脱氢酶的药物(化合物iii)以及德国拜耳公司开发的用于治疗子宫内膜异位 的药物(化合物iv)等等。
[0003][0004]
在现有技术中,在合成上述药物时,通常使用化合物5作为起始原料,然 而纯品的化合物5的熔点仅为58℃
‑
60℃,如果存储温度或运输温度高于40℃, 化合物5就会变成粘稠的油状物,一旦变成粘稠的油状物,后续即使恢复到室 温,化合物5依然会保持油状物的状态而不会重新结晶。由于化合物5具有上 述特性,因此这会导致化合物5难以长时间存储运输,并且在形成油状物后, 化合物5的取用以及称量也会很麻烦。
[0005][0006]
在另一方面,在制备化合物5的过程中,必须将纯度提升至较高的程度才 能使得得到化合物5的晶体,在现有技术中,为了得到化合物5的晶体,往往 都需要通过柱层析的方式来提纯,这加大了化合物5生产工艺化的难题。
[0007]
此外,化合物5在制备药物的过程中,有时候需要先对8号位上的n上进 行修饰,这种情况下需要先对化合物5的3号位上的n进行保护,再脱去8号 位上的boc保护基,最后再对8号位上的n进行修饰,这会导致整个反应流程 十分繁琐,工作量较大。
技术实现要素:
[0008]
本发明是为了解决上述问题而进行的,目的在于提供具有较高熔点、易于 存储运输、易于提纯且能够高效地应用于多种药物合成的多用途的二氮杂双环 类化合物。
[0009]
本发明提供了一种多用途的二氮杂双环类化合物,具有这样的特征,结构 式如式1所示:
[0010][0011]
式中,r为2
‑
硝基苯磺酰基、4
‑
硝基苯磺酰基或2,4
‑
二硝基苯磺酰基中的 任意一种。
[0012]
在本发明提供的多用途的二氮杂双环类化合物中,还可以具有这样的特征: 其中,式中,r为2
‑
硝基苯磺酰基。
[0013]
在本发明提供的多用途的二氮杂双环类化合物中,还可以具有这样的特征: 该化合物通过重结晶得到。
[0014]
在本发明提供的多用途的二氮杂双环类化合物中,还可以具有这样的特征: 该化合物的重结晶包括如下步骤:将粗品溶解在溶剂a中,再加入溶剂b,混 合均匀后,在
‑
50℃~
‑
10℃下静置,在晶体析出后,过滤,即得目标产物,
[0015]
其中,溶剂a为四氯化碳、氯仿、乙酸乙酯、丙酮、乙醇、甲醇、二氯甲 烷、1,2
‑
二氯乙烷、三氯乙烯、四氢呋喃、2
‑
甲基四氢呋喃、甲酸乙酯、乙酸甲 酯、乙酸异丙酯、乙腈、丙腈或二硫化碳中的任意一种,溶液b为石油醚、正 己烷、环己烷、苯、乙醚、异丙醚、正戊烷、正庚烷、丙醚、硝基甲烷、硝基 苯中的任意一种。
[0016]
本发明还提供了一种可以高收率的得到上述多用途的二氮杂双环类化合物 的合成方法,反应方程式如下:
[0017]
[0018]
式中,r为2
‑
硝基苯磺酰基、4
‑
硝基苯磺酰基或2,4
‑
二硝基苯磺酰基中的 任意一种,r1为甲磺酰基或对甲苯磺酰基。
[0019]
在本发明提供的多用途的二氮杂双环类化合物的合成方法中,还可以具有 这样的特征:其中,化合物2的结构式为
[0020]
本发明还提供了另一种可以高收率的得到上述多用途的二氮杂双环类化合 物的合成方法,反应方程式如下:
[0021][0022]
式中,r1为甲磺酰基或对甲苯磺酰基,酰化试剂为为氯或溴,r为2
‑
硝基苯磺酰基、4
‑
硝基苯磺酰基或2,4
‑
二硝基苯磺酰基中的 任意一种。
[0023]
在本发明提供的多用途的二氮杂双环类化合物的合成方法中,还可以具有 这样的特征:其中,化合物2的结构式为
[0024]
在本发明提供的多用途的二氮杂双环类化合物的合成方法中,还可以具有 这样的特征:其中,化合物4为氨气、氨水或溶有氨的有机溶液。
[0025]
本发明还提供了结构式如式1所示的化合物在合成药物中的应用。
[0026]
在本发明提供的化合物在合成药物中的应用中,还可以具有这样的特征: 该药物为brepocitinib(其活性成分为化合物i)。
[0027]
发明的作用与效果
[0028]
根据本发明所涉及的多用途的二氮杂双环类化合物,因为两个氮原子上分 别具有两个不同的保护基,且两个保护基的脱除方法不同,在脱除其中一个保 护基的过程中并不会影响另一个保护基,所以,本发明提供的二氮杂双环类化 合物是一种多用途的能够在多种药物合成中得到应用的中间体。
[0029]
进一步地,因为本发明所涉及的多用途的二氮杂双环类化合物的熔点较高, 易于重结晶得到固体,在高温下不易形成油状物,所以,该二氮杂双环类化合 物适合长途运输以及长时间存储。
附图说明
[0030]
图1是本发明的实施例1中3,8
‑
二氮杂双环[3.2.1]辛烷
‑3‑
(2
‑
硝基苯磺酰 基)
‑8‑
甲酸叔丁酯的氢谱图。
具体实施方式
[0031]
为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解, 以下结合实施例及附图对本发明作具体阐述。
[0032]
在下述实施例中,除另有注明外,各原料均为化学纯的市售产品。
[0033]
在下述实施例中,质谱数据通过waters micromass lct tof质谱仪上获得。
[0034]
在下述实施例中,熔点是通过市售的熔点测定仪测定的,熔点测定仪使用 前未经校正。
[0035]
在下述实施例中,以下缩写可以指:
[0036]
boc:叔丁氧羰基;
[0037]
ms:甲磺酰基;
[0038]
dmf:n,n
‑
二甲基甲酰胺;
[0039]
hatu:1
‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑[4,5
‑
b]吡啶鎓3
‑
氧化物 六氟磷酸盐。
[0040]
<实施例1>
[0041]
化合物1a的合成
[0042]
本实施例提供了一种化合物1a的合成方法,反应方程式如下:
[0043][0044]
反应步骤如下:
[0045]
步骤1,将5g化合物2a(12.9mmol,1eq)、7.3g化合物3a(36.1 mmol,2.8eq)溶于50ml无水乙腈中,85℃回流反应8h,自然回至室温,水 洗2次,取有机相,蒸除溶剂,得油状产品;
[0046]
步骤2,向步骤1中得到的油状产品中滴加乙酸乙酯直至油状产品完全溶 解在乙酸乙酯中,再加入100ml丙醚,混合均匀,
‑
20℃下静置12h,快速抽 滤,即得化合物1a的晶体4.25g,收率82.9%。
[0047]
本实施例制得的化合物1a的氢谱如图1所示。
[0048]
具体的氢谱、质谱以及熔点数据如下:
[0049]1h nmr(400mhz,chloroform
‑
d)δ7.97
–
7.92(m,1h),7.73(pd,j=7.5,1.8 hz,2h),7.65(dd,j=7.4,1.8hz,1h),4.34(s,2h),3.64(dt,j=11.9,1.7hz,2h), 3.08(d,j=11.5hz,2h),1.97(t,j=2.8hz,4h),1.47(s,9h).
[0050]
高分辨ms(esi):m/z[m na]
calcd for c
17
h
23
n3o6sna:420.1205,found: 420.1209.
[0051]
mp:185
‑
187℃.
[0052]
<实施例2>
[0053]
化合物1a的合成
[0054]
本实施例提供了另一种合成化合物1a的合成方法,反应方程式如下:
[0055][0056]
反应步骤如下:
[0057]
步骤1,步骤1,将化合物2a 5g(12.9mmol)加入到15ml乙腈中,再加 入25wt%
‑
28wt%的浓氨水(市售,使用前未经滴定)50ml,加热至70℃,搅 拌反应12小时,得到反应液;
[0058]
步骤2,向反应液中加入20ml二氯甲烷,搅拌,萃取,取有机相,水洗 有机相,加入100ml乙酸乙酯/石油醚混合液进行重结晶,乙酸乙酯/石油醚混 合液为乙酸乙酯与石油醚体积比为1:9的混合液,得2.0g呈类白色固体的化 合物5(即3,8
‑
二氮杂双环[3.2.1]辛烷
‑8‑
甲酸叔丁酯),单步收率72.9%,gc纯 度95.3%,无需进一步提纯即可投入后续反应中;
[0059]
步骤3,将5g化合物5(23.6mmol,1eq)溶解在25ml无水四氢呋喃中, 加入2.86g三乙胺(28.3mmol,1.2eq),混合均匀后,滴加5.74g邻硝基苯甲 酰氯(26.0mmol,1.1eq),室温下搅拌反应3小时,加入30ml水猝灭反应, 水洗2次,取有机相,蒸除溶剂,得油状产品;
[0060]
步骤4,向步骤3中得到的油状产品中滴加乙酸乙酯直至油状产品完全溶 解在乙酸乙酯中,再加入100ml丙醚,混合均匀,
‑
20℃下静置12h,快速抽 滤,即得化合物1a的晶体8.67g,收率92.4%。
[0061]
<实施例3>
[0062]
重结晶溶剂的筛选
[0063]
在本实施例中,对化合物1a重结晶的溶剂进行了筛选,各组实验均使用 同一锅按照实施例1中步骤1记载的反应步骤进行的实验中中得到的1g油状 产品进行重结晶,各组实验均采用混合溶剂重结晶法,表1中的溶剂a为对产 物溶解性较好的溶剂(良性溶剂),溶剂b为对产物溶解性较差的溶剂(不良 溶剂),筛选结果如表1所示:
[0064]
表1重结晶溶剂筛选表
[0065]
序号溶剂a溶剂b重结晶产品性状重结晶收率1四氢呋喃丙醚半固半油状
‑
2四氢呋喃正己烷固体90.4%31,2
‑
二氯甲烷正己烷油状液体
‑
4氯仿环己烷固体83.7%5乙酸乙酯异丙醚固体94.6%6乙酸乙酯丙醚固体96.8%
[0066]
由表1可知,在选用部分溶剂作为重结晶剂时,化合物1a无法重结晶得 到固体,在选用乙酸乙酯作为良性溶剂,醚类化合物作为不良溶剂时,重结晶 的收率最高,可以达到95%左右。
[0067]
<实施例4>
[0068]
化合物1a的脱保护
[0069]
本实施例提供了一种化合物1a脱除boc保护的方法,反应方程式如下:
[0070][0071]
反应步骤如下:
[0072]
将5g化合物1a加入到100ml浓度为3mol/l的盐酸甲醇溶液中,室温下 搅拌反应3h,蒸除溶剂,加入50ml去离子水,滴加1mol/l的氢氧化钠至溶 液ph值为13,加入二氯甲烷萃取(50ml
×
2),合并有机相,饱和食盐水洗涤 一次,蒸除溶剂,即得3.65g化合物6,收率97.6%,化合物6在室温下为黄 色油状液体。
[0073]
<实施例5>
[0074]
化合物1a的脱保护
[0075]
本实施例提供了一种化合物1a脱除邻硝基苯磺酰基的方法,反应方程式 如下:
[0076][0077]
反应步骤如下:
[0078]
将5g化合物1a(12.6mmol,1eq)溶解在50ml dmf中,加入5.1g正十 二烷基硫醇(25.2mol,2eq)以及1.06g氢氧化锂的一水合物(25.2 mmol,2eq),室温下搅拌反应2h,加入100ml乙酸乙酯,使用1mol/l的盐酸 水溶液萃取(50ml
×
2),合并水相,向水相中滴加1mol/l的氢氧化钠至溶液 ph值为13,二氯甲烷萃取(50ml
×
2),合并有机相,蒸除溶剂,得到2.26g 化合物5,收率84.1%。化合物5在室温下为淡黄色固体。
[0079]
<实施例6>
[0080]
brepocitinib中间体的合成
[0081]
本实施例提供了一种brepocitinib中间体的合成方法,反应方程式如下:
[0082][0083]
反应步骤如下:
[0084]
步骤1,将20g化合物6(67.3mmol,1eq)、16.4g化合物7(134.6 mmol,2eq)以及20.4g三乙胺(201.9mmol,3eq)分散到200ml dmf中, 再加入30.7g hatu(80.8mmol,1.2eq),室温下搅拌反应30min,减压浓缩, 快速柱层析,即得21.0g化合物8,收率77.7%;
[0085]
步骤2,将20g化合物8(49.8mmol,1eq)溶解在50ml dmf中,加入 20.2g正十二烷基硫醇(99.6mmol,2eq)以及4.17g氢氧化锂的一水合物 (99.6mmol,2eq),室温下搅拌反应2h,加入100ml乙酸乙酯,使用1mol/l 的盐酸水溶液萃取(50ml
×
2),合并水相,向水相中滴加1mol/l的氢氧化钠 至溶液ph值为13,二氯甲烷萃取(50ml
×
2),合并有机相,蒸除溶剂,得淡 黄色固体,得8.89g化合物9,收率82.5%;
[0086]
步骤3,在冰水浴下,将15g化合物9(69.4mmol,1eq)溶解在500ml 甲醇中,加入11.4g 2,4
‑
二氯嘧啶(76.3mmol,1.1eq)以及9.13g三乙胺 (90.2mmol,1.3eq),自然回至室温,搅拌反应12h,减压浓缩除去溶剂,快 速柱层析,得18.3g化合物10,收率80.2%。
[0087]
实施例的作用与效果
[0088]
根据上述实施例所涉及的多用途的二氮杂双环类化合物,因为两个氮原子 上分别具有boc保护基以及邻硝基苯磺酰基,且两个保护基的脱除方法不同, 在脱除其中一个保护基的过程中并不会影响另一个保护基,所以,本发明提供 的二氮杂双环类化合物是一种多用途的能够在多种药物合成中得到应用的中间 体。
[0089]
进一步地,因为上述实施例所涉及的多用途的二氮杂双环类化合物的熔点 较高,易于重结晶得到固体,在高温下不易形成油状物,所以,该二氮杂双环 类化合物适合长途运输以及长时间存储。
[0090]
进一步地,因为上述实施例所涉及的多用途的二氮杂双环类化合物中3号 位上的保护基为邻硝基苯磺酰基,所以在脱除该保护基时无需使用易使得桥环 断裂的pd/c等强还原试剂,在合成药物中有着更为广泛的应用。
[0091]
进一步地,因为上述实施例所涉及的多用途的二氮杂双环类化合物中3号 位上的保护基为对强酸强碱均稳定的邻硝基苯磺酰基,所以在对8号位上的氮 进行修饰时可以有更多的试剂可以进行选择,适合用于合成一些复杂的药物或 药物中间体。
[0092]
上述实施方式为本发明的优选案例,并不用来限制本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。