一种无pam限制的腺嘌呤碱基编辑器融合蛋白及应用
技术领域
1.本发明属于生物医药领域,涉及一种无pam限制的腺嘌呤碱基编辑器融合蛋白及应用。
背景技术:
2.crispr/cas9系统最初发现于细菌和古细菌中,后经过优化和改造形成强大的基因编辑工具,被广泛应用于dna的敲除、敲入、修饰等研究。crispr/cas9系统由cas9核酸酶和识别靶序列的sgrna两部分组成,sgrna与靶序列互补配对介导cas9核酸酶对基因组的定向切割,造成双链dna断裂(double strand break,dsb)后,利用细胞中的修复机制同源重组(有模板情况下)和非同源末端连接(无模板情况下)实现靶向位点的编辑
[1,2]
。随后,david liu等人构建ruvc结构域失活的nickase cas9(ncas9),并在此基础上开发出单碱基编辑系统即胞嘧啶碱基编辑器(cytosine base editor,cbe)和腺嘌呤碱基编辑器(adenine base editor,abe),两种碱基编辑器在不引起dna双链断裂的情况下可分别实现c:g到t:a、a:t到g:c的碱基转换,极大提升了单碱基编辑的效率和安全性
[2,3]
。
[0003]
abe由腺嘌呤脱氨酶和ncas9融合而成,根据clinvar数据库收录的数据,与人类疾病相关的基因变异有58%是点突变,而其中47%的致病点突变可通过abe介导的a:t到g:c的碱基转换得到修复
[4]
。已有大量研究显示abe在疾病修复领域的应用价值。例如,通过病毒递送abe和相应的sgrna至杜氏肌营养不良的小鼠肌肉中,可修复致病基因dmd的无义突变
[5]
;通过脂质纳米颗粒递送mrna形式的abe至患酪氨酸血症的成年小鼠肝脏中,修复了致病性的剪切位点变异,恢复肝细胞中fah的表达
[6]
。但abe对于位点的编辑受到编辑窗口和pam序列的限制,应用最为广泛的abemax识别的pam序列为ngg,为进一步拓展碱基编辑器的编辑范围,识别不同pam序列的abe相继出现,如识别pam序列为ng的xabe和abe
‑
ng
[7]
,其中pam限制最为宽松的是2020年3月发表的abemax
‑
spry,其pam序列为nrn(r代表a、g)和nyn(y代表c、t)
[8]
。abemax
‑
spry可以靶向基因组的所有序列,但是abemax
‑
spry编辑效率较低,且未解决abe存在的在转录组水平上的脱靶问题,限制了该碱基编辑器的应用,需要对其进行改进和优化。
技术实现要素:
[0004]
一些实施方案中,本发明了提供了一种突变体多肽,所述多肽自n端至c端依次包括spry(d10a)的n端片段、tada8e片段和spry(d10a)多肽的c端片段。
[0005]
一些实施方案中,所述spry(d10a)蛋白n端片段的氨基酸序列如seq id no:1所示,所述tada8e片段的氨基酸序列如seq id no:3所示,所述spry(d10a)蛋白c端片段的氨基酸序列如seq id no:5所示。
[0006]
一些实施方案中,编码所述spry(d10a)蛋白n端片段的核苷酸序列如seq id no:2所示。
[0007]
一些实施方案中,编码所述tada8e片段的核苷酸序列如seq id no:4所示。
[0008]
一些实施方案中,编码所述spry(d10a)蛋白c端片段的核苷酸序列如seq id no:6所示。
[0009]
一些实施方案中,所述突变体多肽用于基因编辑。
[0010]
一些实施方案中,所述基因编辑的编辑窗口为3~10位。
[0011]
一些实施方案中,所述基因编辑的编辑窗口为8~10位。
[0012]
一些实施方案中,所述突变体多肽包含如seq id no:13所示的序列。
[0013]
一些实施方案中,本发明提供了一种融合蛋白,包含所述的突变体多肽。
[0014]
在一些实施方案中,含有所述突变体多肽的融合蛋白可以靶向全基因组,拓宽了基因组的可编辑范围;能够更高效率地引起a:t到g:c的碱基转换,具有很大的应用潜力,包括但不限于基因疾病致病性位点的模拟或修复;一些实施例中,含有所述突变体多肽的融合蛋白拓宽了碱基编辑窗口;且造成转录组水平上的脱靶更低,兼顾效率高和脱靶低的突变体形式。
[0015]
相较于现有的腺嘌呤碱基编辑器突变体,abemax
‑
spry无pam限制,有效提高了基因组的可靶向范围,但是其编辑活性不高。
[0016]
在一些实施方案中,发明人用abe8e中的腺嘌呤脱氨酶tada8e代替abemax
‑
spry中的腺嘌呤脱氨酶二聚体,构建形成8e
‑
spry,8e
‑
spry相比于abemax
‑
spry不仅能够更高效率地引起碱基转换,还拓宽了碱基编辑窗口。
[0017]
在一些实施方案中,发明人又在8e
‑
spry的基础上构建了4种突变体,分别为ce
‑
8e
‑
spry、v106w
‑
spry、8e
‑
spry
‑
hf和v106w
‑
spry
‑
hf。综合评价编辑效率和脱靶后,ce
‑
8e
‑
spry是兼顾效率高和脱靶低的突变体形式。
[0018]
一些实施方案中,所述融合蛋白还包括连接肽,连接肽位于spry(d10a)蛋白n端片段与tada8e片段之间,和/或位于tada8e片段与spry(d10a)蛋白c端片段之间。
[0019]
一些实施方案中,所述连接肽氨基酸序列如seq id no:7所示。
[0020]
一些实施方案中,编码所述连接肽的核苷酸序列如seq id no:8所示。
[0021]
一些实施方案中,所述融合蛋白还包括核定位信号片段。
[0022]
一些实施方案中,所述核定位信号片段位于所述融合蛋白的n端和/或c端。
[0023]
一些实施方案中,所述核定位信号片段的氨基酸序列如seq id no:9和/或seq id no:11所示。
[0024]
一些实施方案中,所述核定位信号的核苷酸序列如seq id no:10或12所示。
[0025]
一些实施方案中,所述核定位信号片段包括两个拷贝。
[0026]
一些实施方案中,所述融合蛋白的氨基酸序列包含如seq id no:13所示的序列。
[0027]
一些实施方案中,所述融合蛋白用于基因编辑。
[0028]
一些实施方案中,所述基因编辑的编辑窗口为3~10位。
[0029]
一些实施方案中,所述基因编辑的编辑窗口为8~10位。
[0030]
一些实施方案中,所述突变体多肽包含如seq id no:13所示的序列。
[0031]
一些实施方案中,本发明提供了一种编码所述的突变体多肽或所述的融合蛋白的多核苷酸。
[0032]
一些实施方案中,所述多核苷酸为核酸构建体。
[0033]
一些实施方案中,本发明提供了一种载体,所述载体包含所述的多核苷酸。
[0034]
一些实施方案中,所述载体为重组表达载体。
[0035]
一些实施方案中,所述载体骨架选自pcmv或其衍生质粒。
[0036]
一些实施方案中,所述pcmv的衍生质粒包括abemax
‑
spry。
[0037]
一些实施方案中,所述载体包括质粒或病毒载体。
[0038]
一些实施方案中,所述载体是用于在高等真核细胞或原核细胞中表达的质粒或病毒载体。
[0039]
一些实施方案中,所述真核细胞选自脑神经瘤细胞或胚胎肾细胞。
[0040]
一些实施方案中,所述人胚胎肾细胞包括hek293t细胞。
[0041]
一些实施方案中,所述脑神经瘤细胞包括n2a细胞。
[0042]
一些实施方案中,本发明提供了一种产所述载体的方法,在骨架质粒中加入编码spry(d10a)蛋白n端片段的多核苷酸、编码tada8e片段的多核苷酸和编码spry(d10a)蛋白c端片段的多核苷酸,由此获得所述的载体。
[0043]
一些实施方案中,所还载体包括质粒或病毒载体。
[0044]
一些实施方案中,所述载体是用于在高等真核细胞或原核细胞中表达的质粒或病毒载体。
[0045]
一些实施方案中,编码所述spry(d10a)蛋白n端片段的核苷酸序列如seq id no:2所示。
[0046]
一些实施方案中,编码所述tada8e片段的核苷酸序列如seq id no:4所示。
[0047]
一些实施方案中,编码所述spry(d10a)蛋白c端片段的核苷酸序列如seq id no:6所示。
[0048]
一些实施方案中,所述骨架质粒包括pcmv或其衍生质粒abemax
‑
spry。
[0049]
一些实施方案中,所述真核细胞选自脑神经瘤细胞或胚胎肾细胞。
[0050]
一些实施方案中,所述人胚胎肾细胞包括hek293t细胞。
[0051]
一些实施方案中,所述脑神经瘤细胞包括n2a细胞。
[0052]
一些实施方案中,所述方法包括从所述衍生质粒abemax
‑
spry中去除tada片段,并用tada8e替换spry(d10a)中1048位至1063位的氨基酸,构建得所述重组表达载体。
[0053]
一些实施方案中,所述载体为ce
‑
8e
‑
spry质粒。
[0054]
一些实施方案中,本发明提供了一种sgrna。
[0055]
一些实施方案中,所述sgrna的序列包括如seq id no:18
‑
65所示的序列。
[0056]
一些实施方案中,本发明提供了一种表达系统,所述表达系统含有所述的表达载体或其基因组中整合有外源的所述的多核苷酸。
[0057]
一些实施方案中,所述表达系统表达所述的融合蛋白或其基因组中整合的外源序列表达所述的融合蛋白或所述表达系统表达含有所述的多核苷酸或其基因组中整合有外源的如上所述的多核苷酸。
[0058]
一些实施方案中,所述表达系统还含有rna。
[0059]
一些实施方案中,所述rna是引导rna。
[0060]
一些实施方案中,所述rna是sgrna。
[0061]
一些实施方案中,所述sgrna的序列包括如seq id no:18
‑
65所示的序列。
[0062]
一些实施方案中,本发明提供了一种宿主细胞,包含所述的多核苷酸或所述的载
体或所述的表达系统。
[0063]
一些实施方案中,本发明提供了一种组合物,包含所述的突变体多肽,所述的融合蛋白,所述的多核苷酸,所述的载体和所述的宿主细胞中的至少一种,或其任意组合。
[0064]
一些实施方案中,所述组合物为试剂盒。
[0065]
一些实施方案中,所述组合物还含有rna。
[0066]
一些实施方案中,所述rna是引导rna。
[0067]
一些实施方案中,所述rna是sgrna。
[0068]
一些实施方案中,所述sgrna的序列包括如seq id no:18
‑
65所示的序列。
[0069]
一些实施方案中,本发明提供了一种任一所述的突变体多肽或所述的融合蛋白或所述的多核苷酸或所述的载体或所述的表达系统或所述的宿主细胞在制备治疗基因疾病的药物中的应用。
[0070]
一些实施方案中,本发明提供了一种任一所述的突变体多肽或所述的融合蛋白或所述的多核苷酸或所述的载体或所述的表达系统或所述的宿主细胞在制备基因编辑试剂中的应用。
[0071]
一些实施方案中,所述基因编辑的编辑窗口为3~10位。
[0072]
一些实施方案中,所述基因编辑的编辑窗口为8~10位。
[0073]
一些实施方案中,本发明提供了一种碱基编辑系统,包含任一所述的突变体多肽或所述的融合蛋白或所述的多核苷酸或所述的载体或所述的表达系统或所述的宿主细胞。
[0074]
一些实施方案中,所述碱基编辑系统还含有rna。
[0075]
一些实施方案中,所述rna是引导rna。
[0076]
一些实施方案中,所述rna是sgrna。
[0077]
一些实施方案中,本发明提供了一种基因编辑方法,通过所述的碱基编辑系统进行基因编辑。
[0078]
一些实施方案中,所述基因编辑的编辑窗口为3~10位。
[0079]
一些实施方案中,所述基因编辑的编辑窗口为8~10位。
[0080]
一些实施方案中,本发明提供了一种方法,用于重组产生任一所述的突变体多肽或所述的融合蛋白,包括步骤:将所述的载体引入宿主细胞以产生转染的或感染的宿主细胞,体外培养所述转染的或感染的宿主细胞,回收细胞培养物并任选地纯化所产生的突变体多肽或融合蛋白。
[0081]
一些实施方案中,本发明提供了一种所述的突变体多肽或所述的融合蛋白的制备方法,包括:(1)在骨架质粒中加入编码spry(d10a)蛋白n端片段的多核苷酸、编码tada8e片段的多核苷酸和编码spry(d10a)蛋白c端片段的多核苷酸,由此获得重组表达载体;(2)转染所述重组表达载体至宿主细胞使其表达所述突变体多肽或所述融合蛋白。
[0082]
一些实施方案中,编码所述spry(d10a)蛋白n端片段的核苷酸序列如seq id no:2所示。
[0083]
一些实施方案中,编码所述tada8e片段的核苷酸序列如seq id no:4所示。
[0084]
一些实施方案中,编码所述spry(d10a)蛋白c端片段的核苷酸序列如seq id no:6所示。
[0085]
一些实施方案中,所述骨架质粒包括pcmv或其衍生质粒abemax
‑
spry。
[0086]
一些实施方案中,所述方法包括从所述衍生质粒abemax
‑
spry中去除tada二聚体,并用tada8e替换spry(d10a)中1048位至1063位的氨基酸,构建得所述重组表达载体。
[0087]
一些实施方案中,所还载体质粒或病毒载体。
[0088]
一些实施方案中,所述载体是用于在高等真核细胞或原核细胞中表达的质粒或病毒载体。
[0089]
一些实施方案中,所述真核细胞选自脑神经瘤细胞或胚胎肾细胞。
[0090]
一些实施方案中,所述人胚胎肾细胞包括hek293t细胞。
[0091]
一些实施方案中,所述脑神经瘤细胞包括n2a细胞。
[0092]
一些实施方案中,本发明提供了一种产生所述载体的方法,包括步骤:将所述载体引入至适宜的细胞系,在适宜条件下培养所述细胞系从而使所述目的载体能够产生,从所述细胞系的培养物回收所产生的质粒和任选地纯化所述质粒。
[0093]
在一些实施方式中,上述的蛋白为分离的多肽。
[0094]
在一些实施方式中,上述的核酸为分离的多核酸。
附图说明
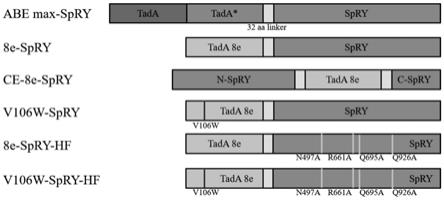
[0095]
图1为abemax
‑
spry和8e
‑
spry及其突变体的示意图。
[0096]
图2
‑
图7为abemax
‑
spry和8e
‑
spry在pam为nnn时的编辑效率。
[0097]
图8为abemax
‑
spry和8e
‑
spry多点编辑效率的统计结果。
[0098]
图9为abemax
‑
spry和8e
‑
spry的编辑窗口。
[0099]
图10
‑
图15为8e
‑
spry及其突变体在pam为nnn时的编辑效率。
[0100]
图16为8e
‑
spry及其突变体多点编辑效率的统计结果。
[0101]
图17为8e
‑
spry及其突变体在pam为nan、ngn、ncn和ntn时多点编辑效率的统计结果。
[0102]
图18为8e
‑
spry及其突变体的编辑窗口。
[0103]
图19为abemax
‑
spry、8e
‑
spry及其突变体的dna靶向编辑效率。
[0104]
图20为abemax
‑
spry、8e
‑
spry及其突变体的rna脱靶量。
[0105]
图21为abemax
‑
spry、8e
‑
spry及其突变体a
‑
to
‑
i的rna脱靶示意图。
[0106]
图22为pku 728g>a细胞模型基因型的sanger测序图以及8种修复sgrna修复效率的sanger测序图。
[0107]
图23为3种有修复效果的sgrna修复效率的柱状图。
[0108]
图24为其他3种abe突变体修复效率的sanger测序图。
具体实施方式
[0109]
以下通过具体的实施例进一步说明本发明的技术方案,具体实施例不代表对本发明保护范围的限制。其他人根据本发明理念所做出的一些非本质的修改和调整仍属于本发明的保护范围。
[0110]
苯丙酮尿症(phenylketonuria,简称pku)是先天代谢性疾病的一种,本症是由于染色体基因突变导致肝脏中苯丙氨酸羟化酶(pah)缺陷从而引起苯丙氨酸(pa)代谢障碍所致。
[0111]
实施例1碱基编辑器质粒的构建
[0112]
首先构建8e
‑
spry以及相应的突变体。参照clonexpress multis one step cloning kit(vazyme,c113
‑
01)说明书设计引物,扩增abe8e(addgene#138489)中的tada8e片段,并用tada8e替代abemax
‑
spry(addgene#140003)中的tada二聚体,构建得到8e
‑
spry质粒。
[0113]
先将8e
‑
spry中的tada8e从原位置删除,再用tada8e替代spry d10a中第1048位至1063位的氨基酸,构建得到ce
‑
8e
‑
spry质粒,从5’端到3’端的顺序依次为spry(d10a)n端、tada8e和spry(d10a)c端,其中spry(d10a)n端的核苷酸序列如seq id no:2(氨基酸序列如seq id no:1所示),tada8e的核苷酸序列如seq id no:4(氨基酸序列如seq id no:3所示),spry(d10a)c端的核苷酸序列如seq id no:6(氨基酸序列如seq id no:5所示)。
[0114]
将8e
‑
spry中的tada8e进行v106w突变,得到v106w
‑
spry,其中tada8e v106w核苷酸序列如seq id no:15,spry d10a核苷酸序列如seq id no:16。
[0115]
将8e
‑
spry中的spry d10a进行n497a,r661a,q695a,和q926a突变,得到8e
‑
spry
‑
hf,其中spry d10a
‑
hf核苷酸序列如seq id no:17。
[0116]
将8e
‑
spry
‑
hf中的tada8e进行v106w突变,得到v106w
‑
spry
‑
hf。
[0117]
8e
‑
spry及其突变体两端均携带核定位信号,核定位信号为bpnls(核定位信号的核苷酸序列如seq id no:10所示;氨基酸序列如seq id no:9所示)或sv40nls(核定位信号的核苷酸序列如seq id no:12所示;氨基酸序列如seq id no:11所示)。8e
‑
spry及其突变体具体图示如图1所示。
[0118]
(1)abemax
‑
spry(融合蛋白)
[0119]
所述abemax
‑
spry(融合蛋白)的氨基酸序列如seq id no:67所示;其组成从n端到c端的顺序依次为bpnls、tada二聚体、spry d10a和bpnls。一些实施例中,两端携带核定位信号也可为sv40nls。
[0120]
(2)8e
‑
spry(融合蛋白)
[0121]
所述8e
‑
spry(融合蛋白)的氨基酸序列如seq id no:68所示;其组成从n端到c端的顺序依次为bpnls、tada8e、spry d10a和bpnls。一些实施例中,两端携带核定位信号也可为sv40nls。
[0122]
(3)ce
‑
8e
‑
spry(融合蛋白)
[0123]
所述ce
‑
8e
‑
spry(融合蛋白)的氨基酸序列如seq id no:13所示(ce
‑
8e
‑
spry融合蛋白的核苷酸序列如seq id no:14所示),其组成为:自n端至c端依次包括bpnls、spry(d10a)的n端片段、tada8e片段和spry(d10a)多肽的c端片段和bpnls,在spry(d10a)n端片段与tada8e片段之间、tada8e片段与spry(d10a)c端片段之间具有连接肽,所述连接肽的氨基酸序列如seq id no:7所示(编码ce
‑
8e
‑
spry连接肽的核苷酸序列如seq id no:8所示)。一些实施例中,两端携带核定位信号也可为sv40nls。
[0124]
(4)v106w
‑
spry(融合蛋白)
[0125]
所述v106w
‑
spry(融合蛋白)的氨基酸序列如seq id no:69所示;其组成从n端到c端的顺序依次为bpnls、tada8ev106w、spry d10a、和bpnls,两端携带核定位信号也可为sv40nls。
[0126]
(5)8e
‑
spry
‑
hf(融合蛋白)
[0127]
所述8e
‑
spry
‑
hf(融合蛋白)的氨基酸序列如seq id no:70所示;其组成从n端到c端的顺序依次为bpnls、tada8e、spry d10a
‑
hf、和bpnls,两端携带核定位信号也可为sv40nls。
[0128]
(6)v106w
‑
spry
‑
hf
[0129]
所述v106w
‑
spry
‑
hf(融合蛋白)的氨基酸序列如seq id no:71所示;其组成从n端到c端的顺序依次为bpnls、tada8ev106w、spry d10a
‑
hf、和bpnls,两端携带核定位信号也可为sv40nls。
[0130]
实施例2
[0131]
在本实施例中,利用abemax
‑
spry、8e
‑
spry及其突变体在293t细胞中进行内源位点的编辑。
[0132]
2.1sgrna质粒的构建
[0133]
参考人基因组序列,根据spry核酸酶的pam特征设计48条sgrna,涵盖16种不同的pam序列,sgrna序列如seq id no:18
‑
65所示,sgrna序列5’端加入accg为上游序列,sgrna反向互补序列的5’端加入aaac为下游序列,合成oligo后上下游序列退火(程序为:95℃,5min;95℃
‑
85℃at
‑
2℃/s;85℃
‑
25℃at
‑
0.1℃/s;hold at 16℃)后与经过bsai(neb:r3733l)酶切后的pgl3
‑
u6
‑
sgrna(addgene#51133)载体连接。酶切体系为:pgl3
‑
u6
‑
sgrna2μg;cutsmart buffer(neb:b7204s)6μl;bsai 1μl;ddh2o补齐到60μl,37℃酶切过夜。连接体系为:solution i(takara:6022q)3μl;酶切后载体1μl;退火产物6μl,16℃连接30min后转化、挑菌、鉴定。对阳性克隆菌摇菌提取质粒(axygene:ap
‑
mn
‑
p
‑
250g)、测定浓度后备用。
[0134]
2.2细胞培养与转染
[0135]
hek293t细胞(购自atcc)接种培养于添加10%血清(gibco:10270
‑
106)的dmem培养基中(gibco:c11995500bt),其中含1%双抗(v/v)(gibco:15140122)。转染前一天铺24孔板,使转染时的细胞密度达到80%左右,转染前2h换液。每孔转染的质粒量分别为碱基编辑器质粒600ng,sgrna质粒(sgrna1
‑
48的序列如seq id no:18~seq id no:65所示)300ng,将质粒稀释于40μl的dmem中,将3μl的ez trans细胞转染试剂(上海李记生物:ac04l092)稀释于40μl的dmem中,最后将稀释好的ez转染试剂加入到稀释好的质粒中,混匀后室温静置15min。将混有质粒和ez的dmem加入24孔板中,6h后用含有10%血清的完全培养基换液,转染48h后显微镜观察绿色荧光蛋白(green fluorescent protein,gfp)的表达,流式细胞分选仪分选gfp阳性细胞。
[0136]
其中gfp是pgl3
‑
u6
‑
sgrna载体上的。
[0137]
2.3检测编辑效率
[0138]
将分选得到的gfp阳性细胞离心去上清后,加入裂解液(50mm kcl,1.5mm mgcl2,10mm tris ph 8.0,0.5%nonidet p
‑
40,0.5%tween 20,100μg/ml protease k),以gfp阳性细胞裂解液为模板,扩增靶向序列,扩增体系为:2
×
buffer(vazyme:p505)25μl;dntp 1μl;forward primer(10μmol/l)1μl;reverse primer(10pmol/l)1μl;细胞裂解产物1μl;dna聚合酶(vazyme:p505)0.5μl;ddh2o补齐到50μl。forward primer和reverse primer序列如seq id no:72~seq id no:167所示(分别对应sgrna1
‑
48)。
[0139]
扩增出来的pcr产物用回收试剂盒进行纯化(axygen:ap
‑
pcr
‑
250g),具体步骤为:扩增产物中加入3倍体积的pcr
‑
a,混匀后加入吸附柱中,12000r/min离心1min;弃废液,吸
附柱中加入700μl w2(需加入指定体积的乙醇),12000r/min离心1min;弃废
‑
液,吸附柱中加入400μl w2(需加入指定体积的乙醇),12000r/min离心1min;弃废液,12000r/min离心2min;开盖晾干乙醇后,加入28μl ddh2o,12000r/min离心1min进行洗脱,纯化后的pcr产物送sanger测序或深度检测,分析编辑效果。
[0140]
相关结果如图2
‑
9所示。结果显示在所有检测位点上,涵盖nan、ngn、ncn和ntn的pam,8e
‑
spry的编辑效率均明显高于abemax
‑
spry;图8的多点编辑效率的统计结果显示,8e
‑
spry显著改善了a到g的编辑效率。图9的编辑窗口结果显示,abemax
‑
spry碱基编辑窗口为5
‑
6位;8e
‑
spry碱基编辑窗口为3
‑
10位,窗口更宽。
[0141]
图10
‑
15显示在nrn(r代表a或g)、nyn(y代表c或t)pam下,8e
‑
spry突变体编辑效率的对比结果;将8e插入到spry中间的ce
‑
8e
‑
spry可以很好的维持其a
‑
to
‑
g的编辑活性,将v106w引入tad8e的v106w
‑
spry同样未明显损伤原来的编辑活性,但是将4种突变引入spry的8e
‑
spry
‑
hf和v106w
‑
spry
‑
hf则使编辑活性明显下降。
[0142]
图16的多点编辑效率的统计结果显示,8e
‑
spry
‑
hf和v106w
‑
spry
‑
hf显著降低了活性,ce
‑
8e
‑
spry编辑效率提高但无显著性差异,v106w
‑
spry编辑效率降低同样无显著性差异。
[0143]
图17在nan、ngn、ncn和ntn的多点编辑效率的统计结果显示,ce
‑
8e
‑
spry在ngn和ntn的编辑效率有所提高,v106w
‑
spry在4种pam下的编辑效率均有所下降,但均无统计学意义。图18的编辑窗口结果显示,v106w
‑
spry维持与8e
‑
spry相同的编辑窗口,均为3
‑
10位,高活编辑窗口(编辑效率大于40%)为3
‑
9位;ce
‑
8e
‑
spry维持相同的编辑窗口即3
‑
10位,高活编辑窗口(编辑效率大于40%)为3
‑
10位,且在8
‑
10位的编辑效率高于8e
‑
spry。
[0144]
表1.实施例2中转染细胞所用的质粒组合(1)
[0145]
[0146][0147]
表2.实施例2中转染细胞所用的质粒组合(2)
[0148]
[0149]
[0150][0151]
实施例3
[0152]
在本实施例中,对比abemax
‑
spry、8e
‑
spry及其突变体在293t细胞中的rna脱靶情况。
[0153]
3.1sgrna的构建
[0154]
用于rna脱靶检测的sgrna序列为5
’‑
ctggaacacaaagcatagac
‑‘
3(seq id no:66),按照2.1所述的质粒构建方法进行构建。
[0155]
3.2细胞培养与转染
[0156]
细胞培养按照2.2所述进行,转染前一天用293t细胞铺6cm dish,使转染时的细胞密度达到80%左右。每皿转染的质粒量为碱基编辑器质粒4μg,sgrna质粒2μg,将质粒稀释于250μl的dmem中,将18μl的ez trans细胞转染试剂(上海李记生物:ac04l092)稀释于250μl的dmem中,最后将稀释好的ez转染试剂加入到稀释好的质粒中,混匀后室温静置15min。将混有质粒和ez的dmem加入6cm dish中,6h后用含有10%血清的完全培养基换液(dmem 10%fbs),转染48h后显微镜观察gfp(gfp是pgl3
‑
u6
‑
sgrna载体上的)表达,流式细胞分选仪分选gfp阳性细胞。取少数阳性细胞按照2.3所述检测编辑效率,其余阳性细胞提取rna后送rna
‑
seq。
[0157]
3.3rna提取
[0158]
gfp阳性细胞3000r/min离心10min后弃上清,加入1ml rnaisolater total rna extraction reagent(vazyme:r401
‑
01
‑
aa)充分裂解细胞;加入200μl氯仿,上下剧烈混匀,室温放置3min后,4℃12000r/min离心15min;取上层水相500μl,加入500μl的异丙醇,上下颠倒混匀,4℃12000r/min离心15min;吸弃上清,加入1ml 75%的乙醇,轻轻颠倒几次清洗沉淀,4℃12000r/min离心5min;吸弃上清,开盖干燥5
‑
10min,待乙醇完全挥发后,加入15μl rnase
‑
free水溶解沉淀,取1μl测浓度。取1μg rna送rna
‑
seq。
[0159]
相关结果如19
‑
21所示。图19显示在dna靶向位点上的第8位a的编辑效率,abemax
‑
spry、8e
‑
spry及其突变体均能引起有效的编辑,其中8e
‑
spry及其突变体引起的dna靶向编辑效率相当,abemax
‑
spry的编辑效率相对较低。图20和图21的rna脱靶结果显示,相对于abemax
‑
spry和8e
‑
spry的其他突变体,ce
‑
8e
‑
spry有效降低了在转录组水平的脱靶编辑。
[0160]
综合编辑效率检测和脱靶检测结果,发明人获得的ce
‑
8e
‑
spry碱基编辑器可以靶向全基因,同时显著提高了a
‑
to
‑
g的编辑效率,并且有效降低了在转录组水平上的脱靶编辑,具有很大的应用潜力。
[0161]
表3.实施例3中转染细胞所用的质粒组合
[0162][0163]
实施例4 ce
‑
8e
‑
spry在修复疾病致病位点中的应用
[0164]
4.1构建人pah 728g>a细胞模型
[0165]
4.1.1突变mut
‑
sgrna构建
[0166]
参考人基因组序列,设计mut
‑
sgrna(seq id no:168),按照2.1所述的质粒构建方法进行构建。
[0167]
4.1.2细胞培养与转染
[0168]
细胞培养按照2.2所述进行,转染前一天铺24孔板,使转染时的细胞密度达到80%左右,转染前2h换液。每孔转染的质粒量分别为碱基编辑器质粒600ng,sgrna质粒300ng,将质粒稀释于40μl的dmem中,将3μl的ez trans细胞转染试剂(上海李记生物:ac04l092)稀释于40μl的dmem中,最后将稀释好的ez转染试剂加入到稀释好的质粒中,混匀后室温静置15min。将混有质粒和ez的dmem加入24孔板中,6h后用含有10%血清的完全培养基换液,转染48h后流式细胞分选仪分选gfp阳性细胞到96孔板中,每孔分选1个阳性细胞,放置于培养箱培养14天后鉴定细胞单克隆基因型。
[0169]
4.1.3单克隆细胞基因型鉴定
[0170]
每孔的单克隆细胞取部分细胞离心后加入裂解液(50mm kcl,1.5mm mgcl2,10mm tris ph 8.0,0.5%nonidet p
‑
40,0.5%tween 20,100μg/ml protease k),以细胞裂解液为模板,扩增靶向序列,扩增体系为:2
×
buffer(vazyme:p505)25μl;dntp 1μl;forward primer(10μmol/l)1μl;reverse primer(10pmol/l)1μl;细胞裂解产物1μl;dna聚合酶(vazyme:p505)0.5μl;ddh2o补齐到50μl。forward primer序列为:5
’‑
gtccctgggcagttatgtgtac
‑3’
(seq id no:177),reverse primer序列为5
’‑
caactggtagctggaggacag
‑3’
(seq id no:178)。扩增产物送sanger测序后挑选pah 728g>a纯和突变的细胞即为人pah 728g>a细胞模型。
[0171]
4.2修复pah 728g>a突变
[0172]
ce
‑
8e
‑
spry在3
‑
10位有较高的编辑效率,且识别pam为nnn,根据ce
‑
8e
‑
spry的编辑窗口和pam特点,发明人围绕需要修复的致病突变设计了8条rec
‑
sgrna(seq id no:169
‑
~seq id no:176),并按照2.1所述的质粒构建方法进行构建。按照2.2所述的细胞培养与转染方法进行转染。按照2.3所述的检测编辑效率的方法进行修复效率的检测。
[0173]
结果如图22和图23所示:mut
‑
sgrna成功造成728g>a的纯和突变;8条rec
‑
sgrna中,rec
‑
sgrna1(即图22和图23中的sg1)对728g>a的修复效率最高,rec
‑
sgrna2(即图22和图23中的sg2)和rec
‑
sgrna3(即图22和图23中的sg3)具有微弱的修复效果。
[0174]
根据x
‑
abemax,abemax
‑
ng和abemax
‑
spry的pam特点和编辑窗口,这3种碱基编辑器的修复sgrna为seq id no:173,按照2.1所述的质粒构建方法进行构建,按照2.2所述的
细胞培养与转染方法进行转染,按照2.3所述的检测编辑效率的方法进行修复效率的检测。结果如图24所示,3种碱基编辑器对728g>a的突变位点均无明显修复效果。
[0175]
本实施例说明由于ce
‑
8e
‑
spry识别的pam为nnn,围绕需要修复的位点有多种sgrna的选择,可经过sgrna的筛选选择最符合修复要求的sgrna,有效提高了可修复位点的范围和修复效果的灵活性。另外,现有的3种碱基编辑器在各自的编辑窗口内均无法修复728g>a的突变位点,发明人提供的ce
‑
8e
‑
spry在突变位点位于编辑窗口第10位时可实现有效的编辑,拓展了现有碱基编辑工具的可编辑范围,显示出独特的编辑特性。
[0176]
参考文献
[0177]
1.jinek m,chylinski k,fonfara i,et al.a programmable dual
‑
rna
‑
guided dna endonuclease in adaptive bacterial immunity.science.2012;337(6096):816
‑
21.
[0178]
2.komor ac,kim yb,packer ms,et al.programmable editing of a target base in genomic dna without double
‑
stranded dna cleavage.nature.2016;533(7603):420
‑
4.
[0179]
3.gaudelli nm,komor ac,rees ha,et al.programmable base editing of a*t to g*c in genomic dna without dna cleavage.nature.2017;551(7681):464
‑
471.
[0180]
4.rees ha and liu dr.publisher correction:base editing:precision chemistry on the genome and transcriptome of living cells.nat rev genet.2018;19(12):801.
[0181]
5.ryu sm,koo t,kim k,et al.adenine base editing in mouse embryos and an adult mouse model of duchenne muscular dystrophy.nat biotechnol.2018;36(6):536
‑
539.
[0182]
6.song cq,jiang t,richter m,et al.adenine base editing in an adult mouse model of tyrosinaemia.nat biomed eng.2020;4(1):125
‑
130.
[0183]
7.huang tp,zhao kt,miller sm,et al.circularly permuted and pam
‑
modified cas9 variants broaden the targeting scope of base editors.nat biotechnol.2019;37(6):626
‑
631.
[0184]
8.walton rt,christie ka,whittaker mn,et al.unconstrained genome targeting with near
‑
pamless engineered crispr
‑
cas9 variants.science.2020;368(6488):290
‑
296.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。