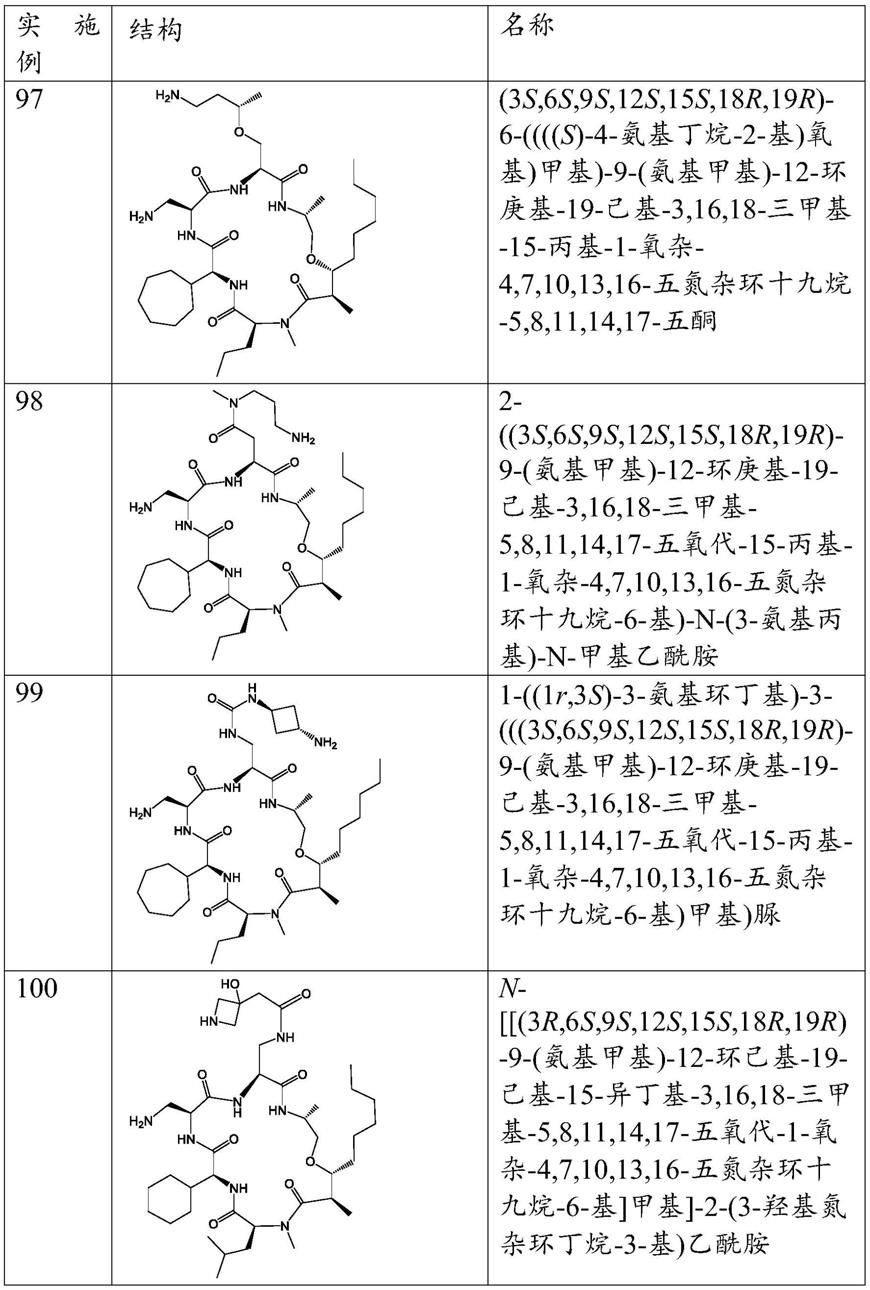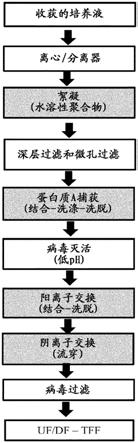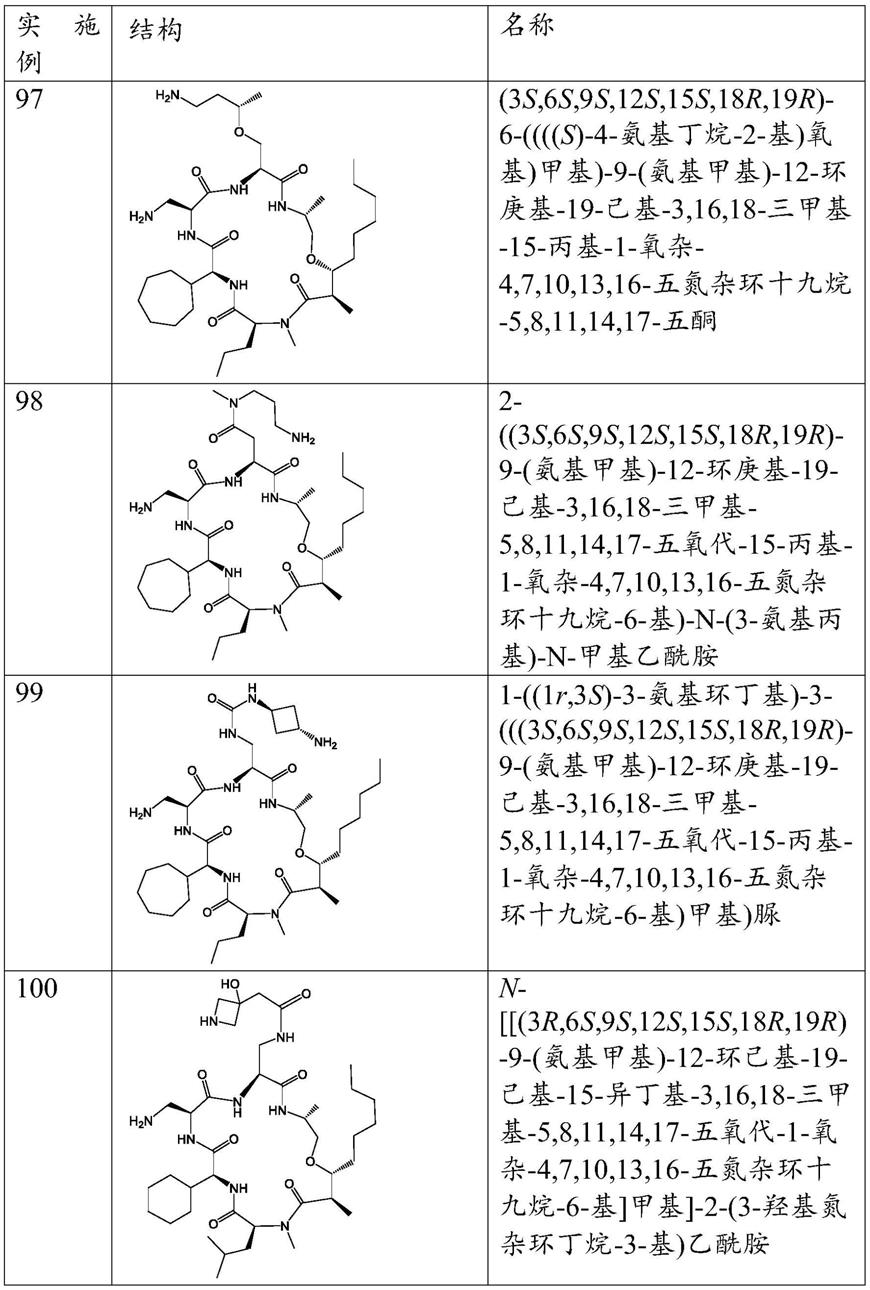
环肽抗生素
1.相关申请的交叉引用
2.本技术享有于2019年3月14日提交的优先权申请pctcn/2019/078189的权益,其公开内容以引用方式并入本文。
背景技术:
3.抗生素耐药性是当代医学中一种严重且日益严重的现象,并已成为21世纪的主要公共卫生问题。因此,需要新型广谱抗生素,尤其是针对新型作用机制的抗生素,来治疗多重耐药病原体。ii型信号肽酶lspa在革兰氏阴性细菌脂蛋白的生物合成中起关键作用。通过抑制lspa功能来阻断脂蛋白合成,不仅会消耗必需的以及毒力相关的脂蛋白,而且可以导致错误定位的未加工脂蛋白的积累,所有这些都会导致细菌细胞死亡。因此,lspa抑制剂提供了一种对抗革兰氏阴性细菌感染的新方法。
技术实现要素:
4.本文描述了用于治疗微生物感染,诸如用于治疗细菌感染的新型大环化合物。在各种实施例中,本公开提供用于治疗细菌感染的环肽、缩肽和含肽化合物。在各种实施例中,本公开提供了多个类别和亚类的结构上与用于治疗细菌感染的球霉素相关的化合物。在各种实施例中,环肽、缩肽和含肽化合物通过抑制脂蛋白信号肽酶ii(lspa)(一种参与细菌中脂蛋白翻译后加工的关键酶)起作用。
5.本文公开了一种式(i)化合物或其药用盐、溶剂化物或立体异构体:
[0006][0007]
其中:
[0008]
r
1a
、r
2a
、r
4a
、r
5a
和r
6a
各自独立地为氢或任选地经取代的c1‑
c6烷基;
[0009]
r
3a
为氢、任选地经取代的c1‑
c
20
烷基、任选地经取代的c2‑
c
20
烯基、任选地经取代的c2‑
c
20
炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的
(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0010]
r
20
为羟基或
‑
nr1r2;
[0011]
r1和r2各自独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基、任选地经取代的(c1‑
c6烷基)杂芳基、
‑
s(=o)2r
a
、
‑
s(=o)2nr
b
r
c
、
‑
c(=o)r
a
、
‑
c(=o)or
b
、
‑
c(=o)nr
b
r
c
或
‑
(c=nr
b
)nr
b
r
c
;
[0012]
或者r1和r2与它们所连接的氮原子一起形成任选地经取代的杂环烷基;
[0013]
r3和r4各自独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0014]
或者r3和r4与它们所连接的碳原子一起形成氧代;
[0015]
r5为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基或任选地经取代的c2‑
c6炔基;
[0016]
r6和r7各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0017]
r8和r9各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0018]
或者r
3a
和r8与它们所连接的原子一起形成任选地经取代的杂环烷基;
[0019]
r
10
和r
11
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c
20
烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0020]
r
12
和r
13
各自独立地为氢、卤素、任选地经取代的c1‑
c
20
烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0021]
r
14
和r
15
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷
基)杂芳基;
[0022]
r
16
和r
17
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0023]
每个r
a
独立地为任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0024]
每个r
b
和r
c
独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0025]
或者r
b
和r
c
与它们所连接的氮原子一起形成任选地经取代的杂环烷基。
[0026]
本文还公开了一种式(ia)化合物或其药用盐、溶剂化物或立体异构体:
[0027][0028]
本文还公开了一种式(ib)化合物或其药用盐、溶剂化物或立体异构体:
[0029][0030]
本文还公开了一种式(ii)化合物或其药用盐、溶剂化物或立体异构体:
烷基)杂芳基;
[0042]
r
12
和r
13
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0043]
或者r
10
和r
12
一起形成任选地经取代的环烷基或任选地经取代的环烯基;
[0044]
r
14
和r
15
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0045]
或者r
14
和r
4a
与它们所连接的原子一起形成任选地经取代的杂环烷基;
[0046]
r
16
和r
17
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0047]
r
18
和r
19
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0048]
或者r
18
和r
19
与它们所连接的碳原子一起形成氧代;
[0049]
每个r
a
独立地为任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0050]
每个r
b
和r
c
独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0051]
或者r
b
和r
c
与它们所连接的氮原子一起形成任选地经取代的杂环烷基;
[0052]
或其药用盐、溶剂化物或立体异构体;
[0053]
其中所述化合物选自由以下项组成的组:
[0054]
[0055]
[0056]
[0057][0058]
本发明还公开了一种药物组合物,该药物组合物包含本文公开的化合物或其药用盐、溶剂化物或立体异构体,以及药用赋形剂。
[0059]
本文还公开了本文公开的化合物或其药用盐、溶剂化物或立体异构体在制备用于治疗患者细菌感染的药物中的用途。
[0060]
本文还公开了一种治疗哺乳动物细菌感染的方法,包括以足以为该哺乳动物提供有益效果的频率和持续时间向该哺乳动物施用有效量的本文公开的化合物或其药用盐、溶
剂化物或立体异构体。
[0061]
以引用方式并入
[0062]
本说明书中提到的所有出版物、专利和专利申请都以引用方式并入本文,所达到的程度如同每个单独的出版物、专利或专利申请都被具体地和单独地指出以引用方式并入。
具体实施方式
[0063]
定义
[0064]
如在本文和所附权利要求中所用,单数形式“一”、“一个/种”和“该/所述”包括复数指代物,除非上下文另外明确规定。因此,例如,提及“一种试剂”包括多种此类试剂,并且提及“该/所述细胞”包括提及本领域技术人员已知的一个或多个细胞(或复数细胞)及其等同物,等等。当范围在本文中用于诸如分子量的物理特性或诸如化学式的化学特性时,旨在将范围的所有组合和子组合以及其中的具体实施例包括在内。当提及数字或数值范围时,术语“大约”表示所指的数字或数值范围是实验变异性(或统计实验误差)内的近似值,因此在某些情况下,数字或数值范围将在规定数量或数值范围的1%到15%之间变化。术语“包含”(以及相关术语,诸如“包含”或“含有”或“具有”或“包括”)并非旨在排除下述情况:在其他某些实施例中,例如,本文所述的任何物质的组成、组合物、方法或过程等“由”或“基本上由”所描述的特征“组成”。
[0065]
如在说明书和所附权利要求中使用的,除非另有说明,否则以下术语具有如下所示的含义。
[0066]“烷基”是指具有1至约10个碳原子或1至6个碳原子的任选地经取代的直链或任选地经取代的支链饱和烃单价基团,其中烷基残基的sp3杂化碳通过单键连接到分子的其余部分。实例包括但不限于甲基、乙基、正丙基、异丙基、2
‑
甲基
‑1‑
丙基、2
‑
甲基
‑2‑
丙基、2
‑
甲基
‑1‑
丁基、3
‑
甲基
‑1‑
丁基、2
‑
甲基
‑3‑
丁基、2,2
‑
二甲基
‑1‑
丙基、2
‑
甲基
‑1‑
戊基、3
‑
甲基
‑1‑
戊基、4
‑
甲基
‑1‑
戊基、2
‑
甲基
‑2‑
戊基、3
‑
甲基
‑2‑
戊基、4
‑
甲基
‑2‑
戊基、2,2
‑
二甲基
‑1‑
丁基、3,3
‑
二甲基
‑1‑
丁基、2
‑
乙基
‑1‑
丁基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、叔戊基和己基,以及更长的烷基基团诸如庚基、辛基等。每当在本文中出现时,诸如“c1‑
c6烷基”的数字范围是指该烷基由1个碳原子、2个碳原子、3个碳原子、4个碳原子、5个碳原子或6个碳原子组成,但本定义也涵盖未指定数字范围的术语“烷基”的出现。在一些实施例中,烷基是c1‑
c
20
烷基、c1‑
c
10
烷基、c1‑
c9烷基、c1‑
c8烷基、c1‑
c7烷基、c1‑
c6烷基、c1‑
c5烷基、c1‑
c4烷基、c1‑
c3烷基、c1‑
c2烷基或c1烷基。除非在说明书中另有具体说明,否则烷基任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,烷基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,烷基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,烷基任选地经卤素取代。
[0067]“烯基”是指具有一个或多个碳
‑
碳双键并具有2至约10个碳原子,更优选2至约6个碳原子的任选地经取代的直链或任选地经取代的支链烃单价基团,其中烯基残基的sp2杂化碳通过单键连接到分子的其余部分。该基团可以是关于一个或多个双键的顺式或反式构象,并且应当理解为包括两种异构体。实例包括但不限于乙烯基(
‑
ch=ch2)、1
‑
丙烯基(
‑
ch2ch=ch2)、异丙烯基[
‑
c(ch3)=ch2]、丁烯基、1,3
‑
丁二烯基等。每当在本文中出现时,诸如“c2‑
c6烯基”的数字范围是指该烯基可以由2个碳原子、3个碳原子、4个碳原子、5个碳原子或6个碳原子组成,但本定义也涵盖未指定数字范围的术语“烯基”的出现。在一些实施例中,烯基是c2‑
c
20
烯基、c2‑
c
10
烯基、c2‑
c9烯基、c2‑
c8烯基、c2‑
c7烯基、c2‑
c6烯基、c2‑
c5烯基、c2‑
c4烯基、c2‑
c3烯基或c2烯基。除非在说明书中另有具体说明,否则烯基任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,烯基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,烯基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,烯基任选地经卤素取代。
[0068]“炔基”是指具有一个或多个碳
‑
碳三键并具有2至约10个碳原子,更优选2至约6个碳原子的任选地经取代的直链或任选地经取代的支链烃单价基团。实例包括但不限于乙炔基、2
‑
丙炔基、2
‑
丁炔基、1,3
‑
丁二炔基等。每当在本文中出现时,诸如“c2‑
c6炔基”的数字范围是指该炔基可以由2个碳原子、3个碳原子、4个碳原子、5个碳原子或6个碳原子组成,但本定义也涵盖未指定数字范围的术语“炔基”的出现。在一些实施例中,炔基是c2‑
c
20
炔基、c2‑
c
10
炔基、c2‑
c9炔基、c2‑
c8炔基、c2‑
c7炔基、c2‑
c6炔基、c2‑
c5炔基、c2‑
c4炔基、c2‑
c3炔基或c2炔基。除非在说明书中另有具体说明,否则炔基任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,炔基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,炔基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,炔基任选地经卤素取代。
[0069]“亚烷基”是指直链或支链二价烃链。除非在说明书中另有具体说明,否则亚烷基可以任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,亚烷基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,亚烷基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,亚烷基任选地经卤素取代。
[0070]“烷氧基”是指式
‑
or
a
的基团,其中r
a
是如所定义的烷基。除非在说明书中另有具体说明,否则烷氧基可以任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,烷氧基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,烷氧基任选地经氧代、卤素、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,烷氧基任选地经卤素取代。
[0071]“芳基”是指衍生自包含氢、6至30个碳原子和至少一个芳环的烃环系统的基团。芳基可以是单环、双环、三环或四环的环系,其可以包括稠合的(当与环烷基或杂环烷基环稠合时,芳基通过芳环原子键合)或桥接的环系。在一些实施例中,芳基为6元至10元芳基。在一些实施例中,芳基为6元芳基。芳基基团包括但不限于,衍生自亚蒽基、亚萘基、亚菲基、蒽、甘菊环、苯、荧蒽、芴、不对称引达省、对称
‑
引达省、茚满、茚、萘、非那烯(phenalene)、菲、七曜烯(pleiadene)、芘和三亚苯的烃环系统的芳基基团。在一些实施例中,芳基为苯基。除非在说明书中另有具体说明,否则芳基可以任选地如下所述经取代,例如经卤素、氨基、腈、硝基、羟基、烷基、烯基、炔基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,芳基任选地经卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,芳基任选地经卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,芳基任选地经卤素或甲基取代。在一些实施例中,芳基任选地经卤素取代。
[0072]“环烷基”是指稳定的、部分或完全饱和的单环或多环碳环,其可包括稠合(当与芳基或杂芳基环稠合时,环烷基通过非芳族环原子键合)或桥环系统.代表性的环烷基包括但不限于,具有三个至十五个碳原子的环烷基(c3‑
c
15
环烷基)、具有三个至十个碳原子的环烷基(c3‑
c
10
环烷基)、具有三个至八个碳原子的环烷基(c3‑
c8环烷基)、具有三个至六个碳原子的环烷基(c3‑
c6环烷基)、具有三个至五个碳原子的环烷基(c3‑
c5环烷基)或具有三个至四个碳原子的环烷基(c3‑
c4环烷基)。在一些实施例中,环烷基为3元至6元环烷基。在一些实施例中,环烷基为5元至6元环烷基。单环环烷基包括例如环丙基、环丁基、环戊基、环己基、环庚基和环辛基。多环环烷基或碳环包括例如金刚烷基、降冰片基、十氢萘基、双环[3.3.0]辛烷、双环[4.3.0]壬烷、顺式
‑
十氢萘、反式
‑
十氢萘、双环[2.1.1]己烷、双环[2.2.1]庚烷、双环[2.2.2]辛烷、双环[3.2.2]壬烷、双环[3.3.2]癸烷和7,7
‑
二甲基
‑
双环[2.2.1]庚烷基。部分饱和的环烷基包括例如环戊烯基、环己烯基、环庚烯基和环辛烯基。除非在说明书中另有具体说明,否则环烷基任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、烷基、烯基、炔基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,环烷基任选地经氧代、卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,环烷基任选地经氧代、卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,环烷基任选地经卤素或甲基取代。在一些实施例中,环烷基任选地经卤素取代。
[0073]“卤代”或“卤素”是指溴、氯、氟或碘。在一些实施例中,卤素是氟或氯。在一些实施例中,卤素是氟。
[0074]“卤代烷基”是指经一个或多个如上文所定义的卤代基团取代的如上文所定义的烷基基团,例如三氟甲基、二氟甲基、氟甲基、三氯甲基、2,2,2
‑
三氟乙基、1,2
‑
二氟乙基、3
‑
溴
‑2‑
氟丙基、1,2
‑
二溴乙基等。
[0075]“杂环烷基”是指稳定的3
‑
至24
‑
元部分或完全饱和的环基团,其包含2至23个碳原子和1至8个选自由氮、氧、磷和硫组成的组的杂原子。除非说明书中另有具体说明,否则杂环烷基基团可以是单环、双环、三环或四环的环系,其可以包括稠合(当与芳基或杂芳基环稠合时,杂环烷基通过非芳族环原子键合)或桥环系统;并且杂环烷基基团中的氮、碳或硫原子可任选地氧化;氮原子可以任选地季铵化。在一些实施例中,杂环烷基为3元至6元杂环烷基。在一些实施例中,杂环烷基为5元至6元杂环烷基。此类杂环烷基的实例包括但不限于氮丙啶基、氮杂环丁烷基、二氧戊环基、噻吩基[1,3]二噻吩基、十氢异喹啉基、咪唑啉基、咪唑烷基、异噻唑烷基、异噁唑烷基、吗啉基、八氢吲哚基、八氢异吲哚基、2
‑
氧代哌嗪基、2
‑
氧代哌啶基、2
‑
氧代吡咯烷基、噁唑烷基、哌啶基、哌嗪基、4
‑
哌啶酮基、吡咯烷基、吡唑烷基、奎宁环基、噻唑烷基、四氢呋喃基、三噻烷基、四氢吡喃基、硫代吗啉基、噻吗啉基、1
‑
氧代
‑
硫代吗啉基、1,1
‑
二氧代
‑
硫代吗啉基、1,3
‑
二氢异苯并呋喃
‑1‑
基、3
‑
氧代
‑
1,3
‑
二氢异苯并呋喃
‑1‑
基、甲基
‑2‑
氧代
‑
1,3
‑
二氧杂环戊烯
‑4‑
基和2
‑
氧代
‑
1,3
‑
二氧杂环戊烯
‑4‑
基。术语杂环烷基还包括所有环形式的碳水化合物,包括但不限于单糖、二糖和寡糖。除非另有说明,否则杂环烷基在环中具有2至10个碳。应理解,当提及杂环烷基中的碳原子数时,杂环烷基中的碳原子数与构成杂环烷基的原子总数(包括杂原子)(即杂环烷基环的骨架原子)
不同。除非在说明书中另有具体说明,否则杂环烷基任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、烷基、烯基、炔基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,杂环烷基任选地经氧代、卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,杂环烷基任选地经氧代、卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,杂环烷基任选地经卤素或甲基取代。在一些实施例中,杂环烷基任选地经卤素取代。
[0076]“杂烷基”是指一种烷基,其中烷基的一个或多个骨架原子选自碳以外的原子,例如氧、氮(例如
‑
nh
‑
、
‑
n(烷基)
‑
)、硫或其组合。杂烷基在杂烷基的碳原子处连接到分子的其余部分。一方面,杂烷基是c1‑
c6杂烷基。除非在说明书中另有具体说明,否则杂烷基任选地如下所述经取代,例如经氧代、卤素、氨基、腈、硝基、羟基、烷基、烯基、炔基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,杂烷基任选地经氧代、卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,杂烷基任选地经氧代、卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,杂烷基任选地经卤素取代。
[0077]“杂芳基”是指包含氢原子、1至13个碳原子、1至6个选自由氮、氧、磷和硫组成的组的杂原子和至少一个芳环的5
‑
至14
‑
元环系统基团。杂芳基基团可以是单环、双环、三环或四环的环系,其可以包括稠合(当与环烷基或杂环烷基环稠合时,杂芳基通过芳族环原子键合)或桥环系统;并且杂环烷基集团中的氮、碳或硫原子可任选地氧化;氮原子可以任选地季铵化。在一些实施例中,杂芳基为5元至10元杂芳基。在一些实施例中,杂芳基为5元至6元杂芳基。实例包括但不限于,吖庚因基、吖啶基、苯并咪唑基、苯并噻唑基、苯并吲哚基、苯并二氧杂环戊烯基、苯并呋喃基、苯并噁唑基、苯并噻唑基、苯并噻二唑基、苯并[b][1,4]二氧杂环庚基、1,4
‑
苯并二氧杂环己基、苯并萘并呋喃基、苯并噁唑基、苯并二氧杂环戊烯基、苯并二噁英基、苯并吡喃基、苯并吡喃酮基、苯并呋喃基、苯并呋喃酮基、苯并噻吩基(benzothienyl)(苯并噻吩基(benzothiophenyl))、苯并三唑基、苯并[4,6]咪唑并[1,2
‑
a]吡啶基、咔唑基、噌啉基、二苯并呋喃基、二苯并噻吩基、呋喃基、呋喃酮基、异噻唑基、咪唑基、吲唑基、吲哚基、吲唑基、异吲哚基、吲哚啉基、异吲哚啉基、异喹啉基、吲哚嗪基、异噁唑基、萘啶基、噁二唑基、2
‑
氧代吖庚因基、噁唑基、氧杂环丙基、1
‑
氧化吡啶基、1
‑
氧化嘧啶基、1
‑
氧化吡嗪基、1
‑
氧化哒嗪基、1
‑
苯基
‑
1h
‑
吡咯基、吩嗪基、吩噻嗪基、吩噁嗪基、酞嗪基、蝶啶基、嘌呤基、吡咯基、吡唑基、吡啶基、吡嗪基、嘧啶基、哒嗪基、喹唑啉基、喹噁啉基、喹啉基、奎宁环基、异喹啉基、四氢喹啉基、噻唑基、噻二唑基、三唑基、四唑基、三嗪基和噻吩基(thiophenyl)(即,噻吩基(thienyl))。除非在说明书中另有具体说明,否则杂芳基任选地如下所述经取代,例如经卤素、氨基、腈、硝基、羟基、烷基、烯基、炔基、卤代烷基、烷氧基、芳基、环烷基、杂环烷基、杂芳基等取代。在一些实施例中,杂芳基任选地经卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh、
‑
ome、
‑
nh2或
‑
no2取代。在一些实施例中,杂芳基任选地经卤素、甲基、乙基、
‑
cn、
‑
cf3、
‑
oh或
‑
ome取代。在一些实施例中,杂芳基任选地经卤素或甲基取代。在一些实施例中,杂芳基任选地经卤素取代。
[0078]
术语“氧代”是指=o。
[0079]
如本文所用,术语“治疗”、“预防”、“改善”和“抑制”以及源自其的词不一定意味着100%或完全治疗、预防、改善或抑制。相反,存在本领域普通技术人员认为其具有潜在益处或治疗效果的不同程度的治疗、预防、改善和抑制。在这方面,所公开的方法可以提供任何
量的任何水平的哺乳动物疾患的治疗、预防、改善或抑制。例如,疾患,包括其症状或病症,可以减少例如约100%、约90%、约80%、约70%、约60%、约50%、约40%、约30%、约20%或约10%。此外,由本文公开的方法提供的治疗、预防、改善或抑制可包括治疗、预防、改善或抑制疾患(例如,癌症或炎性疾病)的一种或多种病症或症状。此外,出于本文的目的,“治疗”、“预防”、“改善”或“抑制”包括延迟疾患或其症状或病症的发作。
[0080]
如本文所用,术语“有效量”或“治疗有效量”是指所施用的本文公开的化合物的足量,其将在一定程度上缓解所治疗的疾病或病症的一种或多种症状,例如、癌症或炎性疾病。在一些实施例中,该结果是疾病的迹象、症状或原因的减轻和/或缓解或生物系统的任何其他所需的改变。例如,用于治疗用途的“有效量”是提供疾病症状的临床显著减少所需的包含本文公开的化合物的组合物的量。在一些实施例中,在任何个别情况下合适的“有效”量是使用诸如剂量递增研究的技术确定的。
[0081]
如本文所用,“个体”(如在治疗对象中)是指哺乳动物和非
‑
哺乳动物。哺乳动物包括,例如人类;非
‑
人类灵长类动物,例如猿和猴;和非灵长类动物,例如狗、猫、牛、马、绵羊和山羊。非
‑
哺乳动物包括例如鱼和鸟。
[0082]
术语“疾病”或“疾患”可互换使用,用于指其中细菌信号肽酶在涉及疾病或病症的生化机制中起作用的疾病或病症,从而可通过作用于酶来实现治疗有益效果。“作用于”信号肽酶可包括结合至信号肽酶和/或抑制信号肽酶的生物活性。
[0083]
化合物
[0084]
在一方面,本文描述了式(i)化合物:
[0085][0086]
其中:
[0087]
r
1a
、r
2a
、r
4a
、r
5a
和r6a各自独立地为氢或任选地经取代的c1‑
c6烷基;
[0088]
r
3a
为氢、任选地经取代的c1‑
c
20
烷基、任选地经取代的c2‑
c
20
烯基、任选地经取代的c2‑
c
20
炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0089]
r
20
为羟基或
‑
nr1r2;
[0090]
r1和r2各自独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任
选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基、任选地经取代的(c1‑
c6烷基)杂芳基、
‑
s(=o)2r
a
、
‑
s(=o)2nr
b
r
c
、
‑
c(=o)r
a
、
‑
c(=o)or
b
、
‑
c(=o)nr
b
r
c
或
‑
(c=nr
b
)nr
b
r
c
;
[0091]
或者r1和r2与它们所连接的氮原子一起形成任选地经取代的杂环烷基;
[0092]
r3和r4各自独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0093]
或者r3和r4与它们所连接的碳原子一起形成氧代;
[0094]
r5为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基或任选地经取代的c2‑
c6炔基;
[0095]
r6和r7各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0096]
r8和r9各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0097]
或者r
3a
和r8与它们所连接的原子一起形成任选地经取代的杂环烷基;
[0098]
r
10
和r
11
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c
20
烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0099]
r
12
和r
13
各自独立地为氢、卤素、任选地经取代的c1‑
c
20
烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0100]
r
14
和r
15
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0101]
r
16
和r
17
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环
烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0102]
每个r
a
独立地为任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0103]
每个r
b
和r
c
独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0104]
或者r
b
和r
c
与它们所连接的氮原子一起形成任选地经取代的杂环烷基;
[0105]
或其药用盐、溶剂化物或立体异构体。
[0106]
在式(i)化合物的一些实施例中,r
20
为羟基。在式(i)化合物的一些实施例中,r
20
为
‑
nr1r2。
[0107]
在式(i)化合物的一些实施例中,r5为氢。
[0108]
在式(i)化合物的一些实施例中,r7为氢。
[0109]
在式(i)化合物的一些实施例中,r
13
为氢。
[0110]
在式(i)化合物的一些实施例中,r
15
为氢。
[0111]
在式(i)化合物的一些实施例中,r
17
为氢。
[0112]
在式(i)化合物的一些实施例中,该化合物具有式(ia)的结构:
[0113][0114]
在式(i)化合物的一些实施例中,该化合物具有式(ib)的结构:
[0115][0116]
在式(i)化合物的一些实施例中,r1为氢。
[0117]
在式(i)化合物的一些实施例中,r2为氢。
[0118]
在式(i)化合物的一些实施例中,r3和r4为氢。
[0119]
在式(i)化合物的一些实施例中,r
1a
为氢或c1‑
c6烷基。在式(i)化合物的一些实施例中,r
1a
为氢。
[0120]
在式(i)化合物的一些实施例中,r
2a
为氢或c1‑
c6烷基。在式(i)化合物的一些实施例中,r
2a
为氢。
[0121]
在式(i)化合物的一些实施例中,r
4a
为氢或c1‑
c6烷基。在式(i)化合物的一些实施例中,r
4a
为氢。
[0122]
在式(i)化合物的一些实施例中,r
5a
为氢或c1‑
c6烷基。在式(i)化合物的一些实施例中,r
5a
为氢。
[0123]
在式(i)化合物的一些实施例中,r
6a
为氢或c1‑
c6烷基。在式(i)化合物的一些实施例中,r
6a
为氢。
[0124]
在式(i)化合物的一些实施例中,r9为氢。
[0125]
在式(i)化合物的一些实施例中,r
10
为氢。
[0126]
在式(i)化合物的一些实施例中,r
11
为氢。
[0127]
在式(i)化合物的一些实施例中,r8为氢或任选地经一个、两个或三个卤素取代的c1‑
c6烷基。
[0128]
在式(i)化合物的一些实施例中,r8为氢或c1‑
c6烷基。
[0129]
在式(i)化合物的一些实施例中,r
3a
和r8与它们所连接的原子一起形成任选地经取代的杂环烷基。
[0130]
在式(i)化合物的一些实施例中,r
3a
和r8与它们所连接的原子一起形成氮杂环丁烷基、吡咯烷基或哌啶基。
[0131]
在式(i)化合物的一些实施例中,r6为任选地经取代的c1‑
c6烷基或任选地经取代的杂环烷基。
[0132]
在式(i)化合物的一些实施例中,r6为任选地经取代的c1‑
c6烷基。
[0133]
在式(i)化合物的一些实施例中,r6为c1‑
c6烷基,其任选地经以下取代:一个、两个或三个卤素、
‑
or
b
、
‑
nr
b
r
c
、
‑
nc(=nr
b
)nr
b
r
c
、
‑
s(=o)2r
a
、
‑
nr
b
s(=o)2r
a
、
‑
s(=o)2nr
b
r
c
、
‑
c(=o)r
a
、
‑
oc(=o)r
a
、
‑
c(=o)or
a
、
‑
oc(=o)or
b
、
‑
c(=o)nr
b
r
c
、
‑
nr
b
c(=o)[c(r
d
)2]1‑
4
nr
b
r
c
、
‑
o[c(r
d
)2]2‑4or
b
、
‑
o[c(r
d
)2]2‑4nr
b
r
c
、
‑
nr
b
[c(r
d
)2]2‑4nr
b
r
c
、
‑
o[c(r
d
)2]2‑4o[c(r
d
)2]2‑4nr
b
r
c
、
‑
o[c(r
d
)2]2‑4nc(=nr
b
)nr
b
r
c
、
‑
oc(=o)nr
b
r
c
、
‑
nr
b
c(=o)nr
b
r
c
、
‑
nr
b
c(=o)r
a
、
‑
nr
b
c(=o)or
b
、
‑
o
‑
(环烷基)nr
b
r
c
、
‑
o
‑
(杂环烷基)nr
b
r
c
、
‑
o
‑
(芳基)nr
b
r
c
、
‑
o
‑
(杂芳基)nr
b
r
c
、
‑
o
‑
(环烷基)(c1‑
c6烷基)nr
b
r
c
、
‑
o
‑
(杂环烷基)(c1‑
c6烷基)nr
b
r
c
、
‑
o
‑
(芳基)(c1‑
c6烷基)nr
b
r
c
、
‑
o
‑
(杂芳基)(c1‑
c6烷基)nr
b
r
c
、芳基或杂芳基;并且每个r
d
独立地为氢、卤素、
‑
oh、
‑
och3或c1‑
c6烷基。
[0134]
在式(i)化合物的一些实施例中,r6为c1‑
c6烷基,其任选地经以下取代:一个、两个或三个
‑
or
b
、
‑
nr
b
r
c
、
‑
c(=o)nr
b
r
c
、
‑
o[c(r
d
)2]2‑4nr
b
r
c
、
‑
nr
b
[c(r
d
)2]2‑4nr
b
r
c
、
‑
o
‑
(环烷基)nr
b
r
c
、
‑
o
‑
(环烷基)(c1‑
c6烷基)nr
b
r
c
;并且每个r
d
独立地为氢、卤素、
‑
oh、
‑
och3或c1‑
c6烷基。
[0135]
在式(i)化合物的一些实施例中,r6为任选地经一个、两个或三个
‑
or
b
取代的c1‑
c6烷基。
[0136]
在式(i)化合物的一些实施例中,r6为为
[0137]
在式(i)化合物的一些实施例中,r6为为为在式(i)化合物的一些实施例中,r6为为
[0138]
在式(i)化合物的一些实施例中,r
12
为氢或任选地经取代的c1‑
c6烷基。在式(i)化合物的一些实施例中,r
12
为任选地经一个、两个或三个卤素或
‑
och3取代的c1‑
c6烷基。在式
(i)化合物的一些实施例中,r
12
为c1‑
c6烷基或c1‑
c6卤代烷基。在式(i)化合物的一些实施例中,r
12
为
[0139]
在式(i)化合物的一些实施例中,r
14
为任选地经取代的c1‑
c6烷基或任选地经取代的(c1‑
c6烷基)环烷基。在式(i)化合物的一些实施例中,r
14
为c1‑
c6烷基。
[0140]
在式(i)化合物的一些实施例中,r
14
为为在式(i)化合物的一些实施例中,r
14
为为
[0141]
在式(i)化合物的一些实施例中,r
16
为任选地经取代的c1‑
c6烷基或任选地经取代的环烷基。在式(i)化合物的一些实施例中,r
16
为c3‑
c7环烷基。在式(i)化合物的一些实施例中,r
16
为环己基。
[0142]
在式(i)化合物的一些实施例中,r
16
为为
[0143]
在式(i)化合物的一些实施例中,r
16
为为在式(i)化合物的一些实施例中,r
16
为
[0144]
在式(i)化合物的一些实施例中,r
3a
为任选地经取代的c1‑
c
20
烷基、任选地经取代的c2‑
c
20
炔基、任选地经取代的(c1‑
c6烷基)环烷基或任选地经取代的(c1‑
c6烷基)芳基。在式(i)化合物的一些实施例中,r
3a
为任选地经一个、两个或三个卤素取代的c1‑
c6烷基。在式(i)化合物的一些实施例中,r
3a
为任选地经一个、两个或三个c1‑
c6烷基取代的(c1‑
c6烷基)芳基。在式(i)化合物的一些实施例中,r
3a
为(c1‑
c6烷基)环烷基。在式(i)化合物的一些实施例中,r
3a
为为为为在式(i)化合物的一些实施例中,r
3a
为为
[0145]
在式(i)化合物的一些实施例中,每个r
a
独立地为c1‑
c6烷基。
[0146]
在式(i)化合物的一些实施例中,每个r
b
和r
c
独立地为氢或c1‑
c6烷基。
[0147]
在一方面,本文描述了式(ii)化合物:
[0148][0149]
其中:
[0150]
r
1a
、r
2a
、r
3a
、r
4a
、r
5a
和r
6a
各自独立地为氢或任选地经取代的c1‑
c6烷基;
[0151]
r1和r2各自独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基、任选地经取代的(c1‑
c6烷基)杂芳基、
‑
s(=o)2r
a
、
‑
s(=o)2nr
b
r
c
、
‑
c(=o)r
a
、
‑
c(=o)or
b
、
‑
c(=o)nr
b
r
c
或
‑
(c=nr
b
)nr
b
r
c
;
[0152]
或者r1和r2与它们所连接的氮原子一起形成任选地经取代的杂环烷基;
[0153]
r3和r4各自独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0154]
或者r3和r4与它们所连接的碳原子一起形成氧代;
[0155]
r5为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基或任选地经取代的c2‑
c6炔基;
[0156]
r6和r7各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0157]
r8和r9各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0158]
r
10
和r
11
各自独立地为氢、卤素、任选地经取代的c1‑
c
20
烷基、任选地经取代的c2‑
c
20
烯基、任选地经取代的c2‑
c
20
炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0159]
r
12
和r
13
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0160]
或者r
10
和r
12
一起形成任选地经取代的环烷基或任选地经取代的环烯基;
[0161]
r
14
和r
15
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0162]
或者r
14
和r
4a
与它们所连接的原子一起形成任选地经取代的杂环烷基;
[0163]
r
16
和r
17
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0164]
r
18
和r
19
各自独立地为氢、卤素、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的(c1‑
c6烷基)环烷基、任选地经取代的杂环烷基、任选地经取代的(c1‑
c6烷基)杂环烷基、任选地经取代的芳基、任选地经取代的(c1‑
c6烷基)芳基、任选地经取代的杂芳基或任选地经取代的(c1‑
c6烷基)杂芳基;
[0165]
或者r
18
和r
19
与它们所连接的碳原子一起形成氧代;
[0166]
每个r
a
独立地为任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0167]
每个r
b
和r
c
独立地为氢、任选地经取代的c1‑
c6烷基、任选地经取代的c2‑
c6烯基、任选地经取代的c2‑
c6炔基、任选地经取代的环烷基、任选地经取代的杂环烷基、任选地经取代的芳基或任选地经取代的杂芳基;
[0168]
或者r
b
和r
c
与它们所连接的氮原子一起形成任选地经取代的杂环烷基;
[0169]
或其药用盐、溶剂化物或立体异构体;
[0170]
其中所述化合物选自由以下项组成的组:
[0171]
[0172]
[0173]
[0174][0175]
在一些实施例中,本文公开的化合物选自表1中的化合物或其药用盐、溶剂化物或立体异构体。
[0176]
表1
[0177]
[0178]
[0179]
[0180]
[0181]
[0182]
[0183]
[0184]
[0185]
[0186]
[0187]
[0188]
[0189]
[0190]
[0191]
[0192]
[0193]
[0194]
[0195]
[0196]
[0197]
[0198]
[0199]
[0200]
[0201]
[0202]
[0203]
[0204]
[0205][0206]
本文公开的化合物的其他形式
[0207]
异构体/立体异构体
[0208]
在一些实施例中,本文所述的化合物作为几何异构体存在。在一些实施例中,本文所述的化合物具有一个或多个双键。本文呈现的化合物包括所有顺式(cis)、反式(trans)、syn、anti、entgegen(e)和zusammen(z)异构体以及它们的相应混合物。在一些情况下,本文所述的化合物具有一个或多个手性中心并且每个中心以r构型或s构型存在。本文所述的化合物包括所有非对映异构、对映异构和差向异构形式以及它们的相应混合物。在本文提供的化合物和方法的其他实施例中,由单个制备步骤、组合或互变产生的对映异构体和/或非对映异构体的混合物可用于本文所述的应用。在一些实施例中,通过使本文所述化合物的外消旋混合物与旋光拆分剂反应以形成一对非对映异构化合物,分离非对映异构体并回收光学纯的对映异构体,将该化合物制备为其单独的立体异构体。在一些实施例中,以可解离的复合物为优选。在一些实施例中,非对映异构体具有不同的物理特性(例如,熔点、沸点、溶解度、反应性等)并且通过利用这些差异性而被分离。在一些实施例中,非对映异构体通过手性色谱分离,或优选通过基于溶解度差异的分离/拆分技术分离。在一些实施例中,然后与拆分剂一起回收光学纯的对映异构体。
[0209]
标记的化合物
[0210]
在一些实施例中,本文所述的化合物以其同位素标记的形式存在。在一些实施例中,本文公开的方法包括通过施用此类同位素标记的化合物来治疗疾病的方法。在一些实施例中,本文公开的方法包括通过将此类同位素标记的化合物作为药物组合物施用来治疗疾病的方法。因此,在一些实施例中,本文公开的化合物包括同位素标记的化合物,其与本文列举的化合物相同,但事实上,一个或多个原子被原子质量或质量数不同于自然界中通
常发现的原子质量或质量数的原子所取代。可并入本文所述的化合物或其溶剂化物或立体异构体中的同位素的实例包括氢、碳、氮、氧、磷、硫、氟和氯的同位素,分别诸如2h、3h、
13
c、
14
c、
l5
n、
18
o、
17
o、
31
p、
32
p、
35
s、
18
f和
36
cl。某些同位素标记的化合物,例如其中并入有放射性同位素(诸如3h、
14
c)的那些化合物,可用于药物和/或底物组织分布测定。尤其优选氚化(即3h)和碳
‑
14(即
14
c)同位素,因为它们易于制备和检测。再者,用重同位素(诸如氘,即2h)取代,由于更高的代谢稳定性产生某些治疗优势,例如,体内半衰期增加或剂量要求减少。在一些实施例中,同位素标记的化合物或其药用盐、溶剂化物或立体异构体通过任何合适的方法制备。
[0211]
在一些实施例中,本文所述的化合物通过其他方式标记,包括但不限于使用生色团或荧光部分、生物发光标记或化学发光标记。
[0212]
药用盐
[0213]
在一些实施例中,本文所述的化合物以其药用盐存在。在一些实施例中,本文公开的方法包括通过施用此类药用盐来治疗疾病的方法。在一些实施例中,本文公开的方法包括通过施用此类药用盐作为药物组合物来治疗疾病的方法。
[0214]
在一些实施例中,本文所述的化合物具有酸性或碱性基团,并且因此与多种无机或有机碱以及无机和有机酸中的任一种反应以形成药用盐。在一些实施例中,这些盐是在本文公开的化合物的最终分离和纯化过程中原位制备的,或者通过将游离形式的纯化化合物与合适的酸或碱单独反应并分离由此形成的盐来制备。
[0215]
药用盐的实例包括通过本文所述的化合物与无机、有机酸或无机碱反应制备的那些盐,此类盐包括乙酸盐、丙烯酸盐、己二酸盐、藻酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、硫酸氢盐、亚硫酸氢盐、溴化物、丁酸盐、丁炔
‑
1,4
‑
二酸盐、樟脑酸盐、樟脑磺酸盐、己酸盐、辛酸盐、氯苯甲酸盐、氯化物、柠檬酸盐、环戊烷丙酸盐、癸酸盐、二葡糖酸盐、磷酸二氢盐、二硝基苯甲酸盐、十二烷基硫酸盐、乙磺酸盐、甲酸盐、富马酸盐、葡庚糖酸盐、甘油磷酸盐、乙醇酸盐、半硫酸盐、庚酸盐、己酸盐、己炔
‑
1,6
‑
二酸盐、羟基苯甲酸盐、γ
‑
羟基丁酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、2
‑
羟基乙磺酸盐、碘化物、异丁酸盐、乳酸盐、马来酸盐、丙二酸盐、甲磺酸盐、扁桃酸盐、偏磷酸盐、甲磺酸盐、甲氧基苯甲酸盐、甲基苯甲酸盐、磷酸一氢盐、1
‑
萘磺酸盐、2
‑
萘磺酸盐、烟酸盐、硝酸盐、棕榈酸盐、果胶酸盐、过硫酸盐、3
‑
苯基丙酸盐、磷酸盐、苦味酸盐、新戊酸盐、丙酸盐、焦硫酸盐、焦磷酸盐、丙炔酸盐、邻苯二甲酸盐、苯乙酸盐、苯丁酸盐、丙磺酸盐、水杨酸盐、琥珀酸盐、硫酸盐、亚硫酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、磺酸盐、酒石酸盐、硫代氰酸盐、甲苯磺酸盐、十一酸盐和二甲苯磺酸盐。
[0216]
再者,本文所述的化合物可以制备为通过将游离碱形式的化合物与药用无机或有机酸反应形成的药用盐,所述无机或有机酸包括但不限于,无机酸,诸如盐酸、氢溴酸、硫酸、硝酸、磷酸、偏磷酸等;以及有机酸,诸如乙酸、丙酸、己酸、环戊烷丙酸、乙醇酸、丙酮酸、乳酸、丙二酸、琥珀酸、苹果酸、马来酸、富马酸、对甲苯磺酸、酒石酸、三氟乙酸、柠檬酸、苯甲酸、3
‑
(4
‑
羟基苯甲酰基)苯甲酸、肉桂酸、扁桃酸、芳基磺酸、甲磺酸、乙磺酸、1,2
‑
乙二磺酸、2
‑
羟基乙磺酸、苯磺酸、2
‑
萘磺酸、4
‑
甲基双环
‑
[2.2.2]辛
‑2‑
烯
‑1‑
甲酸、葡庚糖酸、4,4'
‑
亚甲基双
‑
(3
‑
羟基
‑2‑
烯
‑1‑
甲酸)、3
‑
苯基丙酸、三甲基乙酸、叔丁基乙酸、月桂基硫酸、葡糖酸、谷氨酸、羟基萘甲酸、水杨酸、硬脂酸和粘康酸。
[0217]
在一些实施例中,本文所述的那些包含游离酸基团的化合物与合适的碱诸如药用
金属阳离子的氢氧化物、碳酸盐、碳酸氢盐、硫酸盐反应,与氨反应,或者与药用有机伯胺、仲胺、叔胺或季胺反应。代表性的盐包括碱金属盐或碱土金属盐,如锂盐、钠盐、钾盐、钙盐和镁盐,以及铝盐等。碱的说明性实例包括氢氧化钠、氢氧化钾、氢氧化胆碱、碳酸钠、n
(c1‑4烷基)4等。
[0218]
可用于形成碱加成盐的代表性有机胺包括乙胺、二乙胺、乙二胺、乙醇胺、二乙醇胺、哌嗪等。应当理解,本文所述的化合物还包括它们所含的任何碱性含氮基团的季铵化。在一些实施例中,通过这种季铵化获得水溶性或油溶性或分散性产品。
[0219]
溶剂化物
[0220]
在一些实施例中,本文所述的化合物作为溶剂化物存在。本公开提供了通过施用此类溶剂化物来治疗疾病的方法。本公开进一步提供了通过将此类溶剂化物作为药物组合物施用来治疗疾病的方法。溶剂化物包含化学计量或非化学计量的量的溶剂,并且在一些实施例中,在用药用溶剂诸如水、乙醇等结晶的过程中形成。当溶剂为水时形成水合物,或当溶剂为醇时形成醇合物。可以在本文描述的方法期间方便地制备或形成本文所述化合物的溶剂化物。仅举例而言,本文所述化合物的水合物可通过使用包括但不限于二氧六环、四氢呋喃或甲醇的有机溶剂,从水性/有机溶剂混合物中重结晶而方便地制备。此外,本文提供的化合物可以非溶剂化和溶剂化形式存在。通常,出于本文提供的化合物和方法的目的,溶剂化形式被认为等同于非溶剂化形式。
[0221]
互变异构体
[0222]
在一些情况下,化合物以互变异构体的形式存在。本文所述的化合物包括本文所述式中所有可能的互变异构体。互变异构体是可通过氢原子的迁移而相互转化的化合物,伴随着单键和相邻双键的转换。在可能发生互变异构化的键合排列中,将存在互变异构体的化学平衡。涵盖本文公开的化合物的所有互变异构形式。互变异构体的确切比例取决于几个因素,包括温度、溶剂和ph值。
[0223]
治疗方法
[0224]
本文还公开了治疗需要这种治疗的哺乳动物的方法,包括以足以为该哺乳动物提供有益效果的频率和持续时间向该哺乳动物施用抗菌有效量的任何上述化合物。在一个实施例中,哺乳动物患有对一种或多种临床使用的抗生素治疗具有耐药性的细菌相关感染。在进一步的实施例中,细菌感染的致病菌种是涉及以下细菌的感染:铜绿假单胞菌(pseudomonas aeruginosa)、荧光假单胞菌(pseudomonas fluorescens)、食酸假单胞菌(pseudomonas acidovorans)、产碱假单胞菌(pseudomonas alcaligenes)、恶臭假单胞菌(pseudomonas putida)、嗜麦芽寡养单胞菌(stenotrophomonas maltophilia)、洋葱伯克霍尔德菌(burkholderia cepacia)、嗜水气单胞菌(aeromonas hydrophilia)、大肠杆菌(escherichia coli)、弗氏柠檬酸杆菌(citrobacter freundii)、鼠伤寒沙门氏菌(salmonella typhimurium)、伤寒沙门氏菌(salmonella typhi)、副伤寒沙门氏菌(salmonella paratyphi)、肠炎沙门氏菌(salmonella enteritidis)、痢疾志贺菌(shigella dysenteriae)、弗氏志贺菌(shigella flexneri)、宋内志贺菌(shigella sonnei)、阴沟肠杆菌(enterobacter cloacae)、产气肠杆菌(enterobacter aerogenes)、肺炎克雷伯菌(klebsiella pneumoniae)、产酸克雷伯菌(klebsiella oxytoca)、粘质沙雷氏菌(serratia marcescens)、土拉弗朗西斯菌(francisella tularensis)、摩氏摩根菌
(morganella morganii)、奇异变形杆菌(proteus mirabilis)、普通变形杆菌(proteus vulgaris)、产碱普罗威登斯菌(providencia alcalifaciens)、雷氏普罗威登斯菌(providencia rettgeri)、斯氏普罗威登斯菌(providencia stuartii)、鲍氏不动杆菌(acinetobacter baumannii)、醋酸钙不动杆菌(acinetobacter calcoaceticus)、溶血不动杆菌(acinetobacter haemolyticus)、小肠结肠炎耶尔森菌(yersinia enterocolitica)、鼠疫耶尔森菌(yersinia pestis)、假结核耶尔森菌(yersinia pseudotuberculosis)、中间耶尔森菌(yersinia intermedia)、百日咳博代氏杆菌(bordetella pertussis)、副百日咳博代氏杆菌(bordetella parapertussis)、支气管炎博代氏杆菌(bordetella bronchiseptica)、流感嗜血杆菌(haemophilus influenzae)、副流感嗜血杆菌(haemophilus parainfluenzae)、溶血嗜血杆菌(haemophilus haemolyticus)、副溶血嗜血杆菌(haemophilus parahaemolyticus)、杜克雷嗜血杆菌(haemophilus ducreyi)、多杀性巴氏杆菌(pasteurella multocida)、溶血性巴氏杆菌(pasteurella haemolytica)、粘膜炎布兰汉球菌(branhamella catarrhalis)、幽门螺杆菌(helicobacter pylori)、胎儿弯曲杆菌(campylobacter fetus)、空肠弯曲杆菌(campylobacter jejuni)、大肠弯曲杆菌(campylobacter coli)、伯氏疏螺旋体(borrelia burgdorferi)、霍乱弧菌(vibrio cholerae)、副溶血性弧菌(vibrio parahaemolyticus)、嗜肺军团菌(legionella pneumophila)、单核细胞增多性李斯特氏菌(listeria monocytogenes)、淋病奈瑟氏菌(neisseria gonorrhoeae)、脑膜炎奈瑟氏菌(neisseria meningitidis)、金氏菌属(kingella)、莫拉克斯氏菌属(moraxella)、阴道加德菌(gardnerella vaginalis)、脆弱拟杆菌(bacteroides fragilis)、狄氏拟杆菌(bacteroides distasonis)、拟杆菌属(bacteroides)3452a同源群、普通拟杆菌(bacteroides vulgatus)、椭圆拟杆菌(bacteroides ovalus)、多形拟杆菌(bacteroides thetaiotaomicron)、单形拟杆菌(bacteroides uniformis)、埃氏拟杆菌(bacteroides eggerthii)或内脏拟杆菌(bacteroides splanchnicus)。在一些实施例中,细菌感染是涉及革兰氏阴性细菌的感染。
[0225]
在一个实施例中,是本文所述的化合物展示出可用于治疗细菌感染的抗生素活性,例如仅举例来说,大肠杆菌(e.coli)、阴沟肠杆菌(e.cloaceae)、肺炎克雷伯氏菌(k.pneumoniae)、鲍氏不动杆菌(a.baumannii)或铜绿假单胞菌(p.aeruginosa)的各种菌株。
[0226]
联合疗法
[0227]
本文还公开了治疗需要这种治疗的哺乳动物的方法,包括向该哺乳动物施用针对任何上述治疗方法的第二治疗剂。在另一个实施例中,第二治疗剂不是lspa抑制剂。在另一个实施例中,第二治疗剂为氨基糖苷类抗生素、氟喹诺酮类抗生素、β
‑
内酰胺类抗生素、大环内酯类抗生素、糖肽类抗生素、利福平、氯霉素、氟霉素、粘菌素、莫匹罗星、杆菌肽、达托霉素或利奈唑胺。
[0228]
在一些实施例中是治疗患者(优选人)的细菌感染的方法,其中治疗包括施用治疗或药理学有效量的以下项的组合:1)β
‑
内酰胺抗生素;和2)本文公开的化合物或其药用盐、溶剂化物或立体异构体;和3)药用载体。在一些实施例中,β
‑
内酰胺抗生素是碳青霉烯、头孢菌素、头霉素、单内酰环或青霉素。示例性碳青霉烯类抗生素包括但不限于厄他培南、亚
胺培南、比阿培南和美罗培南。示例性的头孢菌素抗生素包括但不限于头孢吡普、头孢洛林、头孢匹罗、头孢唑兰、头孢吡肟、头孢噻肟和头孢曲松。示例性的青霉素类抗生素包括但不限于氨苄西林、阿莫西林、哌拉西林、苯唑西林、氯唑西林、甲氧西林和萘夫西林。在一些实施例中,β
‑
内酰胺与β
‑
内酰胺酶抑制剂一起施用。在一些实施例中,碳青霉烯与dhp抑制剂(例如,西司他丁)一起施用。
[0229]
在一些实施例中,β
‑
内酰胺类抗生素和本文公开的化合物序贯或同时施用。在一些实施例中,β
‑
内酰胺类抗生素和本文公开的化合物一起施用。在一些实施例中,β
‑
内酰胺类抗生素和本文公开的化合物在相同制剂中或分开的制剂中施用。在一些实施例中,首先施用β
‑
内酰胺或本文公开的化合物。在施用第一种化合物后,例如在1至60分钟内,例如在1、2、3、4、5、10、15、30或60分钟内施用另一种化合物。一方面,当使用β
‑
内酰胺酶抑制剂时,其单独施用,或与本文公开的化合物和/或β
‑
内酰胺类抗生素一起在制剂中施用。一方面,当使用dhp抑制剂来提高碳青霉烯的稳定性时,其单独施用,或与本文公开的化合物和/或碳青霉烯一起在制剂中施用。
[0230]
本文进一步描述了包含本文公开的化合物、药用载体和任选的β
‑
内酰胺类抗生素的药物组合物。在使用组合的实施例中,β
‑
内酰胺类抗生素和本文公开的化合物以使得它们的组合构成治疗有效量的量存在。由于本文公开的化合物的增强作用,组合中存在的β
‑
内酰胺类抗生素的量可能少于单独使用的β
‑
内酰胺类抗生素的量。在某些实施例中,组合物进一步包含β
‑
内酰胺酶类抗生素。
[0231]
在β
‑
内酰胺类抗生素是碳青霉烯的进一步实施例中,提供了包含碳青霉烯类抗生素、dhp抑制剂、本文公开的化合物和药用载体的药物组合物。在β
‑
内酰胺类抗生素是碳青霉烯的一些实施例中,碳青霉烯类抗生素优选地选自厄他培南、亚胺培南和美罗培南组成的组。
[0232]
在一些实施例中是本文公开的化合物用于治疗细菌感染。在一些实施例中是本文公开的化合物与包括β
‑
内酰胺类抗生素的一种或多种另外的治疗剂组合用于治疗细菌感染。在一些实施例中是本文公开的化合物用作治疗细菌感染的药物。在一些实施例中是本文公开的化合物与包括β
‑
内酰胺类抗生素的一种或多种另外的治疗剂组合用作治疗细菌感染的药物。在一些实施例中是本文公开的化合物用于制备治疗细菌感染的药物。在一些实施例中是本文公开的化合物与包括β
‑
内酰胺类抗生素的一种或多种另外的治疗剂组合用于制备治疗细菌感染的药物。
[0233]
在本文描述的一些实施例中,本文公开的化合物可以通过在耐药菌株诸如mrsa中诱导对抗菌剂的敏感性来增强β
‑
内酰胺类抗菌剂的活性。在一些实施例中,本文公开的化合物可以通过降低在药物敏感菌株中达到治疗效果所需的抗菌剂剂量来增强β
‑
内酰胺类抗菌剂的活性。例如,如果本文公开的化合物降低了敏感菌株中抗菌剂的最小抑制浓度(mic)(其中mic是将会完全抑制生长的抗菌剂的最小浓度),那么这种处理可能有利于使减少抗菌剂的施用量(可以减少抗生素的副作用)或减少施用频率成为可能。在一些实施例中,本文公开的化合物可以增强抗菌剂诸如碳青霉烯的活性以防止在具有耐药亚群的异质细菌群中出现耐药亚群。
[0234]
在一些实施例中,增效剂用于增强抗菌剂的活性,抗菌剂的临床功效受到耐药菌株日益流行的限制。在本文描述的一些实施例中,本文公开的化合物用作增效剂,其中本文
公开的化合物可以与β
‑
内酰胺类抗生素一起施用(同时或序贯)以允许有效治疗涉及耐药细菌的感染,或减少治疗感染所需的抗菌剂用量。
[0235]
施用和药物组合物
[0236]
本文所述的药物组合物包含与一种或多种药用载体一起配制的治疗有效量的本文所述的化合物(即,本文公开的任何化合物)。如本文所用,术语“药用载体”是指任何类型的无毒、惰性固体、半固体或液体填充剂、稀释剂、包封材料或制剂助剂。可用作药用载体的材料的一些实例为:糖,诸如乳糖、葡萄糖和蔗糖;淀粉,诸如玉米淀粉和马铃薯淀粉;纤维素及其衍生物,诸如羧甲基纤维素钠、乙基纤维素和乙酸纤维素;黄蓍粉;麦芽;明胶;滑石粉;赋形剂,诸如可可脂和栓剂蜡;油,诸如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;二醇,诸如丙二醇;酯,诸如油酸乙酯和月桂酸乙酯;琼脂;缓冲剂,诸如氢氧化镁和氢氧化铝;海藻酸;无热原水;等渗盐水;林格氏溶液;乙醇;和磷酸盐缓冲溶液;以及其他无毒的相容性润滑剂,诸如月桂基硫酸钠和硬脂酸镁;并且根据配方师的判断,组合物中还可存在着色剂、脱模剂、包衣剂、甜味剂、调味剂和芳香剂、防腐剂和抗氧化剂。本文所述的药物组合物可通过口服、直肠、肠胃外、脑池内、阴道内、腹膜内、局部(如通过粉剂、软膏或滴剂)、口腔、或作为口腔或鼻腔喷雾剂或用于吸入的液体气雾剂或干粉制剂施用至人类和其他动物。
[0237]
用于口服施用的液体剂型可包括药用乳剂、微乳剂、溶液、混悬剂、糖浆和酏剂。除本发明活性化合物以外,液体剂型还任选地包含本领域中常用的惰性稀释剂,诸如水或其他溶剂、增溶剂和乳化剂诸如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3
‑
丁二醇、二甲基甲酰胺、油(特别是棉籽油、花生油、玉米油、胚芽油、橄榄油、蓖麻油和芝麻油)、甘油、四氢糠醇、聚乙二醇和山梨醇脂肪酸酯及其混合物。除惰性稀释剂以外,口服组合物还可包括佐剂,诸如润湿剂、乳化剂和悬浮剂、甜味剂、调味剂和芳香剂。
[0238]
可注射制剂,例如,无菌可注射水性或油性悬浮液任选地根据已知技术使用合适的分散或润湿剂和悬浮剂配制。无菌可注射制剂任选地是无菌可注射溶液、悬浮液或乳液,其处于非毒性的肠胃外可接受的稀释剂或溶剂中,例如,作为1,3
‑
丁二醇的溶液。任选地采用的可接受的媒介物和溶剂是水、林格溶液、u.s.p.和等渗氯化钠溶液。此外,无菌的固定油传统上用作溶剂或悬浮介质。为此,可以使用任何温和的不挥发性油,包括合成的甘油单酯或甘油二酯。此外,脂肪酸诸如油酸用于可注射制剂中。
[0239]
可注射制剂可例如通过以下方式无菌化:通过除菌过滤器过滤,或通过以无菌固体组合物形式并入无菌化剂,该固体组合物可在使用前溶解或分散于无菌水中或其它无菌可注射介质中。
[0240]
为了延长药物的效果,经常希望减缓对来自皮下或肌肉内注射的该药物的吸收。这任选地通过使用水溶性差的晶体或无定形材料的液体悬浮液来实现。然后,药物的吸收速率取决于其溶解速率,并继而可能取决于晶体尺寸和晶形。另选地,任选地通过将药物溶解或悬浮在油媒介物中实现对肠胃外施用的药物的延迟吸收。通过形成药物在可生物降解聚合物诸如聚丙交酯
‑
聚乙交酯中的微胶囊化基质来制备可注射长效剂型。依据药物与聚合物的比率和所采用的具体聚合物的属性,可控制药物的释放速率。其他可生物降解的聚合物的实例包括聚原酸酯和聚酐。也通过将药物入陷到与身体组织相容的脂质体或微乳液中来任选地制备长效可注射制剂。
[0241]
用于直肠和阴道施用的组合物优选为栓,其可通过将本文所述的化合物(即,本文公开的化合物)与合适的非刺激性赋形剂或载体诸如可可脂、聚乙二醇或栓蜡混合来制备,该赋形剂或载体在环境温度下为固体但在身体温度下为液体并因此在直肠或阴道腔体内融化并释放活性化合物。
[0242]
用于口服施用的固体剂型包括胶囊、片、丸、粉末和颗粒。在此类剂型中,将活性化合物与下列物质混合:至少一种惰性的、药用赋形剂或载体诸如柠檬酸钠或磷酸二钙和/或a)填充物或增量剂诸如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸,b)粘合剂诸如(举例而言)羧甲基纤维素、海藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯树胶,c)湿润剂诸如甘油,d)崩解剂,诸如琼脂、碳酸钙、马铃薯或木薯淀粉、海藻酸、某些硅酸盐和碳酸钠,e)溶液阻滞剂诸如石蜡,f)吸收加速剂诸如季铵化合物,g)润湿剂诸如(举例而言)鲸蜡醇和甘油单硬脂酸酯,h)吸收剂诸如高岭土和膨润土和i)润滑剂诸如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠及其混合物。在剂型为胶囊、片剂和丸剂的情况下,剂型任选地包含缓冲剂。
[0243]
相似类型的固体组合物任选地用作软填充明胶胶囊和硬填充明胶胶囊中的填料,该填料使用赋形剂诸如乳糖或奶糖以及高分子量聚乙二醇等。
[0244]
固体剂型片剂、糖衣丸、胶囊、丸剂和颗粒可制备有包衣和外壳,诸如肠溶衣和药物配制领域中已知的其他包衣。它们任选地包含遮光剂,并且也可以是仅或优先地在消化道的某些部分内可选地以延迟方式释放活性成分的组合物。可使用的包埋组合物的实例包括聚合物和蜡。
[0245]
相似类型的固体组合物任选地用作软填充明胶胶囊和硬填充明胶胶囊中的填料,该填料使用赋形剂诸如乳糖或奶糖以及高分子量聚乙二醇等。
[0246]
活性化合物也可以是具有一种或多种上述赋形剂的微囊形式。固体剂型片剂、糖衣丸、胶囊、丸剂和颗粒可制备有包衣和外壳,诸如肠溶衣、控释包衣和药物配制领域中已知的其他包衣。在此类固体剂型中,活性化合物任选地与至少一种惰性稀释剂诸如蔗糖、乳糖或淀粉混合。此类剂型按照惯例任选地包含惰性稀释剂以外的其他物质,例如压片润滑剂及其他压片助剂诸如硬脂酸镁和微晶纤维素。在剂型为胶囊、片剂和丸剂的情况下,剂型任选地包含缓冲剂。它们任选地包含遮光剂,并且也可以是仅或优先地在消化道的某些部分内可选地以延迟方式释放活性成分的组合物。可使用的包埋组合物的实例包括聚合物和蜡。
[0247]
用于外用或透皮施用本文所述化合物的剂型包括油膏、糊剂、乳霜、洗液、凝胶、粉末、溶液、喷雾、吸入剂或贴剂。活性成分在无菌条件下与药用载体和如任选地需要的任何需要的防腐剂或缓冲剂混合。还考虑了眼用制剂、滴耳剂等。
[0248]
除本发明所述的活性化合物以外,软膏、糊剂、乳霜和凝胶可包含赋形剂,该赋形剂诸如动物脂肪和植物脂肪、油、蜡、石蜡、淀粉、黄蓍胶、纤维素衍生物、聚乙二醇、硅氧烷、膨润土、硅酸、滑石粉和氧化锌或其混合物。
[0249]
本文所述的组合物任选地配制用于作为液体气雾剂或可吸入干粉递送。液体气雾剂制剂任选地主要雾化成可递送至终末细支气管和呼吸细支气管的颗粒尺寸,细菌存在于患有支气管感染诸如慢性支气管炎和肺炎的患者的这些细支气管处。病原菌普遍存在于整个气道中,下至支气管、细支气管和肺实质,特别是在终末细支气管和呼吸性细支气管中。
在感染加重期间,细菌也可以存在于肺泡中。液体气雾剂和可吸入干粉药物制剂可在整个支气管树中递送至细支气管末端,并且最终递送至实质组织。
[0250]
本文所述的气雾化制剂任选地使用气雾形成装置递送,所述气雾形成装置诸如喷射、振动多孔板或超声雾化器,优选地经选择以允许形成具有主要在1至5μm之间的质量平均直径的气雾颗粒。此外,该制剂优选具有平衡的渗透压离子强度和氯化物浓度,并且能够将有效剂量的本文所述化合物(即本文公开的化合物)递送至感染部位的最小可气雾化体积。另外,雾化的药物组合物优选对呼吸道的功能无负面影响,并且不引起不良副作用。
[0251]
适合于施用本文所述的气雾剂制剂的雾化装置包括例如喷射器、振动多孔板、超声雾化器和通电的干粉吸入器,其能够将制剂雾化成主要粒径范围为1
‑
5μ的气雾颗粒。在本技术中,“主要”意指所生成的全部气雾剂颗粒中的至少70%,优选超过90%在1
‑
5μ的范围内。喷射雾化器的工作原理是利用气压将液体溶液分解为气雾剂液滴。振动多孔板雾化器的工作原理是利用快速振动的多孔板所产生的声波真空将溶剂液滴从多孔板中挤出。超声雾化器的工作原理是利用压电晶体将液体剪切为小气雾剂液滴。可以使用各种合适的设备,包括例如aeronebtm和aerodosetm振动多孔板雾化器(aerogen,inc.,sunnyvale,california)、雾化器(medic
‑
aid ltd.,west sussex,england)、pari和pari lc喷射雾化器(pari respiratory equipment,inc.,richmond,virginia)以及aerosonictm(devilbiss medizinische produkte(deutschland)gmbh,heiden,germany)和(omron healthcare,inc.,vernon hills,illinois)超声雾化器。
[0252]
在一些实施例中,本文所述的化合物(即,本文公开的化合物)被配制成用作局部用粉剂和喷雾剂,除了本文描述的化合物外,该粉剂和喷雾剂还含有赋形剂诸如乳糖、滑石、硅酸、氢氧化铝、硅酸钙和聚酰胺粉末,或这些物质的混合物。喷雾剂任选地包含常规的推进剂诸如氯氟烃。
[0253]
透皮贴剂具有的另一个优点是实现化合物向人体的受控递送。此类剂型可通过将化合物溶解或分散于适当的介质中制成。吸收增强剂也可用来增加化合物跨越皮肤的通量。可通过提供速率控制膜或通过将化合物分散于聚合物基质或凝胶中来控制速率。
[0254]
根据本文描述的治疗方法,通过以达到预期效果所必需的量和时间向患者施用治疗有效量的本文所述化合物,治疗或预防患者诸如人或低等哺乳动物的细菌感染。“治疗有效量”的本文所述化合物是指以适用于任何医学治疗的合理益处/风险比治疗细菌感染的足够量的化合物。然而,应当理解,本文所述的化合物和组合物的总日用量将由主治医师在合理的医学判断范围内决定。任何特定患者的具体治疗有效剂量水平取决于多种因素,其中包括治疗的疾患和疾患的严重程度;所用特定化合物的活性;所用的特定组合物;患者的年龄、体重、总体健康状况、性别和饮食;施用时间、施用途径和所用特定化合物的排泄率;治疗持续时间;与所用特定化合物组合或同时使用的药物;以及医学领域众所周知的类似因素。
[0255]
以单剂量或分剂量向人或其他哺乳动物施用本文所述化合物(即本文公开的化合物)的本文所述化合物的总日剂量可为例如0.01至50mg/kg体重的量或更通常为0.1至25mg/kg体重。单剂量组合物可包含这样的量或其约数以构成日剂量。一般而言,本文所述的治疗方案包括每天以单剂量或多剂量向需要此类治疗的患者施用约10mg至约2000mg本文所述的化合物。
[0256]
实施例
[0257]
本文公开的化合物通过如下所示的反应方案中描述的方法制备。本文提供的过程与具有本领域普通技术的合成有机化学家的知识相结合,在一些实施例中用于制备如本文公开和要求保护的全部化合物。
[0258]
用于制备这些化合物的起始材料和试剂可从商业供应商处获得,诸如aldrich chemical co.(milwaukee,wis.)、bachem(torrance,calif.)或sigma(st.louis,mo.),或者是通过本领域技术人员已知的方法按照参考文献诸如《fieser和fieser有机合成试剂》(fieser and fieser's reagents for organic synthesis,第1
‑
17卷(john wiley and sons,1991))、《罗德氏碳化合物化学》(rodd’s chemistry of carbon compounds,第1
‑
5卷和增刊(elsevier science publishers,1989))、《有机反应》(organic reactions,第1
‑
40卷(john wiley and sons,1991))、《march高等有机化学》(march’sadvanced organic chemistry(john wiley and sons,第4版))和larock的《综合有机转化》(larock’s comprehensive organic transformations(vch publishers inc.,1989))中所述的过程制备。这些方案仅是对可以通过其在一些实施例中合成本文公开的化合物的一些方法的说明,并且参照该公开内容,可以对这些方案进行多种修改并向本领域技术人员提供建议。如有需要,可以使用常规技术分离和纯化反应的起始材料和中间体以及最终产物,所述常规技术包括但不限于过滤、蒸馏、结晶、色谱等。可以使用常规手段来表征此类材料,包括物理常数和光谱数据。通常通过反相hplc使用ch3cn/h2o以三氟乙酸作为添加剂将化合物分离为三氟乙酸盐。在一些情况下,在不使用三氟乙酸的情况下进行纯化,并以游离碱形式分离化合物。
[0259]
缩写
[0260]
acoh
ꢀꢀꢀꢀꢀ
乙酸
[0261]
dce
ꢀꢀꢀꢀꢀꢀ
1,2
‑
二氯乙烷
[0262]
dcm
ꢀꢀꢀꢀꢀꢀ
二氯甲烷
[0263]
dipea
ꢀꢀꢀꢀ
n,n
‑
二异丙基乙胺
[0264]
dma
ꢀꢀꢀꢀꢀꢀ
n,n
‑
二甲基乙酰胺
[0265]
dmap
ꢀꢀꢀꢀꢀ4‑
二甲基氨基吡啶
[0266]
dmf
ꢀꢀꢀꢀꢀꢀ
n,n
‑
二甲基甲酰胺
[0267]
dmso
ꢀꢀꢀꢀꢀ
二甲基亚砜
[0268]
esi
ꢀꢀꢀꢀꢀꢀ
电喷射离子化
[0269]
etoac
ꢀꢀꢀꢀ
乙酸乙酯
[0270]
h
ꢀꢀꢀꢀꢀꢀꢀꢀ
小时
[0271]
hatu
ꢀꢀꢀꢀꢀ1‑
[双(二甲基氨基)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐
[0272]
hbtu
ꢀꢀꢀꢀꢀ
o
‑
(苯并三唑
‑1‑
基)
‑
n,n,n',n'
‑
四甲基脲阳离子六氟磷酸盐
[0273]
hfip
ꢀꢀꢀꢀꢀ
1,1,1,3,3,3
‑
六氟
‑2‑
丙醇
[0274]
hobt
ꢀꢀꢀꢀꢀ
羟基苯并三唑
[0275]
hplc
ꢀꢀꢀꢀꢀ
高压液相色谱
[0276]
ipac
ꢀꢀꢀꢀꢀ
乙酸异丙酯
[0277]
lcms
ꢀꢀꢀꢀꢀ
液相色谱质谱
[0278]
lda
ꢀꢀꢀꢀꢀꢀ
二异丙基氨基锂
[0279]
m
ꢀꢀꢀꢀꢀꢀꢀꢀ
摩尔
[0280]
min
ꢀꢀꢀꢀꢀꢀ
分钟
[0281]
n
ꢀꢀꢀꢀꢀꢀꢀꢀ
正常
[0282]
nmr
ꢀꢀꢀꢀꢀꢀ
核磁共振
[0283]
pe
ꢀꢀꢀꢀꢀꢀꢀ
石油醚
[0284]
pyaop
ꢀꢀꢀꢀ
(3
‑
羟基
‑
3h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶合
‑
o)三
‑1‑
吡咯烷基
‑
磷六氟磷酸盐
[0285]
r
t
ꢀꢀꢀꢀꢀꢀꢀ
保留时间
[0286]
sfc
ꢀꢀꢀꢀꢀꢀ
超临界流体色谱
[0287]
tea
ꢀꢀꢀꢀꢀꢀ
三乙胺
[0288]
thf
ꢀꢀꢀꢀꢀꢀ
四氢呋喃
[0289]
tfa
ꢀꢀꢀꢀꢀꢀ
三氟乙酸
[0290]
tfe
ꢀꢀꢀꢀꢀꢀ
2,2,2
‑
三氟乙醇
[0291]
tfoh
ꢀꢀꢀꢀꢀ
三氟甲磺酸
[0292]
如通用方案1
‑
6中所示制备本文公开的化合物。
[0293]
方案1.
[0294][0295]
如方案1所述,β
‑
羟基酸4可以从oppolzer磺内酰胺(oppolzer'ssultam)1开始制备,其可以用酰氯酰化以生成中间体2。可以对中间体2进行路易斯酸介导的羟醛烷基化以生成anti
‑
羟醛产物3。化合物3可用氢氧化锂水溶液和过氧化氢水解,得到所需的β
‑
羟基酸化合物4。
[0296]
方案2.
[0297][0298]
如方案2所示,β
‑
羟基酸化合物4可以用经适当取代的2,2
‑
二氧代
‑
噁噻唑烷
‑3‑
甲酸叔丁酯烷基化,得到酯5。可以将这些酯皂化以得到羧酸6。可以通过酸性脱保护,然后用氯甲酸9
‑
芴基甲酯再保护,将氨基保护基团从boc转化为fmoc,得到中间体7。
[0299]
方案3.
[0300][0301]
如方案3所示,氯
‑
(2
‑
氯三苯甲基)
‑
树脂可负载fmoc保护的氨基酸,并在dmf中用哌啶去除fmoc基团,得到中间体8。中间体8可以通过步骤2
‑
4使用标准固相化学技术进行精细加工,利用hobt与fmoc保护的氨基酸和标准侧链保护基团偶联,以产生精细合成的树脂结合肽9。
[0302]
方案4.
[0303][0304]
如方案4所示,树脂结合肽9可以使用标准偶联试剂诸如hatu或pyaop偶联到羧酸6或7,然后进行boc或fmoc脱保护和树脂裂解,以得到受保护肽10。可以使用大环内酰胺化条件(诸如hatu/dipea)将线性肽环化以得到大环11。大环缩肽11的侧链可以使用钯/氢化和/或酸性条件进行全局脱保护,以得到化合物12。
[0305]
方案5.
[0306][0307]
如方案5所示,苄基或甲基酯保护的氨基酸可以进行序贯还原性胺化以得到受保护中间体13。进行boc脱保护,然后进行fmoc保护,得到中间体14。用皂化或氢化条件对羧酸脱保护,得到fmoc保护的氨基酸中间体15。作为另一种选择,可以将中间体13直接转化为羧酸以得到boc保护的氨基酸中间体16。
[0308]
方案6.
[0309][0310]
如方案6所示,树脂结合肽9可以在标准肽偶联条件下与中间体15或16偶联。用哌啶对fmoc基团进行脱保护,然后从树脂上裂解,或从树脂上酸裂解以同时去除boc保护基团,得到线性肽17。可以使用大环内酰胺化条件(诸如hatu/dipea)将线性肽环化以得到大环18。大环缩肽18的侧链可以使用钯/氢化和/或酸性条件进行全局脱保护,以得到化合物19。
[0311]
一般实验条件
[0312]1h nmr光谱是在环境温度下使用带有三重共振5mm探针的varian unity inova(400mhz)光谱仪、avance iii(300mhz)光谱仪或bruker ultrashield(400mhz或500mhz)光谱仪记录的。化学位移以相对于四甲基硅烷的ppm表示。使用了以下缩写:br=宽信号,s=单峰,d=双峰,dd=双双峰,t=三重峰,q=四重峰,m=多重峰。
[0313]
微波实验是使用cem discover、smith synthesizer或biotage initiator进行的,它使用单模谐振器和动态场调谐,两者均给出了可重复性和控制。可以达到40
‑
250℃的温度,并且可以达到高达30bar的压力。
[0314]
使用以下方法之一进行高压液相色谱
‑
质谱(lcms)实验以确定保留时间(rt)和相关的质量离子。光谱仪具有在正离子模式和负离子模式下运行的电喷雾源。在220nm和
254nm采集uv吸光度,并且在所有实验中应用质谱全扫描。使用sedex 85蒸发光散射检测器实现额外检测。thermo scientific带电气溶胶检测器(型号:corona)用于测量对紫外线不敏感的分析物的纯度。cad参数设置如下:
[0315]
·
气体压力:35
‑
40psi
[0316]
·
流量比:0.51
[0317]
·
离子阱:20.3v
[0318]
·
充电器电压:2.91kv
[0319]
·
充电器电流:1.01ua
[0320]
lcms方法a:实验在agilent 1290hplc与使用esi作为电离源的agilent msd质谱仪联用系统上进行。lc分离使用phenomenex xb
‑
c18,1.7mm,50
×
2.1mm色谱柱,并且流速为0.4ml/分钟。溶剂a是具有0.1%甲酸的水,并且溶剂b是具有0.1%甲酸的乙腈。在7分钟内用2%
‑
98%溶剂b构成的梯度,并保持97%b 1.5分钟,然后平衡1.5分钟。lc柱温为40℃。
[0321]
lcms方法b:实验在agilent 1290hplc与使用esi作为电离源的agilent msd质谱仪联用系统上进行。lc分离在agilent zorbax eclipse xdb
‑
c18,3.5mm,100
×
3.0mm色谱柱上以0.7ml/分钟的流速进行。mpa(流动相a)为具有0.1%fa的水,并且mpb(流动相b)为具有0.1%fa的乙腈。梯度以2%mpb开始并且以98%mpb结束,历时25.5min,并且保持98%b 2.5min,然后平衡1.5min。lc柱温为40℃。在220nm和254nm采集uv吸光度,且所有实验均采用质谱全扫描。
[0322]
lcms方法c:实验在与带有pda uv检测器的waters acquity uplc系统相连的waters micromass zq2000四极杆质谱仪上进行。光谱仪具有在正离子模式和负离子模式下运行的电喷雾源。该系统使用保持在40℃的acquity beh c18 1.7um 100x2.1mm色谱柱,或保持在40℃的acquity beh shield rp18 1.7μm 100x2.1mm色谱柱,流速为0.4ml/分钟。在前0.4分钟内的最初溶剂系统是95%的含有0.1%甲酸的水(溶剂a)和5%的含有0.1%甲酸的乙腈(溶剂b),然后在接下来的5.6分钟内,梯度上升到5%的溶剂a和95%的溶剂b。这将保持0.8分钟,之后在接下来的0.2分钟内返回到95%溶剂a和5%溶剂b。
[0323]
lcms方法d:实验在waters acquity uplc与使用esi电离源的waters lct premier xe质谱仪联用系统上进行。lc分离使用acquity uplc beh c18,1.7mm,2.1
×
50mm色谱柱进行,流速为0.6ml/min。mpa(流动相a)为具有0.05%tfa的水,并且mpb(流动相b)为乙腈。梯度以2%mpb开始并且以98%mpb结束,历时18.5min,并且保持98%mpb 1.0min,然后平衡0.5min。lc柱温为40℃。在220nm和254nm采集uv数据,且所有实验均采用质谱全扫描。
[0324]
lcms方法e:实验在dionex ultimate 3000与使用esi作为电离源的thermo scientific q exactive hrms联用系统上进行。lc分离在agilent zorbax eclipse xdb
‑
c18,3.5μm,100
×
3.0mm色谱柱上以0.7ml/分钟的流速进行。mpa(流动相a)为具有0.1%fa的水,并且mpb(流动相b)为具有0.1%fa的乙腈。梯度以2%mpb开始并且以98%mpb结束,历时25.5min,并且保持98%b 2.5min,然后平衡1.5min。lc柱温为40℃。通过dad检测器采集uv吸光度,且所有实验均采用质谱全扫描。
[0325]
lcms方法f:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c18 2.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是95%的含有0.05%三氟乙酸的水(溶剂a)和5%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的2.0分钟内,梯度上升到5%的溶剂a和95%的溶剂b。这将保持0.7分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0326]
lcms方法g:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c18 2.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是95%的含有0.05%三氟乙酸的水(溶剂a)和5%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的2.0分钟内,梯度上升到100%的溶剂b。这将保持0.7分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0327]
lcms方法h:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c18 2.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是70%的含有0.05%三氟乙酸的水(溶剂a)和30%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的3.6分钟内,梯度上升到30%的溶剂a和70%的溶剂b。然后在接下来的0.4分钟内,梯度上升到100%的溶剂b。这将保持0.5分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0328]
lcms方法i:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c18 2.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是95%的含有0.05%三氟乙酸的水(溶剂a)和5%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的3.2分钟内,梯度上升到30%的溶剂a和70%的溶剂b。然后在接下来的0.5分钟内,梯度上升到100%的溶剂b。这将保持0.8分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0329]
lcms方法j:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c18 2.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是95%的含有0.05%三氟乙酸的水(溶剂a)和5%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的3.2分钟内,梯度上升到40%的溶剂a和60%的溶剂b。然后在接下来的0.5分钟内,梯度上升到100%的溶剂b。这将保持0.8分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0330]
lcms方法k:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c18 2.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是80%的含有0.05%三氟乙酸的水(溶剂a)和20%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的3.6分钟内,梯度上升到40%的溶剂a和60%的溶剂b。然后在接下来的0.4分钟内,梯度上升到100%的溶剂b。这将保持0.5分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0331]
lcms方法l:实验在shimadzu uflc与dad检测器、elsd检测器和2020ev ms联用系
统上进行。光谱仪具有在正离子模式下运行的电喷雾源。该系统使用保持在40℃的shim
‑
pack xr
‑
ods c182.2μm 50*3.0mm色谱柱,流速为1.2ml/分钟。在前0.01分钟内的最初溶剂系统是95%的含有0.05%三氟乙酸的水(溶剂a)和5%的含有0.05%三氟乙酸的乙腈(溶剂b),然后在接下来的3.5分钟内,梯度上升到30%的溶剂a和70%的溶剂b。然后在接下来的0.2分钟内,梯度上升到100%的溶剂b。这将保持1.0分钟,之后在接下来的0.3分钟内返回到95%溶剂a和5%溶剂b。
[0332]
中间体1.(2r,3r)
‑3‑
羟基
‑2‑
甲基壬酸
[0333][0334]
步骤1. 1
‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)丙烷
‑1‑
酮
[0335][0336]
在0℃,将丙酰氯(844g,894mmol)加入(3ar,6s,7as)
‑
8,8
‑
二甲基六氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑2,2
‑
二氧化物(175g,813mmol)、dmap(10.5g,81.3mmol)和三乙胺(172ml,1220mmol)在thf(1.4l)中的溶液中。30min后,将反应回温至室温,并且再搅拌3h。减压蒸发反应。将所得残余物用水和乙酸异丙酯稀释。水层用乙酸异丙酯(2x)萃取。合并的有机层用1m naoh(x2)、1m hcl(x2)、盐水洗涤,用硫酸镁干燥,真空蒸发。将粗制固体从dcm和庚烷中重结晶,以给出标题化合物(213.3g,97%收率)。
[0337]
步骤2.(2r,3r)
‑1‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)
‑3‑
羟基
‑2‑
甲基壬烷
‑1‑
酮
[0338][0339]
在0℃,于氮气下向500ml 3颈圆底烧瓶中的三乙基硼烷(91ml,在己烷中的1mol/l溶液)在dcm(50ml)中的溶液中滴加tfoh(13.825g,92.122mmol)。然后将混合物在0℃搅拌30min,然后冷却至
‑
15℃。在
‑
15℃,向其中滴加1
‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)丙烷
‑1‑
酮(10.00g,36.8mmol)和dipea(12.3g)在dcm(50ml)中的溶液。将所得溶液在
‑
10℃于水/盐水浴中搅拌1h(混合物a)。
[0340]
在氮气下,将四氯化钛(110ml,在dcm中的1mol/l溶液,3.0当量)放入250ml 3颈圆
底烧瓶中。在
‑
78℃,向该溶液中滴加庚醛(6.3g,55.3mmol,1.5当量),并将混合物在
‑
50℃搅拌30min。然后将所得溶液转移至附加漏斗(混合物b)。
[0341]
在
‑
78℃,将混合物b滴加到混合物a中,并在
‑
50℃搅拌4h。反应用300ml饱和氯化铵水溶液猝灭,并用2x500ml乙酸乙酯萃取。将有机层合并,用1x300ml盐水洗涤,以无水硫酸钠干燥,过滤并真空蒸发。将残余物施加到硅胶柱上,用乙酸乙酯/石油醚(1∶4)洗脱。合并合适的部分并真空浓缩以得到标题化合物(12g),为85%纯度的淡黄色油状物(含有~15%的起始材料),且未经纯化即用于下一步骤。lcms(esi):[m h]
=386。
[0342]
步骤3.(2r,3r)
‑3‑
羟基
‑2‑
甲基壬酸
[0343]
向250ml圆底烧瓶中放入thf(60ml)、30%h2o2(6.3ml)、lioh(1.96g,81.844mmol)、水(30ml,1.665mol)、(2r,3r)
‑1‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)
‑3‑
羟基
‑2‑
甲基壬烷
‑1‑
酮(6.30g,16.340mmol)。所得溶液在25℃搅拌过夜。然后通过加入100ml饱和na2so3水溶液猝灭反应。将溶液的ph值用1nhcl溶液调节至3。用3x200ml乙酸乙酯提取所得溶液,合并有机层,并经无水硫酸钠干燥,过滤并真空浓缩。将残余物施加到硅胶柱上,用乙酸乙酯/石油醚(1∶2)洗脱。合并适当的级分并在真空下浓缩。这产生2.2g(70%收率)标题化合物,为淡黄色油状物。lcms(esi):[m
‑
h]
‑
=187;1h nmr(400mhz,dmso
‑
d6)δ11.95(s,1h),4.60(d,j=5.2hz,1h),3.59(s,1h),2.43
‑
2.31(m,1h),1.48
‑
1.15(m,10h),0.98(d,j=7.0hz,3h),0.92
‑
0.82(m,3h)。
[0344]
中间体2.(2r,3r)
‑3‑
((r)
‑2‑
((叔丁氧羰基)氨基)丙氧基)
‑2‑
甲基壬酸
[0345][0346]
步骤1.(2s,3r)
‑3‑
((r)
‑2‑
((叔丁氧羰基)氨基)丙氧基)
‑2‑
甲基壬酸(r)
‑2‑
((叔丁氧羰基)氨基)丙酯
[0347][0348]
在室温,向(2r,3r)
‑3‑
羟基
‑2‑
甲基
‑
壬酸(10.13g,53.81mmol,中间体1)在无水thf(100ml)中的溶液中加入(4r)
‑4‑
甲基
‑
2,2
‑
二氧代
‑
噁噻唑烷
‑3‑
甲酸叔丁酯(38.31g,161.46mmol)和四丁基氟化铵(在thf中的1mol/l溶液,80ml,53.81mmol)。在0
°
℃,分批加入氢化钠(在矿物油中,60%,8.61g,215.2mmol)。在氢化钠加入完成且氢气逸出停止后,使反应混合物回温至室温并在室温下搅拌16h。在搅拌下小心地加入hcl水溶液(1mol/l),直至ph~6
‑
7。用乙酸乙酯(3x100ml)对所得溶液进行萃取,并将有机层混合。合并的有机相经无
水硫酸钠干燥,过滤,并减压浓缩。将残余物通过硅胶快速色谱纯化,用乙酸乙酯/石油醚(1/9)洗脱,以得到标题化合物(23.01g,85%收率),为无色油状物。lcms(esi):[m h]
=503.4。
[0349]
步骤3.(2r,3r)
‑3‑
((r)
‑2‑
((叔丁氧羰基)氨基)丙氧基)
‑2‑
甲基壬酸
[0350]
在25℃,向(2s,3r)
‑3‑
((r)
‑2‑
((叔丁氧羰基)氨基)丙氧基)
‑2‑
甲基壬酸(r)
‑2‑
((叔丁氧羰基)氨基)丙酯(5.02g,9.98mmol)在甲醇(50ml)中的溶液中加入氢氧化锂(2.3g,99.87mmol)在水(23ml)中的溶液
°
。将所得溶液在30℃
°
搅拌16h。将所得溶液减压浓缩至大约1/3的体积(蒸发甲醇)。小心地加入hcl(1mol/l)溶液直至ph~6。所得溶液用乙酸乙酯(3x100ml)萃取。合并的有机层以无水硫酸钠干燥并减压浓缩。将残余物通过硅胶快速色谱纯化,用乙酸乙酯/石油醚(1/6)洗脱,以得到标题化合物(3.03g,87%收率),为无色油状物。lcms(esi):[m h]
=346.2;1h nmr(300mhz,dmso
‑
d6)δ12.11(s,1h),6.56(d,j=8.1hz,1h),3.53
–
3.15(m,4h),2.64
–
2.60(m,1h),1.37(s,9h),1.35
–
1.25(m,10h),0.99(d,j=6.9hz,3h),0.95(d,j=7.2hz,3h),0.86(t,j=6.6hz,3h)。
[0351]
中间体3.(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑2‑
甲基壬酸
[0352][0353]
向(2s,3r)
‑3‑
[(2r)
‑2‑
(叔丁氧羰基氨基)丙氧基]
‑2‑
甲基
‑
壬酸(1.15g,3.33mmol,中间体2)在二氧六环(10ml)中的溶液中加入hcl/二氧六环的溶液(40ml,4mol/l)。将混合物在25℃
°
搅拌1h,并减压浓缩。将残余物用水(10ml)和thf(30ml)稀释,并在0℃
°
加入氢氧化锂(320.0mg,13.33mmol)在水(5ml)中溶液,然后在0℃
°
加入氯甲酸9
‑
芴基甲酯(1.03g,3.99mmol)在thf(5ml)中的溶液。将反应混合物在25℃
°
搅拌4h,并减压浓缩。残余物用水(10ml)稀释,小心地加入hcl水溶液(1m)直至ph~6。将所得溶液减压浓缩。将残余物通过硅胶快速色谱纯化,用pe/etoac(2/1)洗脱,以得到标题化合物(1.12g,72%收率),为无色油状物。lcms(esi):[m h]
=468.2;1h nmr(400mhz,dmso
‑
d6)δ12.16(s,1h),7.88(d,j=7.6hz,2h),7.69(d,j=7.6hz,2h),7.43
‑
7.39(m,2h),7.34
‑
7.30(m,2h),7.14(d,j=8.4hz,1h),4.28
‑
4.18(m,3h),3.64
‑
3.47(m,2h),3.36
‑
3.21(m,2h),2.67
‑
2.60(m,1h),1.34
‑
1.18(m,10h),1.04(d,j=6.4hz,3h),0.96(d,j=7.2hz,3h),0.80(t,j=6.4hz,3h)。
[0354]
中间体4.(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑2‑
甲基
‑4‑
(螺[3.3]庚烷
‑2‑
基)丁酸
[0355][0356]
步骤1.2
‑
(螺[3.3]庚烷
‑2‑
基)乙烷
‑1‑
醇
[0357][0358]
在0℃,向膦酰基乙酸三乙酯(11.47g,51.18mmol)在thf(150ml)中的溶液中加入nah(2.23g,55.84mmol,在矿物油中的60%溶液)。将反应混合物在室温搅拌1h并冷却至0℃。在0℃,分批加入螺[3.3]庚烷
‑2‑
酮(5.13g,46.53mmol)。将反应混合物在室温搅拌2h,然后通过加入hcl(100ml,在水中的1mol/l溶液)猝灭。混合物用乙酸乙酯(100ml x 3)萃取,合并的有机相以硫酸钠干燥,并减压蒸发。残余物通过硅胶快速色谱纯化,用石油醚/乙酸乙酯(5/1)洗脱,以得到2
‑
螺[3.3]庚烷
‑2‑
亚基乙酸乙酯(8.02g,95%收率),为黄色油状物。
[0359]
在0℃,向2
‑
螺[3.3]庚烷
‑2‑
亚基乙酸乙酯(8.02g,44.47mmol)在乙酸乙酯(150ml)中的溶液中加入钯(0.8g,碳上负载10%)。将混合物抽真空并再充入氢气,并在氢气球下于室温搅拌5h。过滤混合物并减压蒸发滤液。将所得残余物溶解在thf(150ml)中,然后在0℃加入lialh4(2.48g,65.19mmol)。将反应混合物在室温搅拌3h,并通过小心地加入水(50ml)猝灭。通过过滤移除固体,并且用dcm(100ml x 3)萃取滤液。合并的有机相以硫酸钠干燥,并减压浓缩。将残余物通过硅胶快速色谱纯化,用pe/etoac(2/1)洗脱,以得到标题化合物(5.52g,90%收率),为无色油状物。tlc r
f
=0.4,pe/etoac=2/1。
[0360]
步骤2. 2
‑
(螺[3.3]庚烷
‑2‑
基)乙醛
[0361][0362]
向2
‑
螺[3.3]庚烷
‑2‑
基乙醇(5.52g,39.37mmol)在dcm(300ml)中的溶液中加入氯铬酸吡啶鎓(12.63g,58.74mmol)。将反应混合物在室温搅拌3h。经由过滤移除固体。减压蒸发滤液,并且将粗产物直接用于下一步。tlc r
f
=0.5,pe/ea=4/1。
[0363]
步骤3.(2r,3r)
‑3‑
羟基
‑2‑
甲基
‑4‑
(螺[3.3]庚烷
‑2‑
基)丁酸
[0364][0365]
使用2
‑
(螺[3.3]庚烷
‑2‑
基)乙醛代替庚醛并按照类似于那些针对中间体1描述的过程制备标题化合物。lcms(esi):[m
‑
h]
‑
=211.1。
[0366]
步骤4.(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑2‑
甲基
‑4‑
(螺[3.3]庚烷
‑2‑
基)丁酸
[0367]
按照类似于那些针对中间体3描述的过程,使用(2r,3r)
‑3‑
羟基
‑2‑
甲基
‑4‑
(螺[3.3]庚烷
‑2‑
基)丁酸制备标题化合物。lcms(esi):[m h]
=492.3。
[0368]
中间体5.(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑4‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)
‑2‑
甲基丁酸
[0369][0370]
步骤1. 2
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)乙烷
‑1‑
醇
[0371][0372]
在0℃
°
,向nah(1.93g,48.3mmol,在矿物油中的60%溶液)在1,2
‑
二甲氧基乙烷(30ml)中的悬浮液中加入膦酰基乙酸三乙酯(11.7g,52.3mmol)。将所得混合物在0℃
°
搅拌15min。然后加入双环[3.2.1]辛烷
‑3‑
酮(4.06g,32.7mmol)在1,2
‑
二甲氧基乙烷(20ml)中的溶液。将所得溶液在0℃
°
搅拌15min并在室温搅拌3天,倒入冰冷的hcl溶液(在水中的1mol/l溶液,100ml)中。所得溶液用乙酸乙酯(100ml x 4)萃取,并合并有机层。有机层经无水硫酸钠干燥并真空浓缩。残余物通过硅胶快速色谱法纯化,用etoac/pe(1/4)洗脱,以得到2
‑
(3
‑
双环[3.2.1]辛亚基)乙酸乙酯(2.51g)。将该物质溶解在乙酸乙酯(100ml)中。加入一滴tfa,然后将烧瓶抽真空并用氮气冲洗3次。添加钯(1.3g,碳上负载10%)。将系统抽真空并充入氢气。将所得混合物在氢气球下于室温搅拌过夜。滤除固体,真空浓缩滤液,以得到粗制2
‑
(3
‑
双环[3.2.1]辛烷基)乙酸乙酯(6.53g)。将该物质溶解在thf(120ml)中并在0℃
°
和氮气下用氢化铝锂(2.31g,60.7mmol)分小份处理。使反应混合物回温至室温并在该温度搅拌2小时,在冰浴上冷却,并通过依次小心地加入2.3ml水、2.3ml氢氧化钠水溶液(25%)和2.3ml水猝灭。经由过滤移除沉淀,并真空蒸发滤液。将残余物通过硅胶快速色谱纯化,用乙酸乙酯/石油醚(11
‑
15%乙酸乙酯)洗脱,以得到标题化合物(2.66g,84%收率),为无色油状物。1h nmr(400mhz,dmso
‑
d6)δ4.24(t,j=5.2hz,1h),3.39
–
3.34(m,2h),2.13
–
2.12(m,2h),1.75
–
1.56(m,3h),1.48
–
1.32(m,5h),1.29
–
1.24(m,3h),1.01
–
0.94(m,2h)。1h nmr表明内型和外型异构体的比例为~6:1。
[0373]
步骤2. 2
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)乙醛
[0374][0375]
向2
‑
(3
‑
双环[3.2.1]辛烷基)乙醇(4.21g,27.29mmol)在dcm(150ml)中的溶液中加入氯铬酸吡啶鎓(8.86g,41.21mmol)。将反应混合物在室温搅拌3h。经由过滤移除固体,并减压蒸发滤液。将粗产物不经纯化即直接用于下一步。tlc r
f
=0.5,pe/ea=4/1。
[0376]
步骤3.(2r,3r)
‑4‑
((1r,35,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)
‑3‑
羟基
‑2‑
甲基丁酸
[0377][0378]
在
‑
10℃,将三氟甲磺酸(3.66g,24.4mmol)滴加到三乙基硼烷(24.9ml,24.9mmol,在thf中的1m溶液)在dcm(30ml)中的溶液中,将反应混合物在室温搅拌30min。然后在0℃加入1
‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)丙烷
‑1‑
酮(5.95g,21.92mmol)和dipea(3.63g,28.14mmol)在dcm(30ml)中的溶液。将反应混合物在0℃搅拌30min。然后将反应混合物在
‑
78℃搅拌。加入四氯化钛(28.4ml,在dcm中的1m溶液)。然后加入2
‑
(3
‑
双环[3.2.1]辛烷基)乙醛(3.96g,26.01mmol)在dcm(40ml)中的溶液。将反应混合物在
‑
78℃搅拌2h。将反应混合物倒入饱和氯化铵水溶液(100ml)中,并且用dcm(150ml x 3)萃取。合并的有机相以na2so4干燥并减压蒸发。残余物通过快速硅胶色谱纯化,用石油醚/乙酸乙酯(5/1)洗脱,以得到(2r,3r)
‑4‑
((1r,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)
‑1‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)
‑3‑
羟基
‑2‑
甲基丁烷
‑1‑
酮(7.45g,17.6mmol,80.2%收率),为黄色油状物。
[0379]
在0℃,将lioh(1.67g,69.52mmol)在h2o(40ml)中的溶液加入(2r,3r)
‑4‑
((1r,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)
‑1‑
((3ar,6s,7as)
‑
8,8
‑
二甲基
‑
2,2
‑
二氧化四氢
‑
3h
‑
3a,6
‑
甲桥苯并[c]异噻唑
‑
1(4h)
‑
基)
‑3‑
羟基
‑2‑
甲基丁烷
‑1‑
酮(7.37g,17.39mmol)在乙腈(120ml)
°
中的溶液中。将反应混合物在室温搅拌16h,然后通过加入hcl(在水中的1mol/l溶液)酸化至ph~6,并且用乙酸乙酯(100ml x 3)萃取。合并的有机相以na2so4干燥并减压蒸发。将残余物通过硅胶快速色谱纯化,用石油醚/乙酸乙酯(5/1)洗脱,以得到标题化合物(2.96g,75%收率,内型/外型=6/1),为淡红色固体。将混合物(2.96g)在60℃
°
溶解于石油醚/乙酸乙酯(5/1,100ml)中,并冷却至0℃
°
。过滤收集所得结晶固体,以得到纯内型异构体形式的标题化合物(1.05g)。lcms(esi):[m
‑
h]
‑
=225.2.
[0380]
步骤4.(2r,3r)
‑4‑
[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]
‑3‑
[(2r)
‑2‑
(叔丁氧羰基氨基)丙氧基]
‑2‑
甲基
‑
丁酸[(2r)
‑2‑
(叔丁氧羰基氨基)丙酯]
[0381][0382]
向步骤3的羟基酸(3.6g,15.9mmol)在无水thf(36ml)中的溶液中加入(4r)
‑4‑
甲基
‑
2,2
‑
二氧代
‑
噁噻唑烷
‑3‑
甲酸叔丁酯(11.33g,47.75mmol)和tbaf(24ml,在thf中的1m溶液),然后在0℃
°
于氮气下分小份加入nah(2.55g,63.68mmol,在矿物油中的60%溶液)。将反应混合物在室温搅拌过夜并用hcl(在水中的1m溶液)猝灭至ph~4
‑
5。加入乙酸乙酯
(200ml)并分离各相。水相用乙酸乙酯(2x)萃取。合并有机层,经无水硫酸钠干燥并在真空下浓缩。将残余物通过硅胶快速色谱纯化,用乙酸乙酯和石油醚(1:9)洗脱,以得到标题化合物(3.412g,39.6%收率),为无色油状物。lcms(esi):[m h]
=541.4。
[0383]
步骤5.(2r,3r)
‑4‑
[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]
‑3‑
[(2r)
‑2‑
(叔丁氧羰基氨基)丙氧基]
‑2‑
甲基
‑
丁酸
[0384][0385]
在室温,将lioh(1.45g,63.1mmol)在水中的(15ml)溶液加入(2r,3r)
‑4‑
[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]
‑3‑
[(2r)
‑2‑
(叔丁氧羰基氨基)丙氧基]
‑2‑
甲基
‑
丁酸[(2r)
‑2‑
(叔丁氧羰基氨基)丙酯](3.41g,6.31mmol)在meoh(35ml)中的溶液中。将所得溶液在30℃
°
搅拌过夜。减压蒸馏除去meoh并将残余水溶液用1n hcl溶液酸化至ph 4
‑
5。加入乙酸乙酯(200ml)并分离各相。水相用乙酸乙酯(2x)萃取。合并有机层,经无水硫酸钠干燥并在真空下浓缩。将残余物通过硅胶快速色谱纯化,用乙酸乙酯和石油醚(1:6)洗脱,以得到标题化合物(2.04g,84.3%收率),为无色油状物。lcms(esi):[m h]
=384.3。
[0386]
步骤6.(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑4‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基)
‑2‑
甲基丁酸
[0387]
将hcl/二氧六环的溶液(32ml,4m,160mmol)加入(2r,3r)
‑4‑
[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]
‑3‑
[(2r)
‑2‑
(叔丁氧羰基氨基)丙氧基]
‑2‑
甲基
‑
丁酸(1.84g,4.81mmol)在dcm(8ml)中的溶液中。将混合物在室温搅拌1h并真空浓缩。然后将残余物用水(10ml)和thf(30ml)溶解。在0℃
°
加入lioh(331.8mg,14.43mmol)(溶解于6ml水中)。在0℃,
°
在搅拌下加入芴基甲氧基碳酰氯(1.49g,5.77mmol)(溶解于20ml thf中)。将混合物在25℃
°
搅拌1h。减压蒸馏除去thf并用1n hcl将残余水溶液酸化至ph5
‑
6。加入乙酸乙酯(100ml)并分离各相。水相用乙酸乙酯(2x)萃取。合并有机层,经无水硫酸钠干燥并在真空下浓缩。将残余物通过硅胶快速色谱纯化,用dcm/meoh(49/1)洗脱,以得到标题化合物(2.24g,4.43mmol,92.2%收率),为白色固体。lcms(esi):[m 1]
=506.3;1h nmr(400mhz,dmdo
‑
d6):δ12.17(s,1h),7.88(d,j=7.2hz,2h),7.70(d,j=7.6hz,2h),7.43
‑
7.39(m,2h),7.34
‑
7.30(m,2h),7.15(d,j=8.0hz,1h),4.29
‑
4.18(m,3h),3.64
‑
3.56(m,2h),3.34
‑
3.18(m,2h),2.74
‑
2.70(m,1h),2.20
‑
2.10(m,2h),1.73
‑
1.62(m,1h),1.56
‑
1.41(m,3h),1.39
‑
1.22(m,6h),1.08
‑
0.92(m,9h)。
[0388]
中间体6.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(3
‑
((叔丁氧羰基)氨基)丙基)
‑
l
‑
丝氨酸
[0389][0390]
步骤1.n
‑
((苄氧基)羰基)
‑
o
‑
(3
‑
硝基丙基)
‑
l
‑
丝氨酸甲酯
[0391][0392]
在0℃,在氮气下
°
向(s)
‑
氮杂环丙烷
‑
1,2
‑
二甲酸1
‑
苄酯2
‑
甲酯(11.32g,48.14mmol)和3
‑
硝基丙烷
‑1‑
醇(5.06g,48.12mmol)在氯仿(150ml)中的溶液中加入bf3.et2o(0.1ml)。使反应混合物回温至室温,并在室温搅拌16h在。减压下移除溶剂。将残余物通过硅胶快速色谱纯化,用石油醚/乙酸乙酯(5/1)洗脱,以得到标题化合物(11.02g,67%收率),为无色油状物。lcms(esi):[m h]
=341.1。
[0393]
步骤2.n
‑
((苄氧基)羰基)
‑
o
‑
(3
‑
((叔丁氧羰基)氨基)丙基)
‑
l
‑
丝氨酸甲酯
[0394][0395]
向n
‑
((苄氧基)羰基)
‑
o
‑
(3
‑
硝基丙基)
‑
l
‑
丝氨酸甲酯(11.02g,32.4mmol)在乙醇(80ml)和水(80ml)中的溶液中加入nh4cl(8.75g,162mmol),然后加入铁粉(9.07g,162mmol)。将反应混合物在回流下搅拌4h。滤除固体,并且用乙醇洗涤。减压浓缩滤液以移除大约一半的体积。将残余物分配在水和乙酸乙酯之间。水相用乙酸乙酯(2x)萃取。合并的有机相以硫酸钠干燥并减压浓缩以得到残余物(8.89g),将其重新溶解在dcm(100ml)中,然后加入二碳酸二叔丁酯(9.36g,42.95mmol),然后加入三乙胺(8.68g,85.89mmol)。将反应混合物在室温搅拌2h。减压蒸发溶剂。将残余物通过硅胶快速色谱纯化,用乙酸乙酯/石油醚(0
‑
50%)洗脱,以得到标题化合物(9.22g,78%收率),为黄色油状物。lcms(esi):[m h]
=411.2。
[0396]
步骤3.(3
‑
((叔丁氧羰基)氨基)丙基)
‑
l
‑
丝氨酸甲酯
[0397][0398]
在0℃,在氮气下向n
‑
((苄氧基)羰基)
‑
o
‑
(3
‑
((叔丁氧基羰基)氨基)丙基)
‑
l
‑
丝氨酸甲酯(9.22g,22.48mmol)在乙酸乙酯(100ml)中的溶液中加入钯(碳载,10wt.%,0.93g)。将烧瓶抽真空并回填氢气,然后在氢气球下在25℃搅拌3h。反应混合物经硅藻土过滤,减压浓缩滤液,给出标题化合物。lcms(esi)[m h]
=277.2。
[0399]
步骤4.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(3
‑
((叔丁氧羰基)氨基)丙基)
‑
l
‑
丝氨酸
[0400]
在环境温度,向o
‑
(3
‑
((叔丁氧基羰基)氨基)丙基)
‑
l
‑
丝氨酸甲酯(5.90g,21.35mmol)在thf(150ml)和水(150ml)中的溶液中加入氢氧化锂(2.05g,85.41mmol)。将所得混合物在25℃搅拌3h,冷却至0℃,然后滴加氯甲酸9
‑
芴基甲酯(8.71g,33.74mmol)在thf(10ml)中的溶液。将反应混合物在0℃搅拌2h。通过加入1m hcl将反应混合物酸化至ph~6并用乙酸乙酯(3x100ml)萃取。合并的有机相用na2so4干燥,过滤,并减压浓缩。将残余物通过硅胶快速色谱纯化,用石油醚/乙酸乙酯(5/1)洗脱,以得到标题化合物(6.12g,56%收率),为白色固体。lcms(esi):[m h]
=485.1。1h nmr(300mhz,dmso
‑
d6)δ12.78(s,1h),7.90(d,j=7.5hz,2h),7.75(d,j=7.5hz,2h),7.64(d,j=8.1hz,1h),7.45
‑
7.37(m,2h),7.32
‑
7.30(m,2h),6.79(t,j=5.7hz,1h),4.30
‑
4.09(m,4h),3.67
‑
3.61(m,2h),3.46
‑
3.39(m,2h),3.00
‑
2.94(m,2h),1.63
‑
1.56(m,2h),1.37(s,9h)。
[0401]
中间体7.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
((1r,3s)
‑3‑
((叔丁氧羰基)氨基)环丁基)
‑
l
‑
丝氨酸
[0402][0403]
步骤1.n
‑
((苄氧基)羰基)
‑
o
‑
((1r,3s)
‑3‑
((叔丁氧羰基)氨基)环丁基)
‑
l
‑
丝氨酸甲酯
[0404][0405]
将反式
‑
n
‑
(3
‑
羟基环丁基)氨基甲酸叔丁酯(7.95g,42.46mmol)和bf3.et2o(1.23g,8.54mmol)在氯仿(100ml)中的溶液在0℃
°
搅拌。然后在0℃
°
滴加(s)
‑
氮杂环丙烷
‑
1,2
‑
二甲酸1
‑
苄酯2
‑
甲酯(10.0g,42.51mmol)在氯仿(10ml)中的溶液。然后将反应混合物在25℃
°
搅拌20h并减压浓缩。将残余物通过硅胶快速色谱纯化,用石油醚/乙酸乙酯(2/1)洗脱,以得到标题化合物(2.5g,13%收率),为白色固体。lcms(esi):[m h]
=423.2。
[0406]
步骤2.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
((1r,3s)
‑3‑
((叔丁氧羰基)氨基)环丁基)
‑
l
‑
丝氨酸
[0407]
在氮气下向(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
[3
‑
(叔丁氧羰基氨基)环丁氧基]丙酸甲酯(2.5g,5.92mmol)在乙酸乙酯(30ml)中的溶液中加入钯(碳载,10wt.%,1.0g)。将反应混合物抽真空并用氢气回填,并在氢气球下在25℃
°
搅拌1h。滤除催化剂并用乙酸乙酯冲洗。
减压浓缩滤液,以得到(2s)
‑2‑
氨基
‑3‑
[3
‑
(叔丁氧基羰基氨基)环丁氧基]丙酸甲酯(1.7g,粗制),为黄色油状物。向1.5g的该物质中加入氢氧化锂(250.0mg,10.42mmol)在水(5ml)和二氧六环(15ml)中的溶液。将所得溶液在25℃
°
搅拌1h。然后加入氯甲酸9
‑
芴基甲酯(1.61g,6.24mmol)。将混合物在25℃
°
搅拌1h,用水(20ml)稀释并用1n hcl酸化至ph 6。所得溶液用乙酸乙酯(3x100ml)萃取。合并的有机层以无水硫酸钠干燥,过滤,并减压浓缩。将残余物通过硅胶快速色谱纯化,用石油醚/乙酸乙酯(4/1)洗脱,以得到标题化合物(1.85g,71%收率),为白色固体。lcms(esi):[m h]
=497.2;1h nmr(400mhz,dmso
‑
d6)δ12.76(s,1h),7.89(d,j=7.6hz,2h),7.76(d,j=7.6hz,2h),7.64(d,j=8.4hz,1h),7.44
–
7.40(m,2h),7.35
–
7.31(m,2h),7.16(d,j=7.2hz,1h),4.29
–
4.17(m,4h),4.04
–
3.98(m,2h),3.58
–
3.49(m,2h),2.17
–
2.01(m,4h),1.37(s,9h)。
[0408]
表2.按照类似于那些针对中间体7描述的方法制备下面说明的中间体。
[0409]
[0410][0411]
中间体8.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸
[0412][0413]
步骤1.(s)
‑6‑
(2
‑
(((苄氧基)羰基)氨基)
‑3‑
甲氧基
‑3‑
氧代丙氧基)
‑2‑
氮杂螺[3.3]庚烷
‑2‑
甲酸叔丁酯
[0414][0415]
向200ml烧瓶中加入6
‑
羟基
‑2‑
氮杂螺[3.3]庚烷
‑2‑
甲酸叔丁酯(2.511g,11.18mmol)和氯仿(30ml),然后加入三氟化硼乙醚合物(0.30ml,2.4mmol),然后滴加(s)
‑
氮杂环丙烷
‑
1,2
‑
二甲酸1
‑
苄酯2
‑
甲酯(2.2ml,11mmol)。将所得混合物在室温搅拌6h,然后用甲醇猝灭并真空蒸发。粗产物通过硅胶快速色谱纯化(200g二氧化硅,溶剂梯度:20
‑
80%乙酸异丙酯在庚烷中),以得到1.093g(22%)的标题化合物。lcms(esi):[m h
‑
boc]
=349.15;1h nmr(400mhz,dmso
‑
d6)δ7.69(d,j=8.0hz,1h),7.40
‑
7.27(m,5h),5.04(s,2h),4.25(m,1h),3.85
‑
3.69(m,5h),3.64(s,3h),3.54
‑
3.42(m,2h),2.41
‑
2.29(m,2h),1.99(s,2h),1.35(s,9h)。
[0416]
步骤2.n
‑
((苄氧基)羰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸
[0417][0418]
向(s)
‑6‑
(2
‑
(((苄氧基)羰基)氨基)
‑3‑
甲氧基
‑3‑
氧代丙氧基)
‑2‑
氮杂螺[3.3]庚烷
‑2‑
甲酸叔丁酯(1.093g,2.436mmol)在1,4
‑
二氧六环(12ml)中的溶液中加入氢氧化锂(1.0m水溶液,4ml,4.0mmol)。将反应混合物在室温搅拌90min。将反应混合物倒入10%柠檬
酸水溶液中并用dcm(2x50ml)萃取。合并的dcm萃取液以硫酸镁干燥,过滤,真空蒸发,得到1.0618g(定量)标题化合物。lcms(esi):[m h]
=435.3。
[0419]
步骤3.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸
[0420]
向n
‑
((苄氧基)羰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸在乙醇(10ml)中的溶液中加入钯(碳载,10wt.%)(250.5mg,0.2354mmol)。将反应混合物在氢气球下于室温搅拌4h。将反应混合物通过硅藻土过滤,用2x100ml乙醇冲洗并真空蒸发,以得到定量收率的o
‑
(2
‑
(叔丁氧基羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸。lcms(esi):[m h
‑
tbu]
=245.2。
[0421]
将所得物质溶解在1,4
‑
二氧六环(10ml)和水(10ml)中,并用碳酸氢钠(626.8mg,7.44mmol)和(2,5
‑
二氧代吡咯烷
‑1‑
基)碳酸(9h
‑
芴
‑9‑
基)甲酯(1.192g,3.534mmol)处理。将反应混合物在室温搅拌15h。将反应混合物倒入10%柠檬酸水溶液中并用dcm(3x100ml)萃取。合并的有机萃取液经硫酸镁干燥,过滤并且真空蒸发。粗产物通过硅胶快速色谱纯化(80g二氧化硅,溶剂梯度:0
‑
100%乙酸异丙酯在庚烷中),得到779.1mg(61%)标题化合物,为白色泡沫状物。lcms(esi):[m h]
=523.3。
[0422]
中间体9.(2s)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑4‑
(4
‑
((苄氧基)羰基)哌嗪
‑1‑
基)
‑3‑
((叔丁基二甲基甲硅烷基)氧基)丁酸
[0423][0424]
步骤1.(s)
‑2‑
((叔丁氧羰基)氨基)丁
‑3‑
烯酸苄酯
[0425][0426]
将(s)
‑2‑
氨基丁
‑3‑
烯酸盐酸盐(1.0g,7.3mmol)、二碳酸二叔丁酯(1.6g,7.3mmol)和碳酸氢钠(1.2g,14.6mmol)在thf:水(1∶1,6ml)中的混合物在60℃加热2h。将反应混合物冷却并浓缩。将残余物溶解在dmf(3ml)中并加入苄基溴(1.5g,8.7mmol),并将混合物在室温搅拌过夜。将反应混合物用水稀释,并且用20%庚烷/ipac萃取。有机层用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过硅胶快速色谱纯化,用庚烷/ipac(0
‑
50%)洗脱,以得到标题化合物(0.87g,55%)。1h nmr(400mhz,氯仿
‑
d)δ7.42
‑
7.31(m,5h),5.91(ddd,j=16.3,10.2,5.3hz,1h),5.34(ddd,j=17.2,1.9,0.7hz,1h),5.25(ddd,j=10.3,1.8,
0.6hz,1h),5.23
‑
5.15(m,3h),4.92(s,1h),1.44(s,9h);lcms(esi):[m h]
=292。
[0427]
步骤2.(2s)
‑2‑
((叔丁氧羰基)氨基)
‑2‑
(氧杂环丙烷
‑2‑
基)乙酸苄酯
[0428][0429]
将(2s)
‑2‑
(叔丁氧基羰基氨基)丁
‑3‑
烯酸酯(1.35g,4.6mmol)和m
‑
cpba(2.0g,70%)在dcm(20ml)中的混合物在室温搅拌过夜。将反应混合物浓缩并与10%dcm/庚烷一起研磨。通过过滤移除固体并用10%dcm/庚烷充分洗涤。浓缩滤液,并且将残余物通过快速色谱纯化(硅胶,0
‑
100%庚烷/ipac),得到标题化合物(0.9g,63%)。lcms(esi):[m h]
=308并且[m na]
=330.1;1h nmr(400mhz,dmso
‑
d6)δ7.52(d,j=7.9hz,1h),7.42
‑
7.22(m,5h),5.89(ddd,j=17.0,10.4,6.4hz,1h),5.33(d,j=17.3hz,1h),5.22(d,j=10.4hz,1h),5.14(q,j=12.7hz,2h),4.66(t,j=7.2hz,1h),1.38(d,j=2.4hz,9h)。
[0430]
步骤3. 4
‑
((3s)
‑4‑
(苄氧基)
‑3‑
((叔丁氧羰基)氨基)
‑2‑
羟基
‑4‑
氧代丁基)哌嗪
‑1‑
甲酸苄酯
[0431][0432]
将(2s)
‑2‑
(叔丁氧基羰基氨基)
‑2‑
(氧杂环丙烷
‑2‑
基)乙酸苄酯(0.8g,2.6mmol)和哌嗪
‑1‑
甲酸苄酯(0.58g,2.6mmol)在thf(5ml)中的混合物在70℃加热4h,然后冷却并浓缩。将残余物通过快速色谱纯化(硅胶,0
‑
10%甲醇/dcm),获得标题化合物(0.92g,67%)。lcms(esi):[m h]=528.4;1h nmr(400mhz,dmso
‑
d6)δ7.43
‑
7.25(m,10h),6.59(d,j=9.1hz,1h),5.23
‑
5.12(m,2h),5.06(s,2h),4.30(dd,j=9.1,3.0hz,1h),4.05(q,j=6.3,5.3hz,1h),3.50
‑
3.34(m,4h),2.44
‑
2.22(m,5h),1.38(s,9h)。
[0433]
步骤4.4
‑
[(3s)
‑4‑
苄氧基
‑3‑
(叔丁氧羰基氨基)
‑2‑
[叔丁基(二甲基)甲硅烷基]氧基
‑4‑
氧代
‑
丁基]哌嗪
‑1‑
甲酸苄酯
[0434][0435]
向4
‑
((3s)
‑4‑
(苄氧基)
‑3‑
((叔丁氧基羰基)氨基)
‑2‑
羟基
‑4‑
氧代丁基)哌嗪
‑1‑
甲酸苄酯(0.9g,1.7mmol)在干燥thf(10ml)中的溶液中加入2,6
‑
二甲基吡啶(0.6ml,5.0mmol),然后加入三氟甲磺酸叔丁基二甲基硅酯(0.9g,3.4mmol),将反应混合物搅拌1h,
然后用水稀释并用ipac萃取。有机层用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过快速色谱纯化(硅胶,0
‑
5%meoh/dcm),得到目标化合物(0.53g,50%)。lcms(esi):[m h]=642.5;1h nmr(400mhz,dmso
‑
d6)δ7.49
‑
7.27(m,10h),6.49(d,j=9.3hz,1h),5.17(d,j=6.9hz,2h),5.11(s,2h),4.43(td,j=9.2,3.2hz,1h),4.30
‑
4.14(m,1h),3.48
‑
3.38(m,4h),2.50
‑
2.27(m,6h),1.42(s,9h),0.84(d,j=1.2hz,9h),0.13
‑
0.05(m,6h)。
[0436]
步骤5.(2s)
‑2‑
氨基
‑4‑
(4
‑
((苄氧基)羰基)哌嗪
‑1‑
基)
‑3‑
((叔丁基二甲基甲硅烷基)氧基)丁酸
[0437][0438]
将4
‑
[(3s)
‑4‑
苄氧基
‑3‑
(叔丁氧羰基氨基)
‑2‑
[叔丁基(二甲基)甲硅烷基]氧基
‑4‑
氧代
‑
丁基]哌嗪
‑1‑
甲酸苄酯(0.5g,0.78mmol)溶解在thf:甲醇(4ml;3∶1)中,加入lioh(1n,2当量),并将混合物搅拌1h。将反应混合物用少量柠檬酸水溶液酸化并且用ipac萃取。有机层用硫酸钠干燥并浓缩。将残余物溶解在dcm(2ml)中并加入hcl(4ml,在二氧六环中的4n溶液)并将反应混合物搅拌1h。浓缩反应混合物并原样用于下一步。
[0439]
步骤6.(2s)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑4‑
(4
‑
((苄氧基)羰基)哌嗪
‑1‑
基)
‑3‑
((叔丁基二甲基甲硅烷基)氧基)丁酸
[0440]
将(2s)
‑2‑
氨基
‑4‑
(4
‑
((苄氧基)羰基)哌嗪
‑1‑
基)
‑3‑
((叔丁基二甲基甲硅烷基)氧基)丁酸(从步骤5直接使用)溶解在二氧六环:水(5ml,1∶1)中,并加入碳酸氢钠(0.2g,2.3mmol)和fmoc
‑
osu(0.32g,0.93mmol)。将混合物搅拌过夜,用柠檬酸酸化并用ipac萃取。将有机层用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过快速色谱法(0
‑
10%meoh/dcm)纯化,以获得0.38g的标题化合物。lcms(esi):[m h]
=674.5。
[0441]
中间体10.fmoc
‑
别苏氨酸(bzl)
‑
oh
[0442][0443]
步骤1.o
‑
苄基
‑
n
‑
(叔丁氧羰基)
‑
l
‑
别苏氨酸
[0444][0445]
将(2s,3s)
‑2‑
氨基
‑3‑
羟基丁酸(85g,714mmol)、甲醇(450ml)、水(450ml)和碳酸氢钠(150g,1.79mol)放入用氮气惰性气氛吹扫并保持的2000ml 3颈圆底烧瓶中。随后,历时60min,在0℃分批加入boc2o(187g,856.82mmol,1.20当量)。将所得溶液在25℃搅拌12h。将所得混合物减压浓缩。将所得溶液用500ml水稀释。用3x500ml乙醚萃取所得溶液,并合并水层。将水溶液的ph值用氯化氢(1mol/l)调节至3。用3x500ml乙酸乙酯萃取所得溶液,合并有机层,并以无水硫酸钠干燥,然后减压浓缩。这产生了110g(67%)(2s,3s)
‑2‑
[[(叔丁氧基)羰基]氨基]
‑3‑
羟基丁酸
[0446]
将(2s,3s)
‑2‑
[[(叔丁氧基)羰基]氨基]
‑3‑
羟基丁酸(110g,501.74mmol)和dmf(1100ml)放入用氮气惰性气氛吹扫并保持的2000ml 3颈圆底烧瓶中。随后,历时60min,在0℃分批加入氢化钠(84.4g,3.52mol)。将所得溶液在0℃搅拌2h。在0℃,历时30min,在搅拌下向其中滴加苄基溴(85.4g,499.33mmol)。所得溶液在0℃搅拌5h。然后通过添加1000ml水/冰来猝灭反应。用3x2000ml乙酸乙酯萃取所得溶液,并合并水层。将溶液的ph值用氯化氢水溶液(1mol/l)调节至3。用3x2000ml乙酸乙酯提取所得溶液,合并有机层,并经无水硫酸钠干燥,然后在真空下浓缩。粗产物从比率为10:1的石油醚:乙酸乙酯中重结晶。这产生60g(37%)标题化合物。
[0447]
步骤2.fmoc
‑
别苏氨酸(bzl)
‑
oh
[0448]
将o
‑
苄基
‑
n
‑
(叔丁氧羰基)
‑
l
‑
别苏氨酸(60g,193.95mmol)、二氧六环(600ml)、氯化氢(在二氧六环中的4n溶液,600ml)放入2000ml3颈圆底烧瓶中。将所得溶液在25℃搅拌12h。将反应混合物真空浓缩。这产生50g(粗制)o
‑
苄基
‑
l
‑
别苏氨酸盐酸盐,为白色固体。
[0449]
将o
‑
苄基
‑
l
‑
别苏氨酸盐酸盐(50g,203.50mmol)、二氧六环(500ml)、水(500ml)、碳酸钠(43.3g,408.53mmol)和fmoconsu(69g,1.00当量)放入用氮气惰性气氛吹扫和保持的2000ml3颈圆底烧瓶中。将所得溶液在25℃搅拌12h。将溶液的ph值用氯化氢(1mol/l)调至3。用3x500ml乙酸乙酯提取所得溶液,合并有机层并真空浓缩。将残余物通过硅胶色谱纯化,用乙酸乙酯/石油醚(1∶1)洗脱。这产生53.4g(两步总收率58%)的标题化合物,为白色固体。lcms(esi):[m h]
=432;1h nmr(300mhz,cdcl3)δ12.85(1h,s),7.88
‑
7.90(2h,m),7.74
‑
7.77(2h,m),7.64
‑
7.67(1h,m),7.41
‑
7.44(2h,m),7.22
‑
7.39(7h,m),4.46
‑
4.50(2h,m),4.34
‑
4.38(1h,m),4.20
‑
4.30(3h,m),3.89
‑
3.93(1h,m),1.16(3h,s)。
[0450]
中间体11.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(4
‑
(((叔丁氧羰基)氨基)甲基)
‑2‑
甲基苯基)
‑
l
‑
丝氨酸
[0451][0452]
步骤1.n
‑
[(4
‑
溴
‑3‑
甲基
‑
苯基)甲基]氨基甲酸叔丁酯
[0453][0454]
向(4
‑
溴
‑3‑
甲基
‑
苯基)甲胺(5.00g,25.0mmol)和dipea(6.48g,50.14mmol)在dcm(100ml)中的溶液中滴加boc2o(8.22g,37.7mmol)在10ml的dcm中的溶液。将所得溶液在室温搅拌4h,然后真空浓缩。将残余物通过硅胶快速色谱纯化,用pe/ea(5%of ea)洗脱,以得到标题化合物(6.04g,80.5%收率),为淡黄色固体。1h nmr(400mhz,dmso
‑
d6):δ7.36
‑
7.33(m,3h),7.11(d,j=8.0hz,1h),4.05(d,j=6.0hz,2h),2.25(s,3h),1.39(s,9h)。
[0455]
步骤2.n
‑
[[4
‑
[(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]
‑3‑
甲基
‑
苯基]甲基]氨基甲酸叔丁酯
[0456][0457]
在氮气下,向n
‑
[(4
‑
溴
‑3‑
甲基
‑
苯基)甲基]氨基甲酸叔丁酯(1.00g,3.33mmol)在甲苯(15ml)中的溶液中加入n
‑
[(1s)
‑1‑
(羟基甲基)
‑2‑
[甲氧基(甲基)氨基]
‑2‑
氧代
‑
乙基]氨基甲酸苄酯(943mg,3.34mmol)、[pd(allyl)cl]2(61.0mg,0.167mmol)、t
‑
bubrettphos(162mg,0.334mmol)和cs2co3(2.18g,6.65mmol)。将反应在微波辐射下在60℃
°
搅拌5小时。以相同规模平行重复该反应。合并两次运行并真空浓缩。将残余物通过硅胶快速色谱纯化,用pe/ea(35%of ea)洗脱,以得到标题化合物(895mg,26.7%收率),为淡黄色油状物。lcms(esi):[m na]
=524.2。
[0458]
步骤3.n
‑
[[4
‑
[(2s)
‑2‑
氨基
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]
‑3‑
甲基
‑
苯基]甲基]氨基甲酸叔丁酯
[0459][0460]
在室温,将n
‑
[[4
‑
[(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]
‑3‑
甲基
‑
苯基]甲基]氨基甲酸叔丁酯(950mg,1.89mmol)和10%pd/c(500mg)在乙酸
乙酯(190ml)中的混合物在氢气气氛下搅拌45min。滤除催化剂,并将滤液真空浓缩,以得到标题化合物(668mg,96%收率),为黄色油状物。lcms(esi):[m h]
=368.2。
[0461]
步骤4.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(4
‑
(((叔丁氧羰基)氨基)甲基)
‑2‑
甲基苯基)
‑
l
‑
丝氨酸
[0462]
在0℃
°
将lioh(173mg,7.21mmol)在水(5.0ml)中的溶液加入n
‑
[[4
‑
[(2s)
‑2‑
氨基
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]
‑3‑
甲基
‑
苯基]甲基]氨基甲酸叔丁酯(662.0mg,1.8mmol)在thf(21ml)和h2o(7.0ml)中的溶液中。将反应混合物在室温搅拌过夜并冷却至0℃
°
。然后在0℃
°
滴加芴基甲氧基碳酰氯(930mg,3.6mmol)(溶解在5ml的thf中)。将反应混合物在室温搅拌1h。真空抽除大部分的thf。将残余溶液用水(3.0ml)稀释,并用1m hcl调节至ph 6。将所得溶液真空浓缩,并将残余物通过硅胶快速色谱纯化,用dcm/meoh(10/1)洗脱,以得到标题化合物(648mg,65.8%收率),为白色固体。lcms(esi):[m 1]
=547.3。1h nmr(400mhz,dmso
‑
d6):δ 7.92(d,j=7.6hz,2h),7.85(d,j=8.0hz,2h),7.85(d,j=7.6hz,1h),7.69
–
7.64(m,2h),7.45
–
7.40(m,2h),7.36(d,j=7.2hz,1h),7.32(dd,j=7.2,3.2hz,1h),6.75
–
6.71(m,2h),4.43
–
4.40(m,1h),4.39
–
4.21(m,4h),4.43
–
4.40(m,3h),2.24(s,3h),1.39(s,9h)。
[0463]
中间体12.(2s)
‑4‑
[3
‑
(苄氧敖基氨基)丙氧基]
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丁酸
[0464][0465]
步骤1.(2s)
‑4‑
烯丙氧基
‑2‑
(叔丁氧羰基氨基)丁酸
[0466][0467]
在0℃,
°
在搅拌下向氢化钠(228mg,在矿物油中的60%分散液,5.70mmol)在dmf(20ml)中的悬浮液中滴加boc
‑
l
‑
高丝氨酸(500mg,2.28mmol)在dmf(5ml)中的溶液。当完成加入boc
‑
l
‑
高丝氨酸后,将反应混合物在0
‑
10℃
°
于氮气下再搅拌10min。向该溶液中加入在5.0ml dmf中的烯丙基溴(304mg,2.51mmol)。将反应混合物在室温下搅拌2小时并真空浓缩。加入水(10ml)并用hcl(1m)将混合物酸化至ph~5,并用乙酸乙酯(3x20ml)萃取。合并有机层,经无水硫酸钠干燥并在真空下浓缩。将残余物通过硅胶快速色谱纯化,用在石油醚中的30%乙酸乙酯洗脱,以得到标题化合物(350mg,59.2%收率),为无色油状物。lcms(esi):[m h
‑
boc]
=533.2;1h nmr(300mhz,dmso
‑
d6):δ12.47(s,1h),7.06(d,j=8.1hz,1h),5.94
–
5.80(m,1h),5.29
–
5.10(m,2h),4.07
–
3.90(m,3h),3.40(dd,j=7.2,5.1hz,2h),1.97
–
1.88(m,1h),1.80
–
1.69(m,1h),1.41(s,9h)。
[0468]
步骤2.(2s)
‑4‑
烯丙氧基
‑2‑
(叔丁氧羰基氨基)丁酸甲酯
[0469][0470]
在0℃
°
用在dcm(60ml)和meoh(30ml)中的三甲基甲硅烷基重氮甲烷(13.5ml,在己烷中的2.0m溶液,27.0mmol)处理(2s)
‑4‑
烯丙氧基
‑2‑
(叔丁氧羰基氨基)丁酸(3.50g,13.5mmol)。将反应混合物在室温搅拌30min,然后减压浓缩。将残余物通过快速硅胶快速色谱纯化,用pe/ea 2/1洗脱,以得到标题化合物(3.65g,13.4mmol,99%收率),为无色油状物。1h nmr(300mhz,dmso
‑
d6):δ 7.23(d,j=7.8hz,1h),5.91
–
5.78(m,1h),5.26
–
5.09(m,2h),4.12
–
4.06(m,1h),3.91
–
3.86(m,2h),3.62(s,3h),3.40(dd,j=6.9,5.1hz,2h),1.96
–
1.85(m,1h),1.83
–
1.71(m,1h),1.38(s,9h)。
[0471]
步骤3.(2s)
‑2‑
(叔丁氧羰基氨基)
‑4‑
(3
‑
羟基丙氧基)丁酸甲酯
[0472][0473]
在0℃,
°
在氮气下向(2s)
‑4‑
烯丙氧基
‑2‑
(叔丁氧羰基氨基)丁酸甲酯(4.03g,14.7mmol)在thf(10ml)中的溶液中滴加bh3.thf在thf中的1m溶液(12.5ml,12.5mmol)。将反应混合物在室温搅拌2h。在0℃
°
加入水(0.5ml),然后加入3n naoh(2.5ml,7.5mmol)和30%h2o2(10ml)。将反应混合物在室温搅拌2h。加入50ml水,并加入少量na2so3以消耗过量的h2o2。加入etoac(100ml)并分离各相。水相用etoac(2x)萃取,合并有机相,用na2so4干燥,过滤并减压浓缩。将残余物通过快速硅胶快速色谱纯化,用pe/ea(1/1)洗脱,以得到标题化合物(1.77g,6.09mmol,41.3%收率),为无色油状物。1h nmr(400mhz,dmso
‑
d6):δ 7.21(d,j=7.6hz,1h),4.37(br,1h),4.09
–
4.00(m,1h),3.62(s,3h),3.44
–
3.32(m,6h),1.89
–
1.85(m,1h),1.78
–
1.71(m,1h),1.65
–
1.58(m,2h),1.38(s,9h)。
[0474]
步骤4.(s)
‑4‑
(3
‑
(((苄氧基)敖基)氨基)丙氧基)
‑2‑
((叔丁氧羰基)氨基)丁酸甲酯
[0475][0476]
在0℃
°
将甲磺酰氯(1.19g,10.4mmol)加入(2s)
‑2‑
(叔丁氧羰基氨基)
‑4‑
(3
‑
羟基丙氧基)丁酸甲酯(2.01g,6.91mmol)和三乙胺(2.09g,20.7mmol)在dcm(40ml)中的溶液中。将反应混合物在室温搅拌2小时。加入水(50ml)并分离各相。水相用dcm(2x)萃取,合并有机相,用na2so4干燥,并真空浓缩。
[0477]
将所得残余物溶解在dmf(40ml)中并在0℃
°
用叠氮化钠(2.34g,36.0mmol)处理。将反应混合物在室温搅拌过夜。加入水(30ml),所得混合物用乙酸乙酯(3x)萃取,合并的萃取物用盐水洗涤,用na2so4干燥,过滤并真空浓缩。
[0478]
将所得残余物溶解在乙酸乙酯(40ml)中,并向该混合物中加入10%pd/c(500mg)。将混合物在氢气气氛下于室温搅拌2h。滤除催化剂并减压浓缩滤液。
[0479]
将所得残余物与三乙胺(2.1g,20.8mmol)在dcm(40ml)中合并,并在0℃
°
向该混合物中加入cbz
‑
cl(1.77g,10.4mmol)。将反应在室温搅拌2h,然后真空浓缩。将残余物通过快速硅胶快速色谱纯化,用pe/ea(1/1)洗脱,以得到标题化合物(1.02g,四步总收率34.7%),为无色油状物。lcms(esi):[m h]
=425.4。1h nmr(300mhz,dmso
‑
d6):δ7.39
–
7.27(m,5h),7.25
–
7.21(m,2h),5.00(s,2h),4.11
–
4.01(m,1h),3.62(s,3h),3.44
–
3.26(m,4h),3.07
–
3.00(m,2h),1.94
–
1.84(m,1h),1.81
–
1.71(m,1h),1.65
–
1.58(m,2h),1.38(s,9h)。
[0480]
步骤5.(2s)
‑4‑
[3
‑
(苄氧羰基氨基)丙氧基]
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丁酸
[0481]
在0℃
°
,将hcl/二氧六环的溶液(20ml,80mmol,4m)加入(2s)
‑4‑
[3
‑
(苄氧羰基氨基)丙氧基]
‑2‑
(叔丁氧羰基氨基)丁酸甲酯(950mg,2.24mmol)在dcm(20ml)中的溶液中。将反应在室温搅拌2h,然后减压浓缩。加入thf(60ml),然后在0℃
°
加入lioh(163mg,6.78mmol)在水(20ml)中的溶液。将所得混合物在室温搅拌1h,然后在0℃
°
加入芴基甲氧基碳酰氯(693mg,2.68mmol)在10ml thf中的溶液。将反应混合物在室温搅拌1h,并用1n hcl酸化至ph~5。加入乙酸乙酯(100ml)并分离各相。水相用乙酸乙酯(2x)萃取。将有机相合并,用盐水洗涤,以na2so4干燥,并减压浓缩。将残余物通过硅胶快速色谱纯化,用[dcm]/[meoh](10/1)洗脱,以得到标题化合物(660mg,55.4%收率),为白色固体。lcms(esi):[m h]
=533.2;1h nmr(300mhz,dmso
‑
d6):δ12.59(s,1h),7.89(d,j=7.5hz,2h),7.89(d,j=7.5hz,2h),7.62(d,j=8.4hz,1h),7.44
–
7.37(m,2h),7.34
–
7.21(m,8h),4.99(s,2h),4.30
–
4.19(m,3h),4.09
–
4.02(m,1h),3.41
–
3.31(m,4h),3.07
–
3.01(m,2h),1.96
–
1.88(m,1h),1.81
–
1.73(m,1h),1.66
–
1.57(m,2h)。
[0482]
中间体13.(s)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑2‑
环庚基乙酸
[0483][0484]
向(2s)
‑2‑
氨基
‑2‑
环庚基
‑
乙酸(1.0g,5.84mmol)在dmf(25ml)和水(25ml)中的溶液中加入nahco3(982.0mg,11.69mmol)。在0℃
°
滴加氯甲酸9
‑
芴基甲酯(1.8g,6.98mmol)在thf(5ml)中的溶液。使反应混合物回温至室温并在25℃
°
搅拌24h。小心地加入hcl水溶液(1mol/l)直至ph~6。所得溶液用乙酸乙酯(3x100ml)萃取。将有机层合并,用水(5x)洗涤,以硫酸钠干燥,并减压浓缩。将残余物通过硅胶快速色谱纯化,用dcm/meoh(10/1)洗脱,以得到标题化合物(1.77g,77%收率),为白色固体。lcms(esi):[m h]
=394.2。1h nmr(400mhz,dmso
‑
d6)12.58(s,1h),7.89(d,j=7.6hz,2h),7.76(d,j=7.6hz,2h),7.59(d,j=8.8hz,1h),7.44
‑
7.40(m,2h),7.35
‑
7.30(m,2h),4.31
‑
3.93(m,4h),1.95
‑
1.33(m,13h)。
[0485]
中间体14.(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑2‑
(丁
‑3‑
烯
‑1‑
基)壬
‑8‑
烯酸
[0486][0487]
按照类似于那些针对中间体3描述的方法制备标题化合物。lcms(esi):[m 1] =506.3。
[0488]
中间体15.o
‑
苄基
‑
n
‑
((s)
‑3‑
(((苄氧基)羰基)氨基)
‑2‑
((s)
‑2‑
环己基
‑2‑
((s)
‑4‑
甲基
‑2‑
(甲基氨基)戊酰胺基)乙酰胺基)丙酰基)
‑
l
‑
别苏氨酸
‑
(2
‑
氯三苯甲基树脂)
[0489][0490]
用dmf:dcm的混合物(1∶1,20ml)将2
‑
氯三苯甲基氯树脂(负载0.956mmol/g,5g)溶胀40min。沥干树脂,然后加入(2s,3s)
‑3‑
苄氧基
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丁酸(中间体10)(2.461g,5.70mmol)、dmf(10ml)和dipea(495.6mg,3.84mmol)的溶液。将树脂用氮气鼓泡搅拌4h,沥干,并依次用10ml dcm/meoh/dipea(10/10/1=v/v/v)、10ml dmf、10ml dcm、10ml dmf冲洗。
[0491]
树脂用25ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌1.5h。将树脂沥干并依次用dmf(3x25ml)和dcm(3x25ml)冲洗。用(2s)
‑3‑
(苄氧羰基氨基)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丙酸(4.372g,9.494mmol)的溶液、预先制备的hobt/hatu(1:1)在dmf中的原液(0.4mmol/ml,25ml)和dipea(2.4g,19mmol)处理树脂。将树脂用氮气鼓泡搅拌2.5h,沥干,并依次用10ml dmf、10ml dcm、10ml dmf冲洗。树脂用25ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌1.5h。将树脂沥干并依次用dmf(3x25ml)和dcm(3x25ml)冲洗。用(s)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑2‑
环己基乙酸(3.61g,9.52mmol)的溶液、预先制备的hobt/hatu(1:1)在dmf中的原液(0.4mmol/ml,25ml)和dipea(2.4g,19mmol)处理树脂。将树脂用氮气鼓泡搅拌2.5h,沥干,并依次用10ml dmf、10ml dcm、10ml dmf冲洗。树脂用25ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌1.5h。将树脂沥干并依次用dmf(3x25ml)和dcm(3x25ml)冲洗。用(2s)
‑2‑
{[(9h
‑
芴
‑9‑
基甲氧基)羰基](甲基)氨基}
‑4‑
甲基戊酸(3.492g,9.50mmol)的溶液、预先制备的hobt/hatu(1:1)在dmf中的原液(0.4mmol/ml,25ml)和dipea(2.4g,19mmol)处理树脂。将树脂用氮气鼓泡搅拌2.5h,沥干,并依次用10ml dmf、10ml dcm、10ml dmf冲洗。树脂用25ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌1.5h。将树脂沥干并用dmf(3x25ml)和dcm(3x25ml)冲洗,以得到负载为0.44mmol/g的树脂
结合四肽。
[0492]
中间体t1(方法a).(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(丁基)氨基)己酸
[0493][0494]
步骤1.(2s)
‑2‑
[2
‑
(叔丁氧羰基氨基)乙基氨基]己酸甲酯
[0495][0496]
将(2s)
‑2‑
氨基己酸甲酯盐酸盐(8.02g,44.2mmol)和n
‑
(2
‑
氧代乙基)氨基甲酸叔丁酯(6.68g,42.0mmol)和acoh(2.65g,44.2mmol)在乙酸乙酯(300ml)中的溶液在室温搅拌2h。然后加入三乙酰氧基硼氢化钠(14.1g,66.3mmol)。所得反应混合物在室温搅拌过夜,用水(2x)和盐水洗涤。有机相经无水硫酸钠干燥并且在真空下浓缩。将残余物通过硅胶快速色谱纯化,用石油醚和乙酸乙酯(1/1)洗脱,以得到标题化合物(7.82g,61.4%收率),为黄色油状物。lcms(esi):[m h]
=289.3。
[0497]
步骤2.(2s)
‑2‑
[2
‑
(叔丁氧羰基氨基)乙基
‑
丁基
‑
氨基]己酸甲酯
[0498][0499]
向(2s)
‑2‑
[2
‑
(叔丁氧羰基氨基)乙基氨基]己酸甲酯(7.81g,27.1mmol)在乙酸乙酯(300ml)中的溶液中加入丁醛(2.93g,40.6mmol)和acoh(1.63g,27.1mmol)。将反应混合物在室温搅拌2小时。然后加入三乙酰氧基硼氢化钠(8.62g,40.65mmol)。反应混合物在室温搅拌过夜,先后用水和盐水洗涤。将有机相干燥并真空浓缩。将残余物通过硅胶快速色谱纯化,用石油醚和乙酸乙酯(5/1)洗脱,以得到标题化合物(2.32g,24.9%收率),为黄色油状物。lcms(esi):[m h]
=345.3。
[0500]
步骤3.(2s)
‑2‑
[2
‑
(叔丁氧羰基氨基)乙基
‑
丁基
‑
氨基]己酸
[0501][0502]
在室温,将氢氧化锂(1.13g,47.0mmol)加入(2s)
‑2‑
[2
‑
(叔丁氧羰基氨基)乙基
‑
丁基
‑
氨基]己酸甲酯(4.04g,11.7mmol)在meoh(100ml)和水(30ml)中的溶液中。将反应混合物在室温搅拌过夜,用1n hcl酸化至ph~4,并减压浓缩。将残余物通过硅胶快速色谱纯化,用dcm和meoh(5/1)洗脱,以得到标题化合物酸(2.56g,66.1%收率),为黄色固体。lcms(esi):[m h]
=331.3;1h nmr(300mhz,dmso
‑
d6):δ12.11(s,1h),6.74(t,j=5.9hz,1h),3.40
–
3.38(m,1h),3.03(t,j=6.3hz,2h),2.82
–
2.80(m,1h),2.69
–
2.66(m,3h),1.65
–
1.60(m,2h),1.46
–
1.30(m,17h),0.90
–
0.85(m,6h)。
[0503]
中间体t2(方法b).2
‑
((2
‑
((叔丁氧羰基)氨基)乙基)(甲基)氨基)辛酸
[0504][0505]
在室温,向(2
‑
(甲基氨基)乙基)氨基甲酸叔丁酯(1.75g,10.0mmol)和dipea(3.32ml,19.1mmol)在ch3cn(30ml)中的溶液中加入2
‑
溴辛酸(2.06g,9.22mmol)。将反应混合物在60℃搅拌过夜,然后真空浓缩。将残余物通过硅胶快速色谱纯化,用meoh/dcm(1/9)洗脱,以得到标题化合物(2.065g,70.8%收率),为无色油状物。lcms(esi):[m h]
=317.3;1h nmr(300mhz,dmso
‑
d6):δ10.60(s,1h),6.59(t,j=5.7hz,1h),3.08
–
2.92(m,3h),2.64
–
2.57(m,1h),2.44
–
2.37(m,1h),2.24(s,3h),1.60
–
1.56(m,2h),1.47(s,9h),1.45
–
1.22(m,8h),0.84(t,j=6.3hz,3h)。
[0506]
中间体t12(方法c).(s)
‑2‑
((2
‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)乙基)(丁基)氨基)己酸
[0507][0508]
在室温,向(2s)
‑2‑
[2
‑
(叔丁氧羰基氨基)乙基
‑
丁基
‑
氨基]己酸(2.50g,7.57mmol)在二氧六环(10ml)中的溶液中加入hcl/二氧六环(4.0m,40.ml)。将反应混合物在室温搅拌1h,然后真空浓缩。所得残余物用水(10ml)和thf(30ml)溶解。然后在0℃
°
加入lioh(545mg,22.71mmol)(溶解在水中),然后在该温度加入芴基甲氧基碳酰氯(2.3g,8.91mmol)(溶解在thf中)。将反应混合物在25℃
°
搅拌1h。真空蒸发thf。将残余水溶液用1m hcl水溶液酸化至ph~4
‑
5并真空浓缩。将残余物通过硅胶快速色谱纯化,用dcm/meoh(10/1)洗脱,以得到标题化合物(3.15g,92%收率),为白色固体。lcms(esi):[m h]
=453.3;1h nmr(400mhz,dmso
‑
d6):δ12.09(s,1h),7.88(d,j=7.6hz,2h),7.68(d,j=7.6hz,2h),7.43
‑
7.41(m,2h),7.34
‑
7.30(m,2h),7.19(t,j=5.8hz,1h),4.34
‑
4.18(m,3h),3.07
‑
3.02(m,1h),3.00
‑
2.95(m,2h),2.79
‑
2.72(m,1h),2.60
‑
2.55(m,3h),1.65
‑
1.60(m,1h),1.59
‑
1.53(m,1h),1.45
‑
1.15(m,8h),0.88
‑
0.82(m,6h)。
[0509]
中间体t13(方法d).(s)
‑2‑
(((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙基)
(丁基)氨基)己酸
[0510][0511]
步骤1.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]氨基]己酸甲酯
[0512][0513]
将(4r)
‑4‑
甲基
‑
2,2
‑
二氧代
‑
噁噻唑烷
‑3‑
甲酸叔丁酯(16.5g,69.3mmol)和(2s)
‑2‑
氨基己酸甲酯(40.3g,277mmol)在ch3cn(500ml)中的溶液在60℃
°
搅拌5h。将反应混合物冷却至室温并减压浓缩,将残余物通过硅胶快速色谱纯化,用石油醚和乙酸乙酯(2/1)洗脱,以得到标题化合物(17.6g,83.7%收率),为黄色油状物。lcms(esi):[m h]
=303.3;1h nmr(300mhz,dmso
‑
d6):δ6.50(d,j=8.4hz,1h),3.60(s,3h),3.47
‑
3.42(m,1h),3.13
‑
3.10(m,1h),2.45
‑
2.42(m,1h),2.26
‑
2.20(m,1h),1.50
‑
1.44(m,2h),1.35(s,9h),1.25
‑
1.13(m,4h),0.96(d,j=6.6hz,3h),0.87
‑
0.80(m,3h)。
[0514]
步骤2.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
丁基
‑
氨基]己酸甲酯
[0515][0516]
将(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]氨基]己酸甲酯(17.6g,58.0mmol)和丁醛(8.36g,116mmol)在dcm(200ml)中的溶液在室温下搅拌2h。然后加入三乙酰氧基硼氢化钠(25.0g,118mmol)。将反应混合物在室温搅拌过夜。加入水(100ml)并分离各相。有机相以硫酸钠干燥并且真空浓缩。将残余物通过硅胶快速色谱纯化,用石油醚和乙酸乙酯(5/1)洗脱,以得到标题化合物(20.1g,96.7%收率),为黄色油状物。lcms(esi):[m h]
=359.3。
[0517]
步骤3.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
丁基
‑
氨基]己酸
[0518]
[0519]
将lioh(5.44g,227mmol)在水(100ml)中的溶液加入(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
丁基
‑
氨基]己酸甲酯(20.3g,56.7mmol)的在meoh(300ml)中的溶液中。将反应混合物在室温搅拌过夜并用1nhcl酸化至ph~5,并真空浓缩。将残余物通过硅胶快速色谱纯化,用dcm和甲醇(5/1)洗脱,以得到标题化合物(15.5g,79.2%收率),为黄色固体。lcms(esi):[m h]
=345.3。
[0520]
步骤4.(s)
‑2‑
(((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙基)(丁基)氨基)己酸
[0521]
在0℃将4n hcl/二氧六环(150ml,600mmol)的溶液加入(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
丁基
‑
氨基]己酸(15.5g,44.9mmol)在dcm(150ml)中的溶液中。将反应混合物在室温搅拌2h并真空浓缩。将残余物溶解在thf(150ml)中并在0℃加入lioh(3.02g,125.71mmol)在水(50ml)中的溶液。在0℃加入溶解在20ml thf中的芴基甲氧基碳酰氯(13.0g,50.3mmol)。将反应混合物在室温搅拌1h,然后用1n hcl酸化至ph~5。真空蒸发大部分thf。残余溶液用乙酸乙酯(2x50ml)萃取,合并的有机相用无水硫酸钠干燥并真空浓缩。将残余物通过硅胶快速色谱纯化,用dcm和甲醇(5/1)洗脱,以得到标题化合物(16.5g,84.4%收率),为黄色固体。lcms(esi):[m h]
=467.3;1h nmr(300mhz,dmso
‑
d6):δ12.08(s,1h),7.88(d,j=7.5hz,2h),7.67(d,j=7.5hz,2h),7.43
‑
7.40(m,2h),7.33
‑
7.28(m,2h),7.16(br,1h),4.30
‑
4.17(m,3h),3.76
‑
3.71(m,2h),2.88
‑
2.65(m,4h),1.68
‑
1.64(m,2h),1.58
‑
1.50(m,2h),1.28
‑
1.21(m,6h),1.05(d,j=6.3hz,3h),0.86
‑
0.80(m,6h)。
[0522]
中间体t14(方法e).n
‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙基)
‑
n
‑
(己
‑3‑
炔
‑1‑
基)
‑
l
‑
丙氨酸
[0523][0524]
步骤1.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]氨基]丙酸苄酯
[0525][0526]
将(2s)
‑2‑
氨基丙酸苄酯(11.4g,63.7mmol)和(4r)
‑4‑
甲基
‑
2,2
‑
二氧代
‑
噁噻唑烷
‑3‑
甲酸叔丁酯(5.04g,21.2mmol)和(2s)
‑2‑
氨基丙酸苄酯(11.4g,63.7mmol)的混合物在ch3cn(150ml)中于60℃
°
搅拌过夜。将反应混合物真空浓缩,将残余物通过硅胶快速色谱纯化,用石油醚和乙酸乙酯洗脱,以得到标题化合物(5.66g,79.2%收率),为黄色油状物。lcms(esi):[m h]
=337.2。
[0527]
步骤2.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
己
‑3‑
炔基
‑
氨基]丙酸苄酯
[0528][0529]
在0℃向tf2o(34.5g,122mmol)在dcm(100ml)中的溶液中加入3
‑
己炔
‑1‑
醇(10.0g,102.1mmol)和吡啶(12.1g,154mmol)在dcm(60ml)中的混合物。将反应在该温度搅拌30min。加入水(100ml)并分离各相。有机相(含有三氟甲磺酸己
‑3‑
炔酯)用na2so4干燥,并直接用于下一操作。
[0530]
在0℃
°
向(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]氨基]丙酸苄酯(3.10g,9.21mmol)和吡啶(1.46g,18.4mmol)在dcm(40ml)中的混合物中加入来自前一操作的三氟甲磺酸己
‑3‑
炔酯溶液。将反应在0℃搅拌4h,使其升至室温并在室温搅拌过夜。将反应混合物减压浓缩,并将残余物通过硅胶快速色谱纯化,用石油醚和乙酸乙酯(5/1)洗脱,以得到标题化合物(1.21g,31.7%收率),为黄色固体。lcms(esi):[m h]
=417.4;1h nmr(300mhz,dmso
‑
d6):δ7.38
–
7.30(m,5h),6.43(d,j=8.4hz,1h),5.10(s,2h),3.58(q,j=6.9hz,1h),3.48
–
3.42(m,1h),2.65
–
2.62(m,2h),2.49
–
2.38(m,2h),2.18
–
2.06(m,4h),1.36(s,9h),1.18(d,j=7.2hz,3h),1.04
–
0.94(m,6h)。
[0531]
步骤3.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
己
‑3‑
炔基
‑
氨基]丙酸
[0532][0533]
在0℃
°
将lioh(476mg,19.8mmol)在h2o(20ml)中的溶液加入(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
己
‑3‑
炔基
‑
氨基]丙酸苄酯(2.02g,4.85mmol)在meoh(60ml)中的溶液中。将反应混合物在室温搅拌过夜,然后在0℃用1m hcl酸化至ph~5。真空蒸发大部分meoh。剩余溶液用乙酸乙酯(3x50ml)萃取。合并有机层,用以na2so4干燥并减压浓缩。将残余物通过硅胶快速色谱纯化,用二氯甲烷和甲醇(5/1)洗脱,以得到标题化合物(681mg,43%收率),为黄色油状物。lcms(esi):[m h]
=327.4。
[0534]
步骤4.n
‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙基)
‑
n
‑
(己
‑3‑
炔
‑1‑
基)
‑
l
‑
丙氨酸
[0535]
将(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
己
‑3‑
炔基
‑
氨基]丙酸(980mg,3.01mmol)用tfa(20ml)在dcm(20ml)中的溶液在室温处理1h。然后减压浓缩反应混合物。将thf(30ml)加入残余物中,然后在0℃
°
加入lioh(216mg,9.01mmol)在h2o(10ml)中的溶液。在0℃
°
加入芴基甲氧基碳酰氯(930mg,3.60mmol)在thf(10ml)中的溶液。将反应混合物在室温搅拌1h并用1m hcl酸化至ph~5。加入乙酸乙酯(50ml)并分离各相。将水相用乙酸乙酯(2x50ml)萃取。合并有机层,用以na2so4干燥并减压浓缩。将残余物通过硅胶快速色谱纯化,用dcm和甲醇(5/1)洗脱,以得到标题化合物(1.12g,83.4%收率),为白色固体。lcms(esi):[m h]
=449.2;1h nmr(300mhz,dmso
‑
d6):δ7.89(d,j=7.5hz,2h),7.68(d,j=7.5hz,2h),7.44
–
7.39(m,2h),7.35
–
7.30(m,2h),7.14(br,1h),4.29
–
4.21(m,3h),3.62
–
3.45(m,2h),
2.80
–
2.63(m,3h),2.32
–
2.21(m,3h),2.13
–
2.06(m,2h),1.20(j=6.9hz,3h),1.06
–
0.98(m,6h)。
[0536]
表3.按照类似于那些上述合成方法a至d描述的过程制备表3中描述的中间体。
[0537]
[0538]
[0539]
[0540][0541]
实例1.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
15,16
‑
二丁基
‑9‑
环己基
‑3‑
(羟基甲基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0542][0543]
步骤1.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(丁基)氨基)己酸甲酯
[0544][0545]
向(2s)
‑2‑
氨基己酸甲酯盐酸盐(1.55g,7.68mmol)和甲醇(20ml)的混合物中加入乙酸钠(1.279g,15.59mmol),然后加入丁醛(0.70ml,7.8mmol)和氰基硼氢化钠(1.07g,16.2mmol)。将反应混合物在室温搅拌2h。向反应混合物中加入n
‑
boc
‑2‑
氨基乙醛(1.628g,9.72mmol)和氰基硼氢化钠(1.03g,15.6mmol),反应混合物在室温继续反应2.5h,然后在50℃反应18h。向反应混合物中加入n
‑
boc
‑2‑
氨基乙醛(1.098g,6.556mmol)和氰基硼氢化钠(1.13g,17.1mmol),并将反应混合物在室温下继续搅拌20h。反应混合物用饱和碳酸氢钠水
溶液猝灭并用dcm(3x100ml)萃取。dcm部分以硫酸镁干燥,过滤并真空浓缩。粗产物通过硅胶快速色谱纯化(80g二氧化硅,溶剂梯度:0
‑
50%乙酸乙酯在庚烷中),得到1.491g(56%)标题化合物,为透明无色油状物。lcms(esi):[m h]
=345.2。
[0546]
步骤2.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(丁基)氨基)己酸
[0547][0548]
向(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(丁基)氨基)己酸甲酯(1.488g,4.319mmol)在thf(12ml)中的溶液中加入氢氧化锂(在水中的1.0m溶液,9ml,9mmol)。将所得混合物在室温搅拌20h,然后在50℃搅拌4h。加入另外的氢氧化锂(在水中的1.0m溶液,4ml,4mmol)并将反应混合物在室温搅拌3天。将反应混合物真空蒸发,并将粗产物通过硅胶快速色谱纯化(40g二氧化硅,溶剂梯度:5
‑
10%甲醇在二氯甲烷中的溶液 0.5%甲酸)得到1.3437g(94%)的标题化合物。lcms(esi):[m h]
=331.15。
[0549]
步骤3.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
15,16
‑
二丁基
‑9‑
环己基
‑3‑
(羟基甲基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0550]
使用(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(丁基)氨基)己酸并且按照类似于那些针对中间体15(取代适当的fmoc保护的氨基酸)和实例9步骤2
‑
5描述的过程,将标题化合物制备为三氟乙酸盐。lcms(esi):r
t
(min)=3.503,[m h]
=652.5,方法=a。
[0551]
实例2.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
16
‑
(4
‑
丁基苄基)
‑9‑
环己基
‑3‑
(羟基甲基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0552][0553]
步骤1.(((2s,5s,11s,14s,17s)
‑5‑
((苄氧基)甲基)
‑
10
‑
(4
‑
丁基苄基)
‑
17
‑
环己基
‑
14
‑
异丁基
‑
11,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯
[0554][0555]
按照类似于那些针对实例4所述的过程制备标题化合物。lcms(esi):[m h]
=924.45。
[0556]
步骤2.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
16
‑
(4
‑
丁基苄基)
‑9‑
环己基
‑3‑
(羟基甲基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0557]
在0℃,向(((2s,5s,11s,14s,17s)
‑5‑
((苄氧基)甲基)
‑
10
‑
(4
‑
丁基苄基)
‑
17
‑
环己基
‑
14
‑
异丁基
‑
11,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯(198mg,0.214mmol)在dcm(2.0ml,31mmol)中的溶液中加入三溴化硼(在dcm中的1mol/l溶液)(0.52ml,0.52mmol)。将反应混合物在0℃搅拌2h,然后加入另外的三溴化硼(在二氯甲烷中的1mol/l溶液)(0.30ml,0.30mmol)。将反应混合物在0℃搅拌1h并在室温搅拌2h。反应混合物用甲醇猝灭,用dcm稀释,并用饱和碳酸氢钠水溶液洗涤。分离各层,水层用另外一份dcm萃取。合并的dcm层以硫酸镁干燥,过滤并真空浓缩。粗产物通过反相hplc纯化并冻干,得到36.4mg(24%)标题化合物,为其三氟乙酸盐。lcms(esi):r
t
(min)=4.586,[m h]
=700.5,方法=a。
[0558]
实例3.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
15
‑
(2
‑
氟乙基)
‑
16
‑
己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0559][0560]
步骤1.(叔丁氧羰基)
‑
l
‑
高丝氨酸苄酯
[0561]
[0562]
在0℃,向(3s)
‑4‑
苄氧基
‑3‑
(叔丁氧羰基氨基)
‑4‑
氧代
‑
丁酸(1.016g,3.143mmol)溶解在1,2
‑
二甲氧基乙烷(3ml)中的溶液中加入4
‑
甲基吗啉(0.35ml,3.2mmol),然后缓慢加入氯甲酸异丁酯(0.44ml,3.4mmol)。将反应混合物在0℃搅拌30min,然后通过硅藻土过滤,用1,2
‑
二甲氧基乙烷(3ml)冲洗。将滤液冷却至0℃,并通过缓慢加入硼氢化钠(158mg,4.135mmol)在水(1.5ml)中的溶液进行处理。5min后,加入水(15ml)。用乙酸乙酯萃取所得悬浮液。有机部分以盐水和硫酸镁干燥,过滤并真空浓缩。粗产物通过硅胶快速色谱纯化(40g二氧化硅,溶剂梯度:0
‑
100%乙酸乙酯在dcm中),得到485.6mg(50%)标题化合物,为透明无色油状物。lcms(esi):[m h
‑
叔丁基]
=254.1,[m h
‑
boc]
=210.1。
[0563]
步骤2.(s)
‑2‑
((叔丁氧羰基)氨基)
‑4‑
氟丁酸苄酯
[0564][0565]
向(叔丁氧羰基)
‑
l
‑
高丝氨酸苄酯(550.2mg,1.778mmol)在1,4
‑
二氧六环(3.5ml)中的溶液中加入吡啶
‑2‑
磺酰氟(324.6mg,1.913mmol),然后加入1,3,4,6,7,8
‑
六氢
‑1‑
甲基
‑
2h
‑
嘧啶并[1,2
‑
a]嘧啶(0.52ml,3.6mmol)。在室温,将所得混合物在密封小瓶中于空气中搅拌19h。反应混合物用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(24g二氧化硅,溶剂梯度:0
‑
40%乙酸乙酯在庚烷中),得到356.6mg(64%)标题化合物,为透明无色油状物,其在室温静置过夜后结晶。lcms(esi):[m h
‑
boc]
=212.1;1h nmr(400mhz,dmso
‑
d6)δ7.43
‑
7.30(m,6h),5.23
‑
5.05(m,2h),4.60
‑
4.34(m,2h),4.12(td,j=8.9,4.8hz,1h),2.17
‑
1.87(m,2h),1.37(s,9h)。
[0566]
步骤3.(s)
‑2‑
氨基
‑4‑
氟丁酸苄酯盐酸盐
[0567][0568]
向(2s)
‑2‑
(叔丁氧羰基氨基)
‑4‑
氟
‑
丁酸苄酯(355mg,1.14mmol)在dcm(3ml)中的溶液中加入氯化氢(在二氧六环中的4.0mol/l溶液,3.0ml,12mmol)。将反应混合物在室温搅拌5h。真空蒸发反应混合物,以得到标题化合物,其为hcl盐,并且不经进一步纯化而直接用于下一步。lcms(esi):[m h]
=lcms m/z=212.05。
[0569]
步骤4.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
15
‑
(2
‑
氟乙基)
‑
16
‑
己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0570]
使用(s)
‑2‑
氨基
‑4‑
氟丁酸苄酯盐酸盐并按照类似于那些针对实例4描述的过程,将标题化合物制备为三氟乙酸盐。lcms(esi):r
t
(min)=7.733,[m h]
=684.5,方法=b;1h nmr(400mhz,methanol
‑
d4)δ4.66
‑
4.33(m,4h),4.19
‑
3.96(m,3h),3.69
‑
3.36(m,7h),2.83(s,2h),2.11
‑
1.54(m,12h),1.54
‑
1.13(m,15h),1.13
‑
0.90(m,12h)。
[0571]
实例4.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
15
‑
(2,2,2
‑
三氟乙基)
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0572][0573]
步骤1.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(己基)氨基)
‑
4,4,4
‑
三氟丁酸苄酯
[0574][0575]
向(2s)
‑2‑
氨基
‑
4,4,4
‑
三氟
‑
丁酸苄酯盐酸盐(275.4mg,0.9708mmol)和n
‑
boc
‑2‑
氨基乙醛(175.0mg,1.04mmol)在乙酸乙酯(10.0ml)中的混合物中加入三乙酰氧基硼氢化钠(421mg,1.98mmol)。将反应混合物在室温搅拌3天。向反应混合物中加入己醛(0.25ml,2.1mmol)和三乙酰氧基硼氢化钠(439mg,2.0713mmol)。将反应混合物在50℃加热2h,然后加入己醛(0.30ml,2.5mmol)和三乙酰氧基硼氢化钠(380mg,1.79mmol)。再过3.5h后,反应混合物用乙酸乙酯稀释,用碳酸氢钠水溶液和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(40g二氧化硅,溶剂梯度:0
‑
75%乙酸乙酯在庚烷中),得到350.8mg(76%)标题化合物,为透明无色油状物。lcms(esi):[m h]
=475.25。
[0576]
步骤2.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
15
‑
(2,2,2
‑
三氟乙基)
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0577]
使用(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(己基)氨基)
‑
4,4,4
‑
三氟丁酸苄酯并按照类似于那些针对实例9步骤2
‑
5描述的过程,将标题化合物制备为三氟乙酸盐。lcms(esi):r
t
(min)=3.00,[m h]
=720.47,方法=c;1h nmr(400mhz,dmso
‑
d6)δ9.40(s,1h),8.22(d,j=8.4hz,1h),7.89
‑
7.66(m,3h),7.48
‑
7.21(m,1h),4.44(dd,j=9.8,4.9hz,1h),4.30
‑
3.77(m,3h),3.71(d,j=9.4hz,1h),3.24
‑
2.77(m,7h),2.71(s,2h),2.68
‑
2.57(m,1h),2.41
‑
2.30(m,2h),1.92
‑
0.81(m,36h)。
[0578]
实例5.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
15
‑
丁基
‑9‑
环己基
‑
16
‑
(3
‑
氟丙基)
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0579][0580]
步骤1.(s)
‑2‑
((叔丁氧羰基)氨基)
‑
己酸苄酯
[0581][0582]
向(2s)
‑2‑
(叔丁氧羰基氨基)己酸(5.1549g,22.288mmol)在dmf(50ml)中的溶液中加入碳酸氢钠(2.34g,27.8mmol)和苄基溴(3.0ml,25mmol)。将反应混合物在室温搅拌18h,然后加入碳酸氢钠(1.607g,19.06mmol)和苄基溴(1.5ml,12.5mmol)。将反应混合物在室温再搅拌4h,然后用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(220g二氧化硅,溶剂梯度:0
‑
100%乙酸乙酯在庚烷中),得到5.2931g(74%)标题化合物,为透明无色油状物。lcms(esi):[m h
‑
叔丁基]
=266.15;1h nmr(400mhz,dmso
‑
d6)δ7.53
‑
7.28(m,5h),7.26(d,j=7.7hz,1h),5.15(d,j=12.7hz,1h),5.07(d,j=12.7hz,1h),4.00
‑
3.91(m,1h),1.78
‑
1.48(m,2h),1.37(s,9h),1.28
‑
1.20(m,4h),0.89
‑
0.77(m,3h)。
[0583]
步骤2.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)氨基)已酸苄酯
[0584][0585]
将(s)
‑2‑
(叔丁氧羰基氨基)己酸苄酯(5.2931g,16.47mmol)溶解在dcm(50ml)中并且用氯化氢(在二氧六环中的4m溶液,21ml,84mmol)处理。将所得混合物在室温搅拌15h,然后真空蒸发。将所得残余物与n
‑
boc
‑2‑
氨基乙醛(2.762g,16.5mmol)在乙酸乙酯(50.0ml)中在40℃合并,然后,历时2.5h分3份加入三乙酰氧基硼氢化钠(32mmol,32mmol)。将反应混合物在40℃搅拌16h。反应混合物用乙酸乙酯稀释,用碳酸氢钠水溶液和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(220g二氧化硅,溶剂梯度:0
‑
5%甲醇在dcm中),得到2.2081g(37%)标题化合物,为黄色油状物。lcms(esi):[m h]
=365.2;1h nmr(400mhz,dmso
‑
d6)δ7.40
‑
7.29(m,5h),6.75
‑
6.63(m,1h),5.12(d,j=
2.0hz,2h),3.20(t,j=6.6hz,1h),2.95(h,j=6.6hz,2h),2.55(dd,j=11.6,6.4hz,1h),2.40(dt,j=11.5,6.3hz,1h),1.59
‑
1.44(m,2h),1.37(s,9h),1.28
‑
1.17(m,4h),0.85
‑
0.77(m,3h)。
[0586]
步骤3.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(3
‑
((叔丁基二甲基甲硅烷基)氧基)丙基)氨基)己酸苄酯
[0587][0588]
向(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)氨基)己酸苄酯(0.6066g,1.664mmol)和3
‑
[(叔丁基二甲基甲硅烷基)]
‑1‑
丙醛(0.6ml,3mmol)在乙酸乙酯(8.0ml)中的混合物中加入三乙酰氧基硼氢化钠(855mg,4.034mmol)。将反应混合物在50℃搅拌2h。反应混合物用乙酸乙酯稀释,用碳酸氢钠水溶液和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(40g二氧化硅,溶剂梯度:0
‑
50%乙酸乙酯在庚烷中),得到618.0mg(69%)标题化合物,为透明无色油状物。lcms(esi):[m h]
=537.35。
[0589]
步骤4.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(3
‑
羟基丙基)氨基)已酸苄酯
[0590][0591]
向(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(3
‑
((叔丁基二甲基甲硅烷基)氧基)丙基)氨基)己酸苄酯(618.0mg,1.151mmol)在thf(5ml)中的溶液中加入四丁基氟化铵(在thf中的1mol/l溶液,1.4ml,1.4mmol)。将反应混合物在室温搅拌2h。反应物质用饱和氯化铵水溶液猝灭,用乙酸乙酯稀释并用饱和碳酸氢钠水溶液(2x50ml)洗涤。有机部分以硫酸镁干燥,过滤并真空浓缩。粗产物通过硅胶快速色谱纯化(24g二氧化硅,溶剂梯度:0
‑
5%甲醇在二氯甲烷中的溶液,在210nm监测)得到321.0mg(66%)的标题化合物。lcms(esi):[m h]
=423.25;1h nmr(400mhz,dmso
‑
j6)δ7.41
‑
7.29(m,4h),6.57(t,j=5.7hz,1h),5.11(s,2h),4.30(t,j=5.0hz,1h),3.46
‑
3.33(m,2h),2.97
‑
2.79(m,2h),2.76
‑
2.57(m,2h),2.47
‑
2.32(m,2h),1.68
‑
1.42(m,4h),1.36(s,9h),1.32
‑
1.19(m,3h),0.85(t,j=7.0hz,3h)。
[0592]
步骤5.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(3
‑
氟丙基)氨基)己酸苄酯
[0593][0594]
向(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(3
‑
羟基丙基)氨基)己酸苄酯(321.0mg,0.7596mmol)在1,4
‑
二氧六环(1.5ml)中的溶液中加入吡啶
‑2‑
磺酰氟(150.8mg,0.8890mmol),然后加入1,3,4,6,7,8
‑
六氢
‑1‑
甲基
‑
2h
‑
嘧啶并[1,2
‑
a]嘧啶(0.24ml,1.7mmol)。在室温,将所得混合物在密封小瓶中(于空气中)搅拌63h。反应混合物用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(12g二氧化硅,溶剂梯度:0
‑
80%乙酸乙酯在庚烷中),得到233.1mg(72%)标题化合物,为透明无色油状物。lcms(esi):[m h]
=425.2。
[0595]
步骤6.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
15
‑
丁基
‑9‑
环己基
‑
16
‑
(3
‑
氟丙基)
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0596]
使用(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(3
‑
氟丙基)氨基)己酸苄酯并按照类似于那些针对实例4描述的过程,制备标题化合物。lcms(esi):r
t
(min)=8.210,[m h]
=670.5,方法=b。
[0597]
实例6.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
15
‑
丁基
‑9‑
环己基
‑
16
‑
(2
‑
氟乙基)
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0598][0599]
步骤1.(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(2
‑
氟乙基)氨基)己酸苄酯
[0600][0601]
向(s)
‑2‑
((叔丁氧羰基)氨基)己酸苄酯(实例5,步骤1)(702mg,1.93mmol)在dmf(4.0ml)中的溶液中加入三乙胺(3.0ml,21mmol)和1
‑
氟
‑2‑
碘乙烷(2.0ml,25mmol)。将反应混合物在密封小瓶中于60℃搅拌17h。向反应混合物中加入1
‑
溴
‑2‑
氟乙烷(1.0ml,
13mmol),并将混合物在60℃加热24h。向反应混合物中加入三甲胺(2.0ml,14mmol)和1
‑
溴
‑2‑
氟乙烷(1.00ml,13mmol),并将混合物在60℃搅拌24h。反应混合物用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(40g二氧化硅,溶剂梯度:0
‑
100%乙酸乙酯在dcm中),得到391.7mg(50%)标题化合物,为橙色油状物。lcms(esi):[m h]
=411.25
[0602]
步骤2.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑
15
‑
丁基
‑9‑
环己基
‑
16
‑
(2
‑
氟乙基)
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13
‑
甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0603]
使用(s)
‑2‑
((2
‑
((叔丁氧羰基)氨基)乙基)(2
‑
氟乙基)氨基)己酸苄酯并按照类似于那些针对实例4描述的过程,制备标题化合物。lcms(esi):r
t
(min)=6.69,[m h]
=656.56,方法=d。
[0604]
实例7.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑
15,16
‑
二丁基
‑9‑
环已基
‑3‑
((s)
‑1‑
羟基乙基)
‑
13,18
‑
二甲基
‑
12
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0605][0606]
步骤1.n
‑
me
‑
正缬氨酸
‑
环己基甘氨酸
‑
dap(cbz)
‑
别苏氨酸(bzl)
‑
(2
‑
氯三苯甲基树脂)
[0607][0608]
将市售的2
‑
氯三苯甲基树脂(树脂负载量为0.95mmol/g,5.0g)用40ml dmf/dcm(1:1)溶胀40min。沥干树脂,并用fmoc
‑
别苏氨酸(bzl)
‑
oh(2.46g,5.7mmol)和dipea(2.5g,19.4mmol)在dmf(15ml)中的溶液处理。将树脂在氮气鼓泡下搅拌1.5h,然后沥干并依次用dcm/meoh/dipea(10:10:1,15ml)、dcm(3x15ml)和dmf(3x15ml)冲洗。
[0609]
树脂用25ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌1h。将树脂沥干并用dmf(3x10ml)和dcm(3x10ml)冲洗。用(s)
‑2‑
(fmoc
‑
氨基)
‑3‑
(((苄氧基)羰基)氨基)丙酸
(4.37g,9.49mmol)、hatu(3.61g,9.50mmol)、hobt(1.29g,9.53mmol)和dipea(3.31ml,19.03mmol)在dmf(50ml)中的溶液(在室温下预混15min)处理树脂。将树脂在旋转器上振摇过夜,沥干并用dmf(2x15ml)和dcm(3x15ml)冲洗。
[0610]
树脂用10ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌60min。将树脂沥干并用dmf(3x15ml)和dcm(3x15ml)冲洗。用fmoc
‑
s
‑
环己基甘氨酸
‑
oh(3.61g,9.50mmol)的溶液(在室温预混合15min)、hatu/hobt(1/1)的原液(在dmf中的0.4m溶液,24ml,9.6mmol)和dipea(2.45g,19mmol)处理树脂。将树脂在氮气鼓泡下搅拌2h,然后沥干并用dmf(3x15ml)和dcm(3x15ml)冲洗。
[0611]
树脂用25ml 20%哌啶的dmf溶液处理,并在氮气鼓泡下搅拌60min。将树脂沥干并用dmf(3x15ml)和dcm(3x15ml)冲洗。用(s)
‑
fmoc
‑
n
‑
me
‑
正缬氨酸
‑
oh(3.37g,9.52)、hatu/hobt(1/1)(在dmf中的0.4m溶液,24ml,9.6mmol)和dipea(2.53g,19.6mmol)的预混合溶液处理树脂。将树脂搅拌3h,然后沥干并用dmf(3x15ml)和dcm(3x15ml)冲洗。
[0612]
树脂用25ml 20%哌啶在dmf中的溶液处理,并将小瓶在室温置于旋转器上3h,然后过滤,用dma(3x15ml)和dcm(3x15ml)冲洗并真空干燥,以得到树脂结合肽(7.58g,树脂负载量估计为0.5mmol/g)。
[0613]
步骤2.(6r,9s,12s,15s,18s,21s)
‑
18
‑
((((苄氧基)羰基)氨基)甲基)
‑
21
‑
((s)
‑1‑
(苄氧基)乙基)
‑
8,9
‑
二丁基
‑
15
‑
环已基
‑
2,2,6,11
‑
四甲基
‑
4,10,13,16,19
‑
五氧代
‑
12
‑
丙基
‑3‑
氧杂
‑
5,8,11,14,17,20
‑
六氮杂二十二烷
‑
22
‑
酸
[0614][0615]
将来自前一步骤的树脂结合肽(1.00g,树脂负载量~0.5mmol/g)用(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
丁基
‑
氨基]己酸(447mg,1.29mmol,中间体t5)、pyaop(782mg,1.51mmol)和dipea(387mg,3mmol)在dmf(10ml)中的预混合溶液处理。将树脂在旋转器上振摇24h。将树脂沥干并用dmf(3x10ml)和dcm(3x10ml)冲洗。树脂用hfip/dcm=1/4溶液(足以覆盖树脂1.5倍)处理并在室温下振摇1h,然后过滤。收集液相。用hfip/dcm=1/4重复该过程并收集液相。将树脂用dcm(3x10ml)洗涤。合并滤液并真空浓缩。将残余物通过反相快速色谱在c18柱上纯化,以得到标题化合物(364mg,0.361mmol)。lcms(esi):[m h]
=1008.6。
[0616]
步骤3.(2s,5s,8s,11s,14s)
‑
15
‑
((r)
‑2‑
氨基丙基)
‑5‑
((((苄氧基)羰基)氨基)甲基)
‑2‑
((s)
‑1‑
(苄氧基)乙基)
‑
14
‑
丁基
‑8‑
环己基
‑
12
‑
甲基
‑
4,7,10,13
‑
四氧代
‑
11
‑
丙基
‑
3,6,9,12,15
‑
五氮杂十九烷
‑1‑
酸
[0617][0618]
在0℃
°
将(6r,9s,12s,15s,18s,21s)
‑
18
‑
((((苄氧基)羰基)氨基)甲基)
‑
21
‑
((s)
‑1‑
(苄氧基)乙基)
‑
8,9
‑
二丁基l
‑
15
‑
环己基
‑
2,2,6,11
‑
四甲基
‑
4,10,13,16,19
‑
五氧代
‑
12
‑
丙基
‑3‑
氧杂
‑
5,8,11,14,17,20
‑
六氮杂二十二烷
‑
22
‑
酸(204mg,0.202mmol)溶解在tfa(20ml)中,将所得混合物在0℃
°
搅拌1h,然后用甲苯(40ml)稀释。将所得混合物真空浓缩。加入另外40ml甲苯并将所得混合物真空浓缩,以得到标题化合物,为tfa盐(180mg)。lcms(esi):[m h]
=908.5。
[0619]
步骤4.(((2s,5s,8r,11s,14s,17s)
‑5‑
((s)
‑1‑
(苄氧基)乙基)
‑
10,11
‑
二丁基
‑
17
‑
环己基
‑
8,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
14
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯
[0620][0621]
将(2s,5s,8s,11s,14s)
‑
15
‑
((r)
‑2‑
氨基丙基)
‑5‑
((((苄氧基)羰基)氨基)甲基)
‑2‑
((s)
‑1‑
(苄氧基)乙基)
‑
14
‑
丁基
‑8‑
环己基
‑
12
‑
甲基
‑
4,7,10,13
‑
四氧代
‑
11
‑
丙基
‑
3,6,9,12,15
‑
五氮杂十九烷
‑1‑
酸(184mg,0.202mmol)溶解在thf(160ml)中,并且加入dipea(78mg,0.60mmol)。将所得溶液冷却至0℃
°
。将hatu(76.9mg,0.200mmol)和hobt(27.0mg,0.200mmol)在dmf(2.0ml)中的溶液用thf(20ml)稀释,然后将所得thf/dmf溶液滴加到冷的肽溶液中。反应完成后,真空浓缩反应混合物以抽除thf。残余物用3ml dmf稀释并在剧烈搅拌下滴加到水(60ml)中。将混合物再搅拌15min。通过过滤收集固体,用水洗涤并真空干燥以得到粗制环肽(170mg),其不经进一步纯化而直接使用。lcms(esi):[m h]
=890.5。
[0622]
步骤4.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑
15,16
‑
二丁基
‑9‑
环已基
‑3‑
[(1s)
‑1‑
羟基乙基]
‑
13,18
‑
二甲基
‑
12
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,
11,14
‑
五酮
[0623]
将来自步骤3的粗制环肽(132mg)和10%pd/c(232mg)在乙酸乙酯(20ml)中的混合物在25℃
°
于氢气(1atm)下搅拌24h。经由过滤移除催化剂,并真空浓缩滤液。将残余物通过prep
‑
lcms纯化,以得到作为tfa盐的标题化合物(13.0mg),为白色固体。lcms(esi):[m h]
=666.5,r
t
=2.38min,方法j。1h nmr(400mhz,dmso
‑
d6):δ8.74
‑
8.64(m,2h),8.05(br,3h),7.92
‑
7.04(m,2h),6.09(br,1h),5.22
‑
5.14(m,1h),4.81
‑
4.73(m,2h),4.47
‑
4.41(m,1h),4.33
‑
4.28(m,1h),4.03
‑
3.87(m,3h),3.78
‑
3.73(m,1h),3.21(s,2h),3.19
‑
3.10(m,3h),2.99
‑
2.92(m,1h),2.05
‑
1.95(m,2h),1.88
‑
1.76(m,3h),1.72
‑
1.62(m,6h),1.44
‑
1.06(m,16h),0.97
‑
0.84(m,15h)。
[0624]
实例8.(1r,4s,7s,10s,13s,16r,19r)
‑
10
‑
(氨基甲基)
‑7‑
环己基
‑
13
‑
[(1s)
‑1‑
羟基乙基]
‑4‑
异丁基
‑
3,16
‑
二甲基
‑
18
‑
氧杂
‑
3,6,9,12,15
‑
五氮杂双环[17.8.0]二十七烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0625][0626]
步骤1.n
‑
me
‑
亮氨酸
‑
环已基甘氨酸
‑
dap(b0c)
‑
别苏氨酸(t
‑
bu)
‑
(2
‑
氯三苯甲基树脂)
[0627][0628]
按照类似于那些针对实例7所述的方法制备树脂结合的四肽。
[0629]
步骤2.(2s,5s,8s,11s,14r,15r,18r)
‑
18
‑
氨基
‑
14
‑
(丁
‑3‑
烯
‑1‑
基)
‑2‑
((s)
‑1‑
(叔丁氧基)乙基)
‑5‑
(((叔丁氧羰基)氨基)甲基)
‑8‑
环己基
‑
15
‑
(己
‑5‑
烯
‑1‑
基)
‑
11
‑
异丁基
‑
12
‑
甲基
‑
4,7,10,13
‑
四氧代
‑
16
‑
氧杂
‑
3,6,9,12
‑
四氮杂十九烷
‑1‑
酸
[0630][0631]
将来自步骤1的树脂结合的四肽(1.0g,~0.45mmol/g负载)用thf(5ml)和dipea(308mg,2.39mmol)处理,并令其溶胀30min。独立地将(2r,3r)
‑2‑
丁
‑3‑
烯基
‑3‑
[(2r)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丙氧基]壬
‑8‑
烯酸(364mg,0.720mmol,中间体14)在thf(2.0ml)中的溶液加入三光气(71.5mg,0.240mmol)在thf(2.0ml)中的溶液中。滴加2,4,6
‑
三甲基吡啶(1.25g,10.4mmol),此时混合物放热并形成无色沉淀。将该悬浮液轻轻摇动约1分钟,然后将其添加到预处理的树脂中。将混合物置于室温旋转器上3h,沥干,然后用dmf(10ml)和dcm(2x10ml)冲洗。树脂用10ml 20%哌啶的dmf溶液处理,在氮气鼓泡下搅拌1h,沥干并用dmf(2x10ml)和dcm(2x10ml)冲洗。然后用hfip/dcm=1/4溶液(20ml)处理树脂并于室温旋转器上振摇1h,然后过滤。收集液相。用hfip/dcm=1/4重复该过程并收集液相。将树脂用dcm(3x10ml)洗涤。合并滤液并真空浓缩。将残余物通过反相快速色谱在c18柱上纯化,以得到标题化合物(300mg,62%),为黄色固体。lcms(esi):[m h]
=893.6。
[0632]
步骤3.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑
18
‑
丁
‑3‑
烯基
‑6‑
[(1s)
‑1‑
叔丁氧基乙基]
‑
12
‑
环己基
‑
19
‑
己
‑5‑
烯基
‑
15
‑
异丁基
‑
3,16
‑
二甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸叔丁酯
[0633][0634]
在0℃
°
向来自步骤2的肽(300mg,0.340mmol)在thf(200ml)中的溶液与dipea(175mg,1.35mmol)的混合物中滴加hatu(129mg,0.340mmol)和hobt(129mg,0.34mmol)在dmf (2.0ml)中的溶液。将反应混合物在0℃
°
搅拌2h,然后真空浓缩以抽除thf。残余物用dmf(3ml)稀释并在剧烈搅拌下滴加到60ml水中。过滤收集所得固体,用水(3x10ml)洗涤并干燥,以得到粗产物(271mg),为黄色固体。lcms(esi):[m h]
=875.5。
[0635]
步骤4.n
‑
[[(1r,4s,7s,10s,13s,16r,19r,24z)
‑
13
‑
[(1s)
‑1‑
叔丁氧基乙基]
‑7‑
环己基
‑4‑
异丁基
‑
3,16
‑
二甲基
‑
2,5,8,11,14
‑
五氧代
‑
18
‑
氧杂
‑
3,6,9,12,15
‑
五氮杂双环[17.8.0]二十七碳
‑
24
‑
烯
‑
10
‑
基]甲基]氨基甲酸叔丁酯
[0636][0637]
于氮气下,在0℃
°
向来自步骤3的环肽(110mg,0.130mmol)在dce(70ml)中的溶液中滴加二氯
‑
[(2
‑
异丙氧基苯基)亚甲基]
‑
(三环己基
‑
λ^{5}
‑
膦烷基)钌(43mg,cas 203714
‑
71
‑
0)在dce(1.0ml)中的溶液。将反应在80℃
°
搅拌过夜,然后加入更多催化剂(43mg,cas 203714
‑
71
‑
0)。将反应在80℃
°
再搅拌8h,并加入另一部分催化剂(43mg,cas203714
‑
71
‑
0)。这个过程又重复了2次。添加的总催化剂为5x43mg,总反应时间为3天。将反应混合物减压浓缩。将残余物溶解在2.0ml dce中,并将dce溶液滴加到40ml正己烷中。将混合物在室温搅拌过夜。通过过滤收集固体,用正己烷(3x10ml)洗涤并干燥,以得到粗产物(100mg),为棕色固体,其不经进一步纯化即用于下一步骤。
[0638]
步骤5.n
‑
[[(1r,4s,7s,10s,13s,16r,19r)
‑
13
‑
[(1s)
‑1‑
叔丁氧基乙基]
‑7‑
环己基
‑4‑
异丁基
‑
3,16
‑
二甲基
‑
2,5,8,11,14
‑
五氧代
‑
18
‑
氧杂
‑
3,6,9,12,15
‑
五氮杂双环[17.8.0]二十七烷
‑
10
‑
基]甲基]氨基甲酸叔丁酯
[0639][0640]
将来自步骤4的粗制双环烯烃和10%pd/c(433mg)在乙酸乙酯(50ml)中的混合物在25℃
°
于氢气气氛下搅拌15h。经由过滤移除催化剂并减压浓缩滤液。将残余物通过反相色谱在c18柱上纯化,以得到标题化合物(23.2mg),为黄色油状物。lcms(esi):[m h]
=849.5。
[0641]
步骤6.(1r,4s,7s,10s,13s,16r,19r)
‑
10
‑
(氨基甲基)
‑7‑
环已基
‑
13
‑
[(1s)
‑1‑
羟基乙基]
‑4‑
异丁基
‑
3,16
‑
二甲基
‑
18
‑
氧杂
‑
3,6,9,12,15
‑
五氮杂双环[17.8.0]二十七烷
‑
2,5,8,11,14
‑
五酮
[0642][0643]
将来自步骤5的产物(23.2mg)用tfa(5.0ml)在0℃
°
处理1h,然后减压浓缩。将残余物用prep
‑
lcms纯化(色谱柱:xbridge prep c18obd色谱柱,5μm,19
×
150mm;流动相a:水(0.05%tfa),流动相b:acn;流速:25ml/min;梯度:20%b至50%b在20min内;220nm;rt:18.74min),以得到标题化合物的三氟乙酸盐(5.3mg),为黄色固体。lcms(esi):[m h]
=693.5,r
t
=2.5min,方法h。1h nmr(400mhz,dmso
‑
d6):δ8.72
‑
8.67(m,1h),8.40
‑
8.25(m,1h),7.94
‑
7.88(m,3h),7.64
‑
7.45(m,1h),7.24
‑
6.95(m,1h),4.63
‑
4.54(m,2h),4.18
‑
4.07(m,2h),3.95
‑
3.86(m,2h),3.67
‑
3.60(m,2h),3.50
‑
3.43(m,1h),3.21
‑
3.03(m,4h),2.78(s,2h),2.14
‑
2.07(m,1h),1.83
‑
1.43(m,24h),1.24
‑
1.09(m,6h),1.01
‑
0.88(m,12h)。
[0644]
实例9.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑9‑
环已基
‑
16
‑
(环戊基甲基)
‑
18
‑
乙基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0645][0646]
步骤1.n
‑
((r)
‑2‑
((叔丁氧羰基)氨基)丁基)
‑
n
‑
(环戊基甲基)
‑
l
‑
丙氨酸苄酯
[0647][0648]
向n
‑
[(1r)
‑1‑
(羟基甲基)丙基]氨基甲酸叔丁酯(1.037g,5.480mmol)在dcm(16ml,249.6mmol中的溶液中加入戴斯
‑
马丁氧化剂(dess
‑
martin periodinane)(1.2当量)。将反应混合物在室温搅拌4h。向反应混合物中加入(2s)
‑2‑
氨基丙酸苄酯盐酸盐(911mg,4.22mmol)、三乙酰氧基硼氢化钠(1.816g,8.568mmol)和乙酸乙酯(10ml)。将反应混合物在室温搅拌2h。然后向该混合物中加入环戊烷甲醛(0.80ml,7.6mmol)和三乙酰氧基硼氢化钠(1.65g,7.79mmol),并在室温下继续反应18h。反应混合物用乙酸乙酯稀释,用碳酸氢钠水溶液和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(80g二氧化硅,溶剂梯度:0
‑
70%乙酸异丙酯在庚烷中),以得到1.025g(56%)的标题化合
物。lcms(esi):[m h]
=433.3
[0649]
步骤2.n
‑
((r)
‑2‑
((叔丁氧羰基)氨基)丁基)
‑
n
‑
(环戊基甲基)
‑
l
‑
丙氨酸
[0650][0651]
在氮气下,向n
‑
((r)
‑2‑
((叔丁氧羰基)氨基)丁基)
‑
n
‑
(环戊基甲基)
‑
l
‑
丙氨酸苄酯(1.1878g,2.746mmol)在乙醇(10ml)中的溶液中加入钯(碳载,10wt.%)(154mg,0.1447mmol)。用氮气然后用氢气吹扫反应容器,并在氢气球下在室温下搅拌4h。反应混合物通过硅藻土过滤,用甲醇冲洗并真空蒸发,以得到标题化合物,不经纯化即使用。lcms(esi):[m h]
=343.25。
[0652]
步骤3.n
‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
(((r)
‑2‑
氨基丁基)(环戊基甲基)氨基)
‑
n
‑
甲基丙酰胺基)
‑4‑
甲基戊酰胺基)
‑2‑
环己基乙酰胺基)
‑3‑
(((苄氧基)羰基)氨基)丙酰基)
‑
o
‑
苄基
‑
l
‑
别苏氨酸
[0653][0654]
向过滤管中加入中间体15(0.770g,估计为0.47mmol/g)和8ml dma。使树脂在室温溶胀90min,然后沥干。向树脂中加入n
‑
((r)
‑2‑
((叔丁氧羰基)氨基)丁基1)
‑
n
‑
(环戊基甲基)
‑
l
‑
丙氨酸(231mg,0.674mmol)和pyaop(789mg,1.453mmol)在dmf(4ml)中的溶液,然后加入dipea(0.40ml,2.3mmol)。将所得混合物置于室温旋转器上16h。沥干树脂并用dma(5x)和dcm(5x)冲洗。向树脂中加入6ml 7∶2∶1dcm:acoh:三氟乙醇。将该混合物置于室温旋转器上2h,然后过滤,用dcm冲洗。真空蒸发滤液,与甲苯(3x2ml)共沸。将所得残余物溶解在dcm(5ml)中并用氯化氢(在二氧六环中的4.0mol/l溶液,1.0ml,4.0mmol)处理。将该混合物在室温搅拌90min,然后真空蒸发,得到粗制肽(177.7mg,51%粗收率),其不经纯化即用于下一步。lcms(esi):[m h]
=920.7。
[0655]
步骤4.(((2s,5s,8r,11s,14s,17s)
‑5‑
((s)
‑1‑
(苄氧基)乙基)
‑
17
‑
环已基
‑
10
‑
(环戊基甲基)
‑8‑
乙基
‑
14
‑
异丁基
‑
11,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯
[0656][0657]
在室温,历时1h,在搅拌下向dipea(0.2ml,1mmol)和hatu(167mg,0.430mmol)在thf(100ml)中的溶液中滴加m((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
(((r)
‑2‑
氨基丁基)(环戊基甲基)氨基)
‑
m甲基丙酰胺基)
‑4‑
甲基戊酰胺基)
‑2‑
环己基乙酰胺基)
‑3‑
(((苄氧基)羰基)氨基)丙酰基)
‑
o
‑
苄基
‑
l
‑
别苏氨酸(177.7mg,0.1858mmol)在thf(50ml)中的溶液。又经过90min后,真空蒸发反应混合物。所得残余物用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(12g二氧化硅,溶剂梯度:0
‑
10%甲醇在dcm中),得到76.6mg标题化合物。lcms(esi):[m h]
=902.7。
[0658]
步骤5.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
(环戊基甲基)
‑
18
‑
乙基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0659]
将(((2s,5s,8r,11s,14s,17s)
‑5‑
((s)
‑1‑
(苄氧基)乙基)
‑
17
‑
环己基
‑
10
‑
(环戊基甲基)
‑8‑
乙基
‑
14
‑
异丁基
‑
11,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯(76.6mg,0.085mmol)和乙酸乙酯(5ml)充入烧瓶中,并用氮气吹扫烧瓶。加入三氟乙酸(25μl,0.323mmol)和钯(碳载,10wt.%)(104mg,0.0977mmol)后,将反应混合物抽真空并用氢气回填,然后在室温在氢气球下搅拌18h。反应混合物通过硅藻土过滤,用100ml 5%acoh在乙醇中的溶液冲洗,真空蒸发滤液,与甲苯(3x1ml)共沸。粗产物通过反相hplc纯化并冻干,得到27.6mg(48%)标题化合物,为其三氟乙酸盐。lcms(esi):r
t
(min)=7.920,[m h]
=762.5,方法=b;1h nmr(400mhz,methanol
‑
d4)δ4.93(d,j=15.6hz,1h),4.64
‑
4.17(m,4h),4.15
‑
3.94(m,2h),3.79
‑
3.44(m,4h),3.38(s,1h),3.26(d,j=11.8hz,2h),3.13
‑
2.92(m,1h),2.08(dd,j=42.7,18.2hz,4h),1.99
‑
1.11(m,27h),1.11
‑
0.90(m,11h)。
[0660]
实例10.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
(((1r,3s)
‑3‑
(氨基甲基)环丁氧基)甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0661][0662]
步骤1.n
‑
me
‑
正缬氨酸
‑
环庚基甘胺酸
‑
dap(boc)
‑
反式
‑
叔丁氧羰基)氨基)甲基)环丁基)
‑
丝氨酸
‑
(2
‑
氯三苯甲基树脂)
[0663][0664]
按照类似于那些在实例7中描述的方法制备树脂结合的四肽。
[0665]
步骤2.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[[3
‑
(氨基甲基)环丁氧基]甲基]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0666]
使用来自步骤1的中间体和(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑2‑
甲基
‑4‑
(螺[3.3]庚烷
‑2‑
基)丁酸(中间体4),根据类似于那些在实例13步骤2
‑
4中描述的方法将标题化合物制备为tfa盐。lcms(esi):[m h]
=774.6,r
t
=2.21min,方法h。1h nmr(400mhz,dmso
‑
d6):δ8.52
‑
8.12(m,2h),8.08
‑
8.02(m,3h),7.76(br,3h),7.46
‑
7.38(m,1h),4.68
‑
4.56(m,7h),4.36
‑
4.28(m,2h),4.11
‑
4.02(m,2h),3.96
‑
3.82(m,2h),3.76
‑
3.68(m,1h),3.48
‑
3.40(m,1h),3.38
‑
3.12(m,4h),3.07(s,1h),2.90
‑
2.87(m,3h),2.73(s,2h),2.39
‑
2.28(m,2h),2.07
‑
1.94(m,8h),1.83
‑
1.70(m,5h),1.67
‑
1.27(m,15h),1.02
‑
0.87(m,8h)。
[0667]
实例11.(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(((2
‑
氮杂螺[3.3]庚烷
‑6‑
基)氧基)甲基)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0668][0669]
步骤1.n
‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
((2r,3r)
‑3‑
((r)
‑2‑
氨基丙氧基)
‑
n,2
‑
二甲基壬酰胺基)戊酰胺基)
‑2‑
环己基乙酰胺基)
‑3‑
(((苄氧基)羰基)氨基)丙酰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸
[0670][0671]
将市售的2
‑
氯三苯甲基氯树脂(树脂负载量为1.5mmol/g,1.255g)用10ml dma溶胀4.5h。沥干树脂,并用n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸(中间体8)(0.775g,1.48mmol)和dipea(0.60ml,3.4mmol)在dma(5ml)中的溶液处理。将树脂置于室温旋转器上过夜。将树脂沥干并用dma(2x8ml)冲洗,然后通过用6ml 2∶1dcm:甲醇处理15min来封盖未反应的树脂。沥干树脂并用dma(3x8ml)冲洗。
[0672]
fmoc
‑
别苏氨酸(bzl)
‑
oh(3.267g,7.571mmol)、hbtu(2.904g,7.657mmol)、dipea(1.9ml,11mmol)和dma(25ml)(在室温下预混15min)。将树脂在氮气鼓泡下搅拌2.5hr,然后沥干并用dma冲洗。通过用25ml dma、1ml dipea和1ml乙酸酐处理树脂来封端未反应的起始材料。将树脂在氮气鼓泡下搅拌5min,然后沥干并用dma(2x25ml)和dcm(3x25ml)冲洗。树脂用8ml 20%哌啶在dmf中的溶液处理2.5h。将树脂沥干并依次用dma(3x8ml)、dcm(3x8ml)和dma(2x8ml)冲洗。
[0673]
用fmoc
‑
dap(cbz)
‑
oh(1.384g,3.005mmol)、hbtu(1.14g,3.01mmol)和dipea(0.8ml,5mmol)在dma(5ml)中的溶液(在室温下预混20min)处理树脂。将树脂置于室温旋转器上1.75h,然后沥干并依次用3x50ml dma和3x50ml dcm冲洗。树脂用8ml 20%哌啶在dmf中的溶液处理90min。将树脂沥干并依次用dma(4x8ml)、dcm(4x8ml)冲洗。
[0674]
用(2s)
‑2‑
环庚基
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)乙酸(中间体11,824mg,2.09mmol)、hbtu(1.14g,3.01mmol)和dipea(0.8ml,5mmol)在dma(5ml)中的溶液(在室温下预混15min)处理树脂。将树脂置于室温旋转器上2h,然后沥干并用dma(3x8ml)冲洗。树脂用8ml20%哌啶在dmf中的溶液处理1h。将树脂沥干并依次用dma(3x8ml)、dcm(3x8ml)和dma
(2x8ml)冲洗。
[0675]
用fmoc
‑
n
‑
me
‑
正缬氨酸(1.06g,3.00mmol)、hbtu(1.12g,2.95mmol)和dipea(0.8ml,5mmol)在dma(5ml)中的溶液(在室温下预混15min)处理树脂。将树脂置于室温旋转器上2h,然后沥干并用dma(3x8ml)和dcm(3x8ml)冲洗。树脂用8ml 20%哌啶在dmf中的溶液处理2h。将树脂沥干并依次用dma(4x8ml)、dcm(4x8ml)冲洗。
[0676]
将树脂在dma中溶胀,然后沥干并用(2r,3r)
‑3‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙氧基)
‑2‑
甲基壬酸(中间体3)(644.0mg,1.377mmol)、pyaop(980.4mg,1.805mmol)和dipea(0.45ml,2.6mmol)在dmf(5ml,64.4mmol)中的混合物处理。将所得混合物置于室温旋转器上18h。过滤树脂并用dma(3x8ml)和dcm(3x8ml)冲洗。
[0677]
向树脂中加入8ml 20%哌啶在dmf中的溶液。将反应容器置于室温旋转器上2.5hr,然后沥干并依次用dma(4x8ml)和dcm(4x8ml)冲洗。
[0678]
向树脂中加入dcm(6ml,93.61mmol)和hfip(2ml,19.07mmol)。将反应容器置于室温旋转器上2.5h。过滤树脂,用dcm冲洗,并真空蒸发滤液,得到粗制肽。lcms(esi):[m h]
=1014.8。
[0679]
步骤2. 6
‑
(((3r,6s,9s,12s,15s,18r,19r)
‑9‑
((((苄氧基)羰基)氨基)甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)甲氧基)
‑2‑
氮杂螺[3.3]庚烷
‑2‑
甲酸叔丁酯
[0680][0681]
在室温,历时90min,在搅拌下向dipea(0.20ml,1.1mmol)和hatu(314mg,0.809mmol)在thf(200ml)中的溶液中滴加n
‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
((2r,3r)
‑3‑
((r)
‑2‑
氨基丙氧基)
‑
n,2
‑
二甲基壬酰胺基)戊酰胺基)
‑2‑
环庚基乙酰胺基)
‑3‑
(((苄氧基)羰基)氨基)丙酰基)
‑
o
‑
(2
‑
(叔丁氧羰基)
‑2‑
氮杂螺[3.3]庚烷
‑6‑
基)
‑
l
‑
丝氨酸(412.0mg,0.4062mmol)在thf(100ml)中的溶液。又经过1h后,真空蒸发反应混合物。所得残余物用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(24g二氧化硅,溶剂梯度:0
‑
10%甲醇在dcm中),得到157.4mg(39%)标题化合物。lcms(esi):[2m h]
=1992.15。
[0682]
步骤3.(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(((2
‑
氮杂螺[3.3]庚烷
‑6‑
基)氧基)甲基)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0683]
将6
‑
(((3r,6s,9s,12s,15s,18r,19r)
‑9‑
((((苄氧基)羰基)氨基)甲基)
‑
12
‑
环庚
基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)甲氧基)
‑2‑
氮杂螺[3.3]庚烷
‑2‑
甲酸叔丁酯(157.4mg,0.1580mmol)和乙酸乙酯(5ml)充入烧瓶中,并用氮气吹扫该烧瓶。加入三氟乙酸(25μl,0.323mmol)和钯(碳载,10wt.%)(187mg,0.176mmol)后,将反应混合物抽真空并用氢气回填,然后在室温在氢气球下搅拌16h。反应混合物通过硅藻土过滤,用100ml 5%乙酸在乙醇中的溶液冲洗,真空蒸发滤液,与甲苯(3x1ml)共沸。在0℃向所得残余物中加入dcm(2.0ml)和三氟乙酸(0.2ml,3mmol)。将该混合物在0℃搅拌5h,然后真空蒸发,与甲苯(2x2ml)共沸,将所得残留物通过反相hplc纯化并冻干,得到30.1mg(21%)标题化合物,为其三氟乙酸盐。lcms(esi):r
t
(min)=10.073,[m h]
=762.5,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.66
‑
8.27(m,3h),8.18
‑
7.69(m,5h),7.37(d,j=8.4hz,1h),4.56(dd,j=10.6,4.7hz,1h),4.47
‑
4.13(m,2h),4.04(d,j=14.5hz,1h),3.99
‑
3.72(m,8h),3.64(d,j=13.2hz,2h),3.29
‑
2.70(m,9h),2.11(d,j=70.9hz,4h),1.87
‑
1.11(m,25h),1.11
‑
0.79(m,12h)。
[0684]
实例12.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑3‑
((3
‑
氨基丙氧基)甲基)
‑
15,16
‑
二丁基
‑9‑
环己基
‑
12
‑
异丁基
‑
13,18
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0685][0686]
步骤1.n
‑
me
‑
亮氨酸
‑
环己基甘氨酸
‑
dap(boc)
‑3‑
((叔丁氧羰基)氨基)丙基)
‑
丝氨酸
‑
(2
‑
氯三苯甲基树脂)
[0687][0688]
按照类似于那些针对实例7步骤1所述的方法制备树脂结合的四肽。
[0689]
步骤2.(2s,5s,8s,11s,14s)
‑
15
‑
((r)
‑2‑
氨基丙基)
‑5‑
(((叔丁氧羰基)氨基)甲基)
‑2‑
((3
‑
((叔丁氧羰基)氨基)丙氧基)甲基)
‑
14
‑
丁基
‑8‑
环已基
‑
11
‑
异丁基
‑
12
‑
甲基
‑
4,7,10,13
‑
四代
‑
3,6,9,12,15
‑
五氮十九烷
‑1‑
酸
[0690][0691]
将来自步骤1的树脂结合的四肽(1.50g,树脂负载值~0.6mmol/g)用(2s)
‑2‑
[丁基
‑
[(2r)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丙基]氨基]己酸(672mg,1.44mmol,中间体t13)、pyaop(1.88g,3.60mmol)和dipea(0.94ml,5.4mmol)在10ml dmf中的混合物(在室温预混合10min)处理。将树脂在旋转器上振摇16h,沥干并用dmf(4x10ml)和dcm(2x10ml)冲洗。然后,树脂用15ml 20%哌啶的dmf溶液处理,在旋转器上振摇1h,然后沥干并用dmf(4x10ml)和dcm(2x10ml)冲洗。树脂用10ml hfip/dcm(1/4)混合物处理,并在氮气鼓泡下搅拌1h。收集液相并将树脂用10ml hfip/dcm(1/4)处理两次以上,每次1h。合并液相并用dcm(2x10ml)洗涤树脂。合并滤液并真空浓缩。将残余物通过快速色谱在c18上纯化,用ch3cn/h2o洗脱,以得到标题化合物(330mg,39%收率),为黄色固体。lcms(esi):[m h]
=941.7。
[0692]
步骤3.n
‑
[3
‑
[[(2s,5r,8s,11s,14s,17s)
‑
17
‑
[(叔丁氧羰基氨基)甲基]
‑
7,8
‑
二丁基
‑
14
‑
环己基
‑
11
‑
异丁基
‑
5,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基]甲氧基]丙基]氨基甲酸酯
[0693][0694]
使用来自步骤2的物质,按照类似于那些在实例7步骤3中描述的方法制备标题化合物。lcms(esi):[m h]
=923.7。
[0695]
步骤4.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑3‑
(3
‑
氨基丙氧基甲基)
‑
15,16
‑
二丁基
‑9‑
环己基
‑
12
‑
异丁基
‑
13,18
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0696]
使用来自步骤3的物质并且按照类似于那些针对实例7步骤4所述的方法,将标题化合物制备为三氟乙酸盐。lcms(esi):[m h]
=723.6,r
t
=2.19min,方法=j。1h nmr(400mhz,dmso
‑
d6):δ9.01
‑
8.64(m,2h),8.21
‑
8.05(m,3h),7.85
‑
7.72(m,3h),7.60
‑
7.22
(m,2h),4.82
‑
4.72(m,1h),4.44
‑
4.31(m,2h),4.09
‑
3.92(m,2h),3.49
‑
3.32(m,10h),3.23
‑
3.12(m,4h),2.92
‑
2.80(m,3h),2.21
‑
2.11(m,1h),2.06
‑
1.93(m,2h),1.81
‑
1.73(m,3h),1.70
‑
1.55(m,8h),1.42
‑
1.26(m,6h),1.19
‑
1.10(m,5h),1.07
‑
0.83(m,15h)。
[0697]
实例13.(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(((1r,3s)
‑3‑
氨基环丁氧基)甲基)
‑9‑
(氨基甲基)
‑
19
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0698][0699]
步骤1.n
‑
me
‑
正缬氨酸
‑
环庚基甘胺酸
‑
dap(boc)
‑
反式
‑
叔丁氧羰基)氨基
‑
环丁基)
‑
丝氨酸
‑
(2
‑
氯三苯甲基树脂)
[0700][0701]
按照类似于那些在实例7步骤1描述的方法制备树脂结合的四肽。
[0702]
步骤2.(2s,5s,8s,11s,14r,15r,18r)
‑
18
‑
氨基
‑
15
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基甲基)
‑2‑
(((1r,3s)
‑3‑
((叔丁氧羰基)氨基)环丁氧基)甲基)
‑5‑
(((叔丁氧羰基)氨基)甲基)
‑8‑
环庚基
‑
12,14
‑
二甲基
‑
4,7,10,13
‑
四氧代
‑
11
‑
丙基
‑
16
‑
氧杂
‑
3,6,9,12
‑
四氮杂十九烷
‑1‑
酸
[0703][0704]
将来自步骤1的树脂结合的四肽(600mg,~0.5mmol/g)用thf(3ml)和dipea(0.31ml,1.8mmol)处理,并令其溶胀30min。独立地将(2r,3r)
‑4‑
[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]
‑3‑
[(2r)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丙氧基]
‑2‑
甲基
‑
丁酸(243mg,0.478mmol,中间体5)在thf(6.0ml)中的溶液加入三光气(55.2mg,0.19mmol)在thf(3.0ml)中的溶液中。滴加2,4,6
‑
三甲基吡啶(0.87ml,6.6mmol),此时混合物放热并形成无色沉淀。将该悬浮液轻轻摇动约1min,然后将其添加到预处理的树脂中。将混合物置于室温旋转器上3h,沥干,然后用dmf(10ml)和dcm(2x10ml)冲洗。
[0705]
树脂用10ml 20%哌啶的dmf溶液处理,在氮气鼓泡下搅拌1h,然后沥干并用dmf(2x10ml)和dcm(2x10ml)冲洗。然后用hfip/dcm=1/4溶液(20ml)处理树脂并于室温旋转器上振摇1h,然后过滤。收集液相。用hfip/dcm=1/4重复该过程并收集液相。将树脂用dcm(3x10ml)洗涤。合并液相和滤液并真空浓缩。将残余物通过反相快速色谱在c18柱上纯化,以得到标题化合物(262mg),为黄色固体。lcms(esi):[m h]
=992.7。
[0706]
步骤3.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑
19
‑
[[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]甲基]
‑6‑
[[3
‑
(叔丁氧羰基氨基)环丁氧基]甲基]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸叔丁酯
[0707][0708]
将来自步骤2的产物(240mg,0.242mmol)溶解在含有dipea(0.17ml,0.97mmol)的thf(240ml)中。将溶液冷却至0℃
°
。在0℃
°
向该溶液中滴加hatu(138mg,0.36mmol)和hobt(49.1mg,0.36mmol)在dmf(2ml)和thf(8ml)中的预混合溶液。将混合物在0℃
°
搅拌1h。将混
合物真空浓缩以移除thf。残余物用dmf(3ml)稀释并在剧烈搅拌下滴加到水(300ml)中。过滤收集所得固体,用水(2x10ml)洗涤并干燥,以得到标题化合物(185mg,粗制),为无色固体。lcms(esi):[m h]
=974.6。
[0709]
步骤4.(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(((1r,3s)
‑3‑
氨基环丁氧基)甲基)
‑9‑
(氨基甲基)
‑
19
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0710]
将来自步骤3的产物(175mg,粗制)用三氟乙酸(3.0ml)处理,并在0℃
°
搅拌2h。真空浓缩反应混合物。将残余物在以下条件下通过prep
‑
hplc纯化:柱:xselect csh prep c18 obd色谱柱,19*250mm,5um;流动相a:水(0.05%tfa),流动相b:acn;流速:25ml/min;梯度:20%b至40%b在20min内;210nm;rt:19.57min,以得到作为tfa盐的标题化合物(46.1mg),为白色固体。lcms(esi):[m h]
=774.6,r
t
=1.97min,方法h。1h nmr(400mhz,dmso
‑
d6):δ8.52
‑
8.12(m,2h),8.03
‑
7.93(m,6h),7.65
‑
7.33(m,1h),4.56
‑
4.15(m,4h),3.84
‑
3.60(m,4h),3.28
‑
2.82(m,9h),2.74(s,2h),2.24
‑
2.02(m,8h),1.79
‑
1.58(m,10h),1.52
‑
1.39(m,10h),1.37
‑
1.26(m,5h),1.03
‑
0.93(m,11h)。
[0711]
实例14.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[(3
‑
氨基丙基氨基)甲基]
‑
12
‑
环庚基
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0712][0713]
步骤1.(((3r,6s,9s,12s,15s,18r,19r)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
6,9
‑
二基)双(亚甲基))二
‑
氨基甲酸苄酯叔丁酯
[0714][0715]
按照类似于那些在实例13步骤1
‑
3描述的方法制备标题化合物。
[0716]
步骤2.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸叔丁酯
[0717][0718]
在室温,将来自步骤1的产物(905mg,1.01mmol)和10%pd/c(450mg)在异丙醇(100ml)中在氢气(1atm)下搅拌16h。经由过滤移除催化剂,并减压浓缩滤液。将残余物通过反相色谱在c18柱上纯化(0
‑
46%乙腈/0.05%tfa水溶液),以得到标题化合物(350mg),为白色固体。lcms(esi):[m h]
=766.6。
[0719]
步骤3.n
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
[(叔丁氧羰基氨基)甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基氨基]丙基]氨基甲酸叔丁酯
[0720][0721]
向来自步骤2的产物(420mg,0.548mmol)在dcm(400ml)中的溶液中加入n
‑
(3
‑
氧代丙基)氨基甲酸叔丁酯(95.7mg,0.55mmol)。将溶液在室温搅拌2h,然后加入三乙酰氧基硼氢化钠(233.6mg,1.1mmol)。所得混合物在室温搅拌2h,然后用水和盐水洗涤。有机相经无水硫酸钠干燥并且在真空下浓缩。残余物通过反向硅胶快速色谱在c18色谱柱上纯化,用ch3cn/h2o(45/55)洗脱,以得到标题化合物(235mg,46.3%收率),为白色固体。lcms(esi):[m h]
=923.6。
[0722]
步骤4.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[(3
‑
氨基丙基氨基)甲基]
‑
12
‑
环庚基
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0723]
将来自步骤3的产物(100.4mg,0.11mmol)用tfa(5.0ml)在0℃
°
处理30min,然后真空浓缩。将残余物通过prep
‑
lcms纯化,以得到作为其tfa盐的标题化合物(45.5mg),为白色固体。lcms(esi):[m h]
=723.6,r
t
=2.2min,方法=k。1h nmr(400mhz,dmso
‑
d6):δ8.93
‑
8.56(m,3h),8.38
‑
7.90(m,7h),7.52
‑
7.44(m,1h),4.62
‑
4.55(m,2h),4.28
‑
4.09(m,2h),
3.91
‑
3.74(m,3h),3.65
‑
3.48(m,4h),3.23
‑
3.03(m,5h),2.98
‑
2.79(m,6h),2.72(s,1h),2.13
‑
1.90(m,5h),1.80
‑
1.62(m,6h),1.57
‑
1.26(m,16h),1.05
‑
0.84(m,11h)。
[0724]
实例15.(3s,6s,9s,12s,15s,17r)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
(环戊基甲基)
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13,15,17
‑
三甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0725][0726]
步骤1.(2s,5s,8s,11s,14s,16s)
‑
17
‑
氨基
‑2‑
((s)
‑1‑
(叔丁氧基)乙基)
‑5‑
(((叔丁氧羰基)氨基)甲基)
‑8‑
环己基
‑
15
‑
(环戊基甲基)
‑
11
‑
异丁基
‑
12,14,16
‑
三甲基
‑
4,7,10,13
‑
四氧代
‑
3,6,9,12,15
‑
五氮杂十七烷
‑1‑
酸
[0727][0728]
按照类似于那些在实例12步骤2中描述的方法,从n
‑
me
‑
亮氨酸
‑
环己基甘氨酸
‑
dap(boc)
‑
别苏氨酸(t
‑
bu)
‑
(2
‑
氯三苯甲基树脂)(实例8,步骤1)和(2s)
‑2‑
[环戊基甲基
‑
[(1s)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)
‑1‑
甲基
‑
乙基]氨基]丙酸(中间体t21)制备标题化合物。lcms(esi):[m h]
=838.6。
[0729]
步骤2.(((2s,5s,9s,11s,14s,17s)
‑5‑
((s)
‑1‑
(叔丁氧基)乙基)
‑
17
‑
环已基
‑
10
‑
(环戊基甲基)
‑
14
‑
异丁基
‑
9,11,13
‑
三甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸叔丁酯
[0730][0731]
使用步骤1的产物并且按照类似于那些在实例12步骤3中描述的方法,制备标题化合物。lcms(esi):[m h]
=820.6。
[0732]
步骤3.(3s,6s,9s,12s,15s,17r)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
(环戊基甲基)
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13,15,17
‑
三甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0733]
将来自步骤2的产物(192mg,0.234mmol)用tfa(10ml)在0℃。处理1h。减压蒸发混合物,并将残余物通过制备型lcms纯化:柱:xbridge prep c18obd色谱柱,5μm,19
×
150mm;流动相a:水(0.05%tfa),流动相b:acn;流速:25ml/min;梯度:10%b至35%b在20min内;210nm;rt:14.86min,以得到作为其tfa盐的标题化合物(14.6mg),为白色固体。lcms(esi):[m h]
=664.5,r
t
=2.18min,方法=j。1h nmr(300mhz,dmso
‑
d6):δ9.00
‑
8.59(m,1h),8.09
‑
7.95(m,3h),7.84
‑
7.24(m,2h),4.98
‑
4.86(m,1h),4.65
‑
4.57(m,1h),4.06
‑
3.99(m,3h),3.91
‑
3.65(m,1h),3.21
‑
2.69(m,11h),2.35
‑
2.12(m,4h),1.81
‑
1.47(m,14h),1.36
‑
0.88(m,20h)。
[0734]
实例16.n
‑
[[(2s,5r,8s,11s,14s,17s)
‑
17
‑
(氨基甲基)
‑
7,8
‑
二丁基
‑
14
‑
环己基
‑
5,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
11
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基]甲基]
‑2‑
氮杂螺[3.3]庚烷
‑6‑
甲酰胺(三氟乙酸盐)
[0735][0736]
步骤1.n
‑
[[(2s,5r,8s,11s,14s,17s)
‑5‑
(苄氧羰基氨基甲基)
‑
10,11
‑
二丁基
‑
17
‑
环己基
‑
8,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
14
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基]甲基]氨基甲酸叔丁酯
[0737][0738]
按照类似于那些在实例7步骤1
‑
4描述的方法制备标题化合物。lcms(esi):[m h]
=885.5。
[0739]
步骤2.(((2s,5r,8s,11s,14s,17s)
‑5‑
(氨基甲基)
‑
10,11
‑
二丁基
‑
17
‑
环已基
‑
8,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
14
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸叔丁酯
[0740][0741]
将来自步骤1的产物(181mg,0.205mmol)和pd/c(200mg,碳上负载10%pd)在乙酸乙酯(17ml)中的混合物在氢气气氛下于40℃
°
搅拌6h。经由过滤移除催化剂,并减压浓缩滤液。粗产物直接用于下一步骤。lcms(esi):[m h]
=751.5。
[0742]
步骤3. 6
‑
[[(2s,5r,8s,11s,14s,17s)
‑
17
‑
[(叔丁氧羰基氨基)甲基]
‑
7,8
‑
二丁基
‑
14
‑
环已基
‑
5,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
11
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基]甲基氨基甲酰基]
‑2‑
氮杂螺[3.3]庚烷
‑
2.甲酸叔丁酯
[0743][0744]
在0℃
°
向(((2s,5r,8s,11s,14s,17s)
‑5‑
(氨基甲基)
‑
10,11
‑
二丁基
‑
17
‑
环己基
‑
8,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
14
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸叔丁酯(100mg,0.130mmol)和2
‑
叔丁氧羰基
‑2‑
氮杂螺[3.3]庚烷
‑6‑
甲酸(52.7mg,0.220mmol)和dipea(65.8mg,0.510mmol)在thf(10ml)中的溶液中滴加hatu(77.5mg,0.204mmol)和hobt(21.4mg,0.159mmol)在dmf(1.0ml)中的溶液。将混合物在0℃
°
搅拌1h。在搅拌下加入水(30ml)。收集所得固体并用水(3x)洗涤以得到粗制标题化合物(120.5mg),为黄色固体。lcms(esi):[m h]
=974.6。
[0745]
步骤4.n
‑
[[(2s,5r,8s,11s,14s,17s)
‑
17
‑
(氨基甲基)
‑
7,8
‑
二丁基
‑
14
‑
环己基
‑
5,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
11
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基]甲基]
‑2‑
氮杂螺[3.3]庚烷
‑6‑
甲酰胺
[0746]
将步骤3的产物(110mg,0.113mmol)用tfa(6.0ml)在0℃
°
处理1h,然后真空浓缩。将残余物通过制备型hplc纯化,以得到作为其tfa盐的标题化合物(15.9mg),为白色固体。lcms(esi):[m h]
=774.6,r
t
=2.05min,方法=j。1h nmr(400mhz,dmso
‑
d6):δ8.91
‑
8.49(m,3h),8.16
‑
8.03(m,4h),7.96
‑
7.86(m,1h),7.62
‑
6.96(m,2h),4.79
‑
4.70(m,1h),4.58
‑
4.42(m,2h),4.31
‑
4.15(m,2h),4.02
‑
3.87(m,8h),3.22
‑
3.01(m,8h),2.91
‑
2.82(m,2h),2.35
‑
2.21(m,5h),2.04
‑
1.84(m,3h),1.67
‑
1.54(m,7h),1.47
‑
1.23(m,9h),1.15
‑
1.06(m,4h),1.02
‑
0.93(m,8h),0.90
‑
0.83(m,4h)。
[0747]
实例17和18.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环已基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑6‑
(哌嗪
‑1‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)和(3r,6s,9s,12s,15s,18r,19r)39
‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑6‑
(哌嗪
‑1‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐),单一的未知立体异构体
[0748][0749]
步骤1. 4
‑
(((3r,9s,12s,15s,18r,19r)
‑9‑
((((苄氧基)羰基)氨基)甲基)
‑
12
‑
环己基
‑
19
‑
已基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)甲基)哌嗪
‑1‑
甲酸苄酯
[0750][0751]
按照类似于那些针对实例25和11所述的过程制备标题化合物。两种非对映异构体通过硅胶快速色谱分离(12g二氧化硅,溶剂梯度:0
‑
10%甲醇在dcm中),得到两种单一的未知立体异构体。
[0752]
立体异构体1:产量=46.5mg;lcms(esi):[m h]
=1003.7
[0753]
立体异构体2:产量=41.2mg;lcms(esi):[m h]
=1003.8
[0754]
步骤2.(3r,6s,9s,12s,15s,18r,19r)
‑
9(氨基甲基)
‑
12
‑
环已基
‑
19
‑
已基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑6‑
(哌嗪
‑1‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)和(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
已基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑6‑
(哌嗪
‑1‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0755]
使用在步骤1中分离的异构体并按照类似于那些针对实例11所述的过程,将标题化合物各自制备为单一的未知立体异构体。
[0756]
实例17:lcms(esi):r
t
(min)=9.98,[m h]
=735.6,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.53(s,2h),8.15(d,j=4.7hz,1h),7.98
‑
7.81(m,4h),7.68(d,j=8.2hz,1h),4.75
‑
4.60(m,1h),4.28
‑
4.14(m,1h),3.98
‑
3.76(m,3h),3.15(s,4h),3.10
‑
2.57(m,13h),1.90
‑
0.74(m,41h)。
[0757]
实例18:lcms(esi):r
t
(min)=10.12,[m h]
=735.6,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.63(d,j=93.7hz,3h),8.34
‑
8.09(m,1h),8.02
‑
7.59(m,5h),4.75
‑
3.64(m,8h),3.23
‑
2.55(m,17h),2.21
‑
0.71(m,39h)。
[0758]
实例19.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
((3
‑
(((3
‑
氨基丙基)氨基)甲基)苯氧基)甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0759][0760]
步骤1.n
‑
[3
‑
[[3
‑
[(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]苯基]甲基氨基]丙基]氨基甲酸叔丁酯
[0761][0762]
在0℃
°
向(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
羟基
‑
丙酸(10.0g,41.9mmol)在dmf(120ml)中的溶液中加入hatu(23.9g,63.0mmol)、n,o
‑
二甲基羟胺盐酸盐(6.15g,63.0mmol)和dipea(10.8g,83.9mmol)。将反应混合物在室温搅拌2h。将系统用乙酸乙酯(500ml)稀释,依次用0.5mnaoh水溶液(3
×
250ml)和盐水(250ml)洗涤。有机层用无水硫酸钠干燥并真空浓缩,并将残留物通过硅胶色谱纯化,用(etoac/pe=8/2)洗脱,给出n
‑
[(1s)
‑1‑
(羟基甲基)
‑2‑
[甲氧基(甲基)氨基]
‑2‑
氧代
‑
乙基]氨基甲酸苄酯(11.0g,93.1%收率),为淡黄色油状物。lcms(esi):[m h]
=283.1。
[0763]
在氮气下向n
‑
[(1s)
‑1‑
(羟基甲基)
‑2‑
[甲氧基(甲基)氨基]
‑2‑
氧代
‑
乙基]氨基甲酸苄酯(860mg,3.05mmol)在甲苯(10ml)中的溶液中加入3
‑
溴苯甲醛(673mg,3.64mmol)、cs2co3(1.99g,6.11mmol)、t
‑
bubrettphos(148mg,0.305mmol)和[pd(allyl)cl]2(55.5mg,0.152mmol)。将反应混合物在60℃搅拌过夜,并真空浓缩。将残余物通过硅胶柱快速色谱纯化,用(etoac/pe=4/6)洗脱,以得到n
‑
[(1s)
‑1‑
[(3
‑
甲酰基苯氧基)甲基]
‑2‑
[甲氧基(甲基)氨基]
‑2‑
氧代
‑
乙基]氨基甲酸苄酯(140mg,11.9%收率),为黄色油状物。lcms(esi):[m na]
=409.1。
[0764]
在0℃向(3
‑
氨基丙基)氨基甲酸叔丁酯(336mg,1.93mmol)在dcm(12ml)中的溶液中加入n
‑
[(1s)
‑1‑
[(3
‑
甲酰基苯氧基)甲基]
‑2‑
[甲氧基(甲基)氨基]
‑2‑
氧代
‑
乙基]氨基甲酸苄酯(670mg,1.73mmol)在dcm(3.0ml)中的溶液。将混合物在室温下搅拌2h。在0℃分批加入三乙酰氧基硼氢化钠(818mg,3.86mmol)。将混合物在室温搅拌1h,然后真空浓缩。将残余物加载到硅胶柱上,用(dcm/meoh=91/9)洗脱,给出标题化合物(810mg,77.2%收率),为黄色油状物。lcms(esi):[m h]
=545.4。
[0765]
步骤2.n
‑
[[3
‑
[(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧
基]苯基]甲基]
‑
n
‑
[3
‑
(叔丁氧羰基氨基)丙基]氨基甲酸叔丁酯
[0766][0767]
在0℃
°
向n
‑
[3
‑
[[3
‑
[(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]苯基]甲基氨基]丙基]氨基甲酸叔丁酯(710mg,1.3mmol)和dipea(336.7mg,2.61mmol)在dcm(20ml)中的溶液中加入(boc)2o(569.7mg,2.61mmol)在dcm(5ml)中的溶液。将反应混合物在25℃
°
搅拌1.5h,然后真空浓缩。将残余物通过硅胶快速色谱纯化,用pe/etoac(2/1)洗脱,以得到标题化合物(692mg,82.3%收率),为黄色油状物。lcms(esi):[m h]
=645.3。
[0768]
步骤3.n
‑
[[3
‑
[(2s)
‑2‑
氨基
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]苯基]甲基]
‑
n
‑
[3
‑
(叔丁氧羰基氨基)丙基]氨基甲酸叔丁酯
[0769][0770]
将n
‑
[[3
‑
[(2s)
‑2‑
(苄氧基(甲基)氨基)
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]苯基]甲基]
‑
n
‑
[3
‑
(叔丁氧羰基氨基)丙基]氨基甲酸叔丁酯(588mg,0.909mmol)和pd(碳载,10wt%,600mg)在etoac(120ml)中的混合物在氢气氛下在25℃
°
搅拌45min。经由过滤移除催化剂,并真空浓缩滤液,以得到标题化合物(460mg),为黄色油状物。lcms(esi):[m h]
=511.3。
[0771]
步骤4.(2s)
‑3‑
[3
‑
[[叔丁氧羰基
‑
[3
‑
(叔丁氧羰基氨基)丙基]氨基]甲基]苯氧基]
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丙酸
[0772][0773]
在室温将lioh(108mg,4.5mmol)在水(2ml)中的溶液加入n
‑
[[3
‑
[(2s)
‑2‑
氨基
‑3‑
[甲氧基(甲基)氨基]
‑3‑
氧代
‑
丙氧基]苯基]甲基]
‑
n
‑
[3
‑
(叔丁氧羰基氨基)丙基]氨基甲酸叔丁酯(460mg,0.90mmol)在thf(24ml)和water(8ml)中的溶液中。将反应混合物在25℃
°
搅拌3天,然后冷却至0℃
°
。在0℃
°
滴加溶解在5ml thf中的氯甲酸9
‑
芴基甲酯(465.4mg,1.8mmol)。将反应混合物在25℃
°
搅拌0.5h。真空蒸发thf。将残余物用水(2ml)稀释,并用1m hcl水溶液调节至ph~6。将所得溶液真空浓缩,并将残余物通过硅胶快速色谱纯化,用dcm/meoh(10/1)洗脱,以得到标题化合物(241.5mg,38.9%收率),为白色固体。lcms(esi):[m h]
=690.2。1h nmr(300mhz,dmso
‑
d6):δ13.16(s,1h),7.88(d,j=8.1hz,2h),7.73
‑
7.62(m,3h),7.44
‑
7.41(m,2h),7.33
‑
7.20(m,3h),6.84
‑
6.77(m,4h),4.38
‑
4.21(m,8h),3.10
‑
3.07(m,2h),2.88
‑
2.85(m,2h),1.60
‑
1.57(m,2h),1.35(s,18h)。
[0774]
步骤5.o
‑
(3
‑
(((叔丁氧羰基)(3
‑
((叔丁氧羰基)氨基)丙基)氨基)甲基)苯基)
‑
m((s)
‑3‑
((叔丁氧羰基)氨基)
‑2‑
((s)
‑2‑
环庚基
‑2‑
((s)
‑2‑
(甲基氨基)戊酰胺基)乙酰胺基)丙酰基)
‑
l
‑
丝氨酸
‑
(2
‑
氯三苯甲基树脂)
[0775][0776]
按照类似于那些在实例7步骤1描述的过程制备树脂结合的四肽。
[0777]
步骤6.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
((3
‑
(((3
‑
氨基丙基)氨基)甲基)苯氧基)甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0778]
使用来自步骤5的树脂结合的四肽并且按照类似于那些在实例13步骤2
‑
4中描述的过程,制备标题化合物。lcms(esi):[m h]
=829.6,r
t
=2.78min,方法=j。1h nmr(400mhz,dmso
‑
d6):δ9.05
‑
8.34(m,4h),8.10
‑
7.90(m,7h),7.53
‑
6.96(m,5h),4.62
‑
4.58(m,1h),4.39
‑
4.37(m,1h),4.28
‑
4.23(m,1h),4.14
‑
4.05(m,2h),3.88
‑
3.82(m,2h),3.29
‑
3.15(m,4h),3.11(s,1h),3.01
‑
2.91(m,4h),2.87
‑
2.78(m,4h),2.70(s,2h),2.07
‑
1.91(m,4h),1.79
‑
1.23(m,25h),1.04
‑
0.83(m,12h)。
[0779]
实例20.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
18
‑
(2
‑
氟乙基)
‑
16
‑
己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0780][0781]
步骤1.(叔丁氧羰基)
‑
d
‑
高丝氨酸苄酯
[0782][0783]
将boc(d)
‑
高丝氨酸(1.0g,4.3mmol)、苄基溴(1.1g,6.5mmol)和碳酸钾(0.8g,8.7mmol)在dmf(5ml)中的混合物搅拌过夜。用水稀释反应混合物,并且用ipac/庚烷混合物萃取。有机层用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过快速色谱法(硅胶,0
‑
100%ipac/庚烷)纯化,以获得1.2g(91%)的标题化合物。
[0784]
步骤2.(r)
‑2‑
((叔丁氧羰基)氨基)
‑4‑
氟丁酸苄酯
[0785][0786]
向(叔丁氧羰基)
‑
d
‑
高丝氨酸苄酯(1.2g,3.9mmol)在干燥1,4
‑
二氧六环(5ml)中的溶液中加入吡啶
‑2‑
磺酰氟(0.7g,3.9mmol)和1,3,4,6,7,8
‑
六氢
‑1‑
甲基
‑
2h
‑
嘧啶并[1,2
‑
a]嘧啶(1.2g,7.8mmol),并将混合物在室温搅拌过夜。用水稀释反应混合物,并且用ipac/庚烷混合物萃取。有机层用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过快速色谱法(硅胶,0
‑
50%ipac/庚烷)纯化,以获得0.87g(72%)的标题化合物。1h nmr(400mhz,dmso
‑
d6)δ7.47
‑
7.32(m,5h),5.29
‑
4.94(m,2h),4.50(dt,j=46.9,5.3hz,2h),4.24
‑
4.01(m,1h),2.25
‑
1.83(m,2h),1.38(s,9h)。
[0787]
步骤3.(r)
‑
(4
‑
氟
‑1‑
羟基丁烷
‑2‑
基)氨基甲酸叔丁酯
[0788][0789]
向(r)
‑2‑
((叔丁氧羰基)氨基)
‑4‑
氟丁酸苄酯(2.2g,7.1mmol)的冰冷溶液中加入
氢化铝锂(3.5ml,在thf中的2m溶液),并将混合物搅拌1h。用饱和氯化铵水溶液猝灭反应混合物,过滤移除固体并用ipac充分洗涤。滤液以硫酸钠干燥,并浓缩。将残余物通过快速色谱法(硅胶,0
‑
5%meoh/dcm)纯化,以获得1.0g(68%)的标题化合物。1h nmr(400mhz,氯仿
‑
d)δ5.12
‑
4.89(m,1h),4.56(dt,j=47.2,5.8hz,2h),3.87
‑
3.59(m,3h),2.77(s,1h),2.09
‑
1.77(m,2h),1.45(d,j=4.2hz,9h)。
[0790]
步骤4.(r)
‑
(4
‑
氟
‑1‑
氧代丁烷
‑2‑
基)氨基甲酸叔丁酯
[0791][0792]
将(r)
‑
(4
‑
氟
‑1‑
羟基丁
‑2‑
基)氨基甲酸叔丁酯(0.5g,2.4mmol)、tempo(75mg,0.42mmol)和碘苯二乙酸酯(1.2g,3.6mmol)在dcm(20ml)中的混合物在室温搅拌4h。将反应混合物浓缩并与20%dcm/庚烷一起研磨。通过过滤移除固体并用20%dcm/庚烷充分洗涤。浓缩滤液并将残余物通过快速色谱法(硅胶,0
‑
5%meoh/dcm)纯化,以获得0.35g(70%)的标题化合物。1h nmr(400mhz,氯仿
‑
d)δ1h nmr(400mhz,氯仿
‑
d)δ9.61(d,j=2.1hz,1h),5.34(s,1h),4.59(dt,j=47.0,6.0hz,2h),4.31(s,1h),2.49
‑
2.08(m,2h),1.46(s,9h)。
[0793]
步骤5.n
‑
((r)
‑2‑
((叔丁氧羰基)氨基)
‑4‑
氟丁基)
‑
n
‑
己基
‑
l
‑
丙氨酸苄酯
[0794][0795]
将(r)
‑
(4
‑
氟
‑1‑
氧代丁烷
‑2‑
基)氨基甲酸叔丁酯(0.25g,1.2mmol)和(2s)
‑2‑
氨基丙酸苄酯盐酸盐(0.25g,1.2mmol)在3ml乙酸乙酯中的混合物搅拌1h,然后加入三乙酰氧基硼氢化物(0.38g,1.8mmol),并将混合物在室温搅拌过夜。向该混合物中加入己醛(0.24g,2.4mmol)和三乙酰氧基硼氢化物(0.5g,2.4mmol),并将混合物在50℃加热过夜。冷却反应混合物,用ipac稀释并在碳酸氢钠水溶液上搅拌。分离有机层,用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过快速色谱法(硅胶,0
‑
50%ipac/庚烷)纯化,以获得0.2g(30%)的标题化合物。lcms(esi):[m h]
=453.5。
[0796]
步骤6.n
‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑4‑
氟丁基)
‑
n
‑
已基
‑
l
‑
丙氨酸苄酯
[0797][0798]
将((r)
‑2‑
((叔丁氧羰基)氨基)
‑4‑
氟丁基)
‑
l
‑
丙氨酸酯(0.4g,0.9mmol)溶解在2ml的dcm中。加入hcl(1ml,在二氧六环中的4n溶液),并将反应混合物搅拌1h,然后浓缩。将
残余物溶于二氧六环:水(1∶1,6ml)中,加入fmoc
‑
osu(0.3g,0.9mmol)和碳酸氢钠(0.22g,2.7mmol),并将混合物搅拌1h。反应混合物用ipac稀释,用水、盐水洗涤,用硫酸钠干燥并浓缩。将残余物通过硅胶色谱法(溶剂梯度0
‑
50%ipac/庚烷)纯化,以获得0.46g(90%)的标题化合物。1h nmr(400mhz,dmso
‑
j6)δ7.89(d,j=7.6hz,2h),7.68(d,j=7.5hz,2h),7.48
‑
7.26(m,9h),7.04(dd,j=32.9,8.8hz,1h),5.16
‑
5.00(m,2h),4.52
‑
4.14(m,5h),3.55(d,j=9.5hz,2h),2.60
‑
2.27(m,2h),2.05
‑
1.44(m,2h),1.37
‑
1.04(m,13h),0.80(m,3h)。lcms(esi):[m h]
=575.2。
[0799]
步骤7.n
‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑4‑
氟丁基)
‑
n
‑
己基
‑
l
‑
丙氨酸
[0800][0801]
将(2s)
‑2‑
[[(2r)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)
‑4‑
氟
‑
丁基]
‑
己基
‑
氨基]丙酸苄酯(0.46g,0.8mmol)溶解在乙醇(5ml)中,并加入pd(0.05g,碳载,10%)。反应混合物用氢气吹扫并抽真空三次,然后在氢气球下搅拌30min。过滤移除催化剂并浓缩滤液,给出标题化合物(0.40g):1h nmr(400mhz,dmso
‑
d
6 d2o)δ7.88(t,j=6.7hz,2h),7.69(dd,j=7.5,2.3hz,2h),7.42(t,j=7.5hz,2h),7.37
‑
7.29(m,2h),7.04(d,j=8.9hz,1h),4.54
‑
4.46(m,1h),4.42
‑
4.18(m,3h),3.61(s,2h),2.68
‑
2.41(m,2h),1.42
‑
1.33(m,2h),1.28
‑
1.09(m,13h),0.91
‑
0.76(m,3h)。lcms(esi):[m h]
=485.4。
[0802]
步骤8.n
‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
(((r)
‑2‑
氨基
‑4‑
氟丁基)(己基)氨基)
‑
n
‑
甲基丙酰胺基)
‑4‑
甲基戊酰胺基)
‑2‑
环已基乙酰胺基)
‑3‑
(((苄氧基)羰基)氨基)丙酰基)
‑
o
‑
苄基
‑
l
‑
别苏氨酸
[0803][0804]
向过滤管中加入中间体15(0.75g,估计为0.45mmol/g)和8ml dmf。使树脂在室温溶胀90min,然后沥干。向树脂中加入n
‑
((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑4‑
氟丁基)
‑
n
‑
己基
‑
l
‑
丙氨酸(0.33g,0.68mmol)和pyaop(0.36mg,0.68mmol)在dmf(4ml)中的溶液,然后加入dipea(0.35ml,2.0mmol)。将所得混合物置于室温旋转器上16h。沥干树脂并用
dma(5x)和dcm(5x)冲洗。向树脂中加入6ml20%哌啶/dmf溶液并搅拌1h,然后沥干并用dcm(5x)冲洗。然后将树脂悬浮在6ml 1∶3hftp:dcm混合物中并搅拌1h。过滤该混合物,用dcm冲洗。真空蒸发滤液,与甲苯(3x2ml)共沸,得到粗制肽140mg(粗收率43%),其不经纯化即用于下一步骤。lcms(esi):[m h]
=940.9。
[0805]
步骤9.(((2s,5s,8r,11s,14s,17s)
‑5‑
((s)
‑1‑
(苄氧基)乙基)
‑
17
‑
环己基
‑8‑
(2
‑
氟乙基)
‑
10
‑
己基
‑
14
‑
异丁基
‑
11,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯
[0806][0807]
在室温,历时4h,在搅拌下向dipea(0.2ml,1mmol)和hatu(0.0.26mg,0.43mmol)在thf(10ml)中的溶液中滴加n
‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
((s)
‑2‑
(((r)
‑2‑
氨基
‑4‑
氟丁基)(己基)氨基)
‑
n
‑
甲基丙酰胺基)
‑4‑
甲基戊酰胺基)
‑2‑
环己基乙酰胺基)
‑3‑
(((苄氧基)羰基)氨基)丙酰基)
‑
o
‑
苄基
‑
l
‑
别苏氨酸(0.14g,0.15mmol)在thf(100ml)中的溶液,并搅拌过夜。真空蒸发反应混合物。所得残余物用乙酸乙酯稀释,用水(2x)和盐水洗涤,以硫酸钠干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(12g二氧化硅,溶剂梯度:0
‑
10%甲醇在dcm中),得到60mg标题化合物。lcms(esi):[m h]
=922.9。
[0808]
步骤10.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
18
‑
(2
‑
氟乙基)
‑
16
‑
己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0809]
将上述残余物(60mg,0.065mmol)和乙酸乙酯(5ml)充入烧瓶,并用氮气吹扫烧瓶。加入三氟乙酸(25μl,0.323mmol)和钯(碳载,10 wt.%)(50mg)后,将反应混合物抽真空并用氢气回填,然后在室温在氢气球下搅拌18h。反应混合物通过硅藻土过滤,用100ml 5%acoh在甲醇中的溶液冲洗,真空蒸发滤液,与甲苯(3x1ml)共沸。粗产物通过反相hplc纯化并冻干,得到19.6mg(37%,纯度87%)标题化合物,为其三氟乙酸盐。lcms(esi):r
t
(min)=8.33,[m h]
=698.5,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.69
‑
8.55(m,1h),8.41
‑
8.01(m,1h),8.01
‑
7.74(m,3h),7.66
‑
7.41(m,1h),5.38
‑
4.69(m,1h),4.69
‑
4.18(m,4h),4.18
‑
3.44(m,7h),3.25
‑
3.03(m,4h),3.05
‑
2.85(m,1h),2.16
‑
1.89(m,2h),1.89
‑
1.36(m,11h),1.36
‑
0.77(m,25h)。
[0810]
实例21. 4
‑
(2
‑
氨基乙基)
‑
n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]苯甲酰胺(三氟乙酸盐)
[0811][0812]
步骤1.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
[[[4
‑
[2
‑
(叔丁氧羰基氨基)乙基]苯甲酰基]氨基]甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸叔丁酯
[0813][0814]
在0℃向4
‑
(2
‑
((叔丁氧羰基)氨基)乙基)苯甲酸(82.4mg,0.31mmol)、hatu(117.8mg,0.31mmol)、hobt(41.8mg,0.31mmol)和dipea(0.11ml,0.63mmol)在thf(120ml)中的溶液中加入n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸酯(121mg,0.158mmol,实例14步骤2)在2ml dmf中的溶液。将混合物搅拌1h然后真空浓缩。将残余物通过反相色谱纯化(乙腈0
‑
80/0.05%tfa水溶液),以得到标题化合物(95.4mg,59.8%收率),为白色固体。lcms(esi):[m h]
=1013.7。
[0815]
步骤2. 4
‑
(2
‑
氨基乙基)
‑
n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]苯甲酰胺
[0816]
将步骤1的产物(95.2mg,0.090mmol)用tfa(5.0ml)在0℃
°
处理30min,然后真空浓缩。将残余物通过制备型lcms纯化,以得到作为其tfa盐的标题化合物(25.5mg),为白色固体。lcms(esi):[m h]
=813.6,r
t
=2.89min,方法=k;1h nmr(400mhz,dmso
‑
d6):δ8.85
‑
8.12(m,2h),7.99
‑
7.90(m,3h),7.88
‑
7.72(m,5h),7.56
‑
7.04(m,4h),4.62
‑
4.43(m,1h),4.22
‑
4.03(m,1h),3.98
‑
3.82(m,2h),3.32
‑
3.15(m,4h),3.12
‑
3.05(m,5h),3.02
‑
2.89(m,5h),2.80(s,2h),2.21
‑
1.94(m,3h),1.79
‑
1.52(m,8h),1.48
‑
1.25(m,17h),1.03
‑
0.90(m,7h),0.88
‑
0.82(m,4h)。
[0817]
实例22.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑3‑
((3
‑
氨基丙氧基)甲基)
‑
15
‑
丁基
‑9‑
环己基
‑
16
‑
(环丙基甲基)
‑
12
‑
异丁基
‑
13,18
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0818][0819]
步骤1.(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
(环丙基甲基)氨基]己酸苄酯
[0820][0821]
将(2s)
‑2‑
氨基己酸苄酯盐酸盐(1.0g,3.88mmol)和(r)
‑
(1
‑
氧代丙烷
‑2‑
基)氨基甲酸叔丁酯(0.63g,3.5mmol)在乙酸乙酯(10ml)中的混合物搅拌2h,然后加入三乙酰氧基硼氢化钠(1.23g,5.8mmol),并将混合物在室温搅拌过夜。然后向该混合物中加入环丙基甲醛(0.81g,5.82mmol)和三乙酰氧基硼氢化钠(1.0g,3.5mmol),并将混合物在室温搅拌24h。反应混合物用ipac稀释并在碳酸氢钠溶液上搅拌。分离有机层,用盐水洗涤,以硫酸钠干燥并浓缩。将残余物通过柱色谱法(硅胶,0
‑
50%ipac/庚烷)纯化,以得到1.1g(65%)的标题化合物。lcms(esi):[m h]
=433.3。1h nmr(400mhz,dmso
‑
d6)δ7.46
‑
7.26(m,5h),6.32(d,1h),5.18
‑
4.99(m,2h),3.52(t,j=7.4hz,2h),2.60(dd,j=13.4,8.5hz,1h),2.45(dd,j=12.4,5.9hz,1h),2.38
‑
2.17(m,2h),1.65
‑
1.53(m,2h),1.36(s,10h),1.34
‑
1.18(m,3h),0.98(d,3h),0.90
‑
0.83(m,3h),0.82
‑
0.73(m,1h),0.48
‑
0.39(m,1h),0.38
‑
0.29(m,1h),0.12
‑
0.02(m,2h)。
[0822]
步骤2.(s)
‑2‑
(((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙基)(环丙基甲基)氨基)己酸
[0823][0824]
向(2s)
‑2‑
[[(2r)
‑2‑
(叔丁氧羰基氨基)丙基]
‑
(环丙基甲基)氨基]己酸苄酯(1.1g,2.5mmol)在4ml dcm中的溶液中加入hcl(4ml,在二氧六环中的4m溶液),并将所得混合物搅拌2h,然后浓缩。将残余物溶解在二氧六环:水的1∶1混合物中,加入fmoc
‑
osu(1.7g,
4.65mmol)和碳酸钠(0.65g,5.00 mmol),并将混合物在室温搅拌过夜。反应混合物用ipac稀释,分离有机层,用盐水洗涤,用硫酸钠干燥并浓缩。将残余物通过色谱纯化(硅胶,0
‑
50%ipac/庚烷),得到苄酯(1.4g)。lcms(esi)[m h]
=556.8。
[0825]
将上述残余物溶解在含有钯(0.12g,碳载,10wt%)的乙醇(30ml)中,并在h2气球下氢化30min。通过硅藻土过滤移除催化剂并浓缩滤液。将残余物在40℃真空烘箱中干燥过夜以获得标题化合物(1.1g)。lcms(esi)[m h]
=465.1;1h nmr(400mhz,dmso
‑
d6)δ7.96
‑
7.85(m,2h),7.74
‑
7.66(m,2h),7.41(m,2h),7.32(m,2h),6.92(d,j=8.3hz,1h),4.35
‑
4.12(m,3h),3.61(d,j=14.4hz,1h),3.36(q,j=8.8,7.4hz,1h),2.75
‑
2.58(m,1h),2.48
‑
2.26(m,3h),1.66
‑
1.00(m,10h),0.90
‑
0.76(m,4h),0.54
‑
0.28(m,2h),0.17
‑
0.01(m,2h)。
[0826]
步骤3.(11s,14r,17s,20s,23s,26s)
‑
11
‑
氨基
‑
26
‑
((((苄氧基)羰基)氨基)甲基)
‑
17
‑
丁基
‑
23
‑
环己基
‑
16
‑
(环丙基甲基)
‑
20
‑
异丁基1
‑
2,2,14,19
‑
四甲基
‑
4,12,18,21,24
‑
五氧代
‑
3,9
‑
二氧杂
‑
5,13,16,19,22,25
‑
六氮杂二十七烷
‑
27
‑
酸
[0827][0828]
将(s)
‑3‑
(((苄氧基)羰基)氨基)
‑2‑
((s)
‑2‑
环己基
‑2‑
((s)
‑4‑
甲基
‑2‑
(甲基氨基)戊酰胺基)乙酰胺基)丙酸
‑
(2
‑
氯苯基三苯甲基
‑
树脂)(0.6g,0.47mmol/g,使用类似于那些在中间体15的合成中描述的方法制备)在dmf(3ml)中溶胀30min,然后用(s)
‑2‑
(((r)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)丙基)(环丙基甲基)氨基)己酸(0.26g,0.56mmol)、pyaop(0.3g,0.56mmol)和dipea(0.15ml,0.85mmol)在dmf(4ml)中的预混合溶液处理。将树脂和溶液搅拌20h,然后沥干并用dmf和dcm充分洗涤。将树脂悬浮在20%哌啶/dmf混合物(8ml)中并搅拌1h。然后沥干树脂并用dmf和dcm充分洗涤。树脂用中间体6(0.26g,0.6mmol)、pyaop(0.31g,0.6mmol)和dipea(0.1ml,0.8mmol)在dmf(4ml)中的预混溶液处理并搅拌3h,然后沥干并用dcm充分洗涤。树脂用20%哌啶/dmf(8ml)处理并搅拌1h。滤除液体,树脂用dcm充分洗涤并悬浮在hfip和dcm的1∶3混合物(8ml)中并搅拌1h。通过过滤收集液体,同时用dcm洗涤树脂。在与甲苯共沸的同时浓缩合并的滤液(3次)以获得粗制肽(0.2g)。lcms(esi):[m h]
=974.8。
[0829]
步骤4.(3
‑
(((2s,5r,8s,11s,14s,17s)
‑
17
‑
((((苄氧基)羰基)氨基)甲基)
‑8‑
丁基
‑
14
‑
环己基
‑7‑
(环丙基甲基)
‑
11
‑
异丁基
‑
5,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲氧基)丙基)氨基甲酸叔丁酯
[0830][0831]
历时3h,将在无水thf(175ml)中的来自步骤3的粗制肽(0.2g,0.2mmol)滴加到hatu(0.16g,0.4mmol)和dipea(0.11ml,0.62mmol)的混合物中,然后搅拌过夜。将反应混合物浓缩并溶解在ipac中,用水、盐水洗涤,用硫酸钠干燥并浓缩。将残余物通过柱色谱纯化(硅胶,o
‑
10%meoh/dcm),得到环肽(60mg,30%)。lcms(esi):[m h]
=955.8。
[0832]
步骤5.(3s,6s,9s,12s,15s,18r)
‑6‑
(氨基甲基)
‑3‑
((3
‑
氨基丙氧基)甲基)
‑
15
‑
丁基
‑9‑
环己基
‑
16
‑
(环丙基甲基)
‑
12
‑
异丁基
‑
13,18
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0833]
将步骤4的产物溶解在乙酸乙酯(3ml)中,加入pd(碳载,10wt%,60mg)和三氟乙酸(0.2μl),将烧瓶抽真空并用氢气吹扫,然后在氢气球下搅拌3h。过滤移除催化剂并用5%acoh/甲醇洗涤。浓缩滤液,所得残余物用1ml hcl(在二氧六环中的4n溶液)处理1h,然后浓缩,与甲苯共沸(3次)。所得残余物通过反相hplc纯化并冻干,得到10.3mg(6%,纯度87%)标题化合物,为其三氟乙酸盐。lcms(esi):r
t
(min)=6.00,[m h]
=721.5,方法=b;1h nmr(400mhz,dmso
‑
d6)δ9.01
‑
8.61(m,2h),8.00(t,j=32.4hz,5h),7.69(d,j=23.8hz,3h),7.33(d,j=144.9hz,2h),4.89
‑
4.57(m,2h),4.57
‑
3.87(m,7h),3.59(d,j=18.1hz,6h),3.28
‑
3.01(m,3h),2.83(s,2h),2.05(d,j=50.5hz,2h),1.91
‑
1.41(m,11h),1.41
‑
0.60(m,25h),0.43(d,j=41.0hz,2h)。
[0834]
实例23. 2
‑
((3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)乙酰胺(三氟乙酸盐)
[0835][0836]
步骤1.
[0837]2‑
[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]乙酸叔丁酯
[0838][0839]
按照类似于那些针对实例8步骤1
‑
3所述的方法制备标题化合物。lcms(esi):[m h]
=885.6。
[0840]
步骤2. 2
‑
((3r,6s,9s,12s,15s,18r,19r)
‑9‑
((((苄氧基)羰基)氨基)甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)乙酸
[0841][0842]
将来自步骤1的产物(1.04g,1.17mmol)用tfa(15ml)在0℃
°
处理1h,然后真空浓缩。残余物通过反相色谱纯化(溶剂梯度:0
‑
70%乙腈/0.05%tfa水溶液),以得到标题化合物(403mg,41.4%收率)。lcms(esi):[m h]
=829.5。
[0843]
步骤3.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(2
‑
氨基
‑2‑
氧代
‑
乙基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸苄酯
[0844][0845]
向2
‑
((3r,6s,9s,12s,15s,18r,19r)
‑9‑
((((苄氧基)羰基)氨基)甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)乙酸(101mg,0.120mmol)在dmf(2.0ml)中的溶液中加入hatu(92.0mg,0.240mmol)和dipea(62.0mg,0.48mmol)。将反应混合物搅拌5min。向混合物中加入氨(1.0ml,在thf中的0.5m溶液)和氯化铵(13.0mg,0.24mmol)。将反应混合物搅拌1h并真空浓缩。残余物通过反相色谱纯化(溶剂梯度:0
‑
68%乙腈/0.05%tfa水溶液),以得到标题化合物(67.0mg,66.4%收率)。lcms(esi):[m h]
=828.6。
[0846]
步骤4.2
‑
[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]乙酰胺
[0847]
将n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(2
‑
氨基
‑2‑
氧代
‑
乙基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸苄酯(67.0mg,0.080mmol)和pd/c(100mg,碳上负载10%pd)在2
‑
丙醇(10ml)中的混合物在室温于氢气氛下搅拌2.5h。经由过滤移除催化剂,并真空蒸发滤液。将残余物通过制备型hplc纯化并冻干,以得到作为其tfa盐的标题化合物(12.3mg),为白色固体。lcms(esi):[m h]
=694.5,r
t
=2.43min,方法=h。1h nmr(400mhz,dmso
‑
d6):δ8.62
‑
8.18(m,2h),8.02
‑
7.92(m,4h),7.48
‑
7.21(m,2h),6.93
‑
6.86(m,1h),4.62
‑
4.41(m,1h),4.32
‑
4.22(m,1h),4.18
‑
4.10(m,1h),3.86
‑
3.78(m,1h),3.30
‑
3.22(m,3h),3.10(s,2h),3.02
‑
2.91(m,2h),2.71(s,1h),2.21
‑
2.02(m,2h),1.76
‑
1.52(m,8h),1.48
‑
1.22(m,20h),1.05
‑
0.85(m,13h)。
[0848]
实例24.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
((3
‑
((3
‑
氨基丙基)氨基)丙氧基)甲基)
‑
19
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0849][0850]
步骤1.n
‑
me
‑
正缬氨酸
‑
环庚基甘氨酸
‑
dap(cbz)3
‑
((叔丁氧羰基)氨基)丙基)
‑
丝氨酸
‑
(2
‑
氯三苯甲基树脂)
[0851][0852]
按照类似于那些在实例7步骤1描述的方法制备树脂结合的四肽。
[0853]
步骤2.n
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
19
‑
[[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]甲基]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]丙基]氨基甲酸叔丁酯
[0854][0855]
使用来自步骤1的树脂结合的四肽和(2r,3r)
‑4‑
[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]
‑3‑
[(2r)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)丙氧基]
‑2‑
甲基
‑
丁酸(中间体5)并且按照类似于那些在实例13中描述的方法,制备标题化合物。lcms(esi):[m h]
=996.6。
[0856]
步骤3.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(3
‑
氨基丙氧基甲基)
‑
19
‑
[[(1s,
5r)
‑3‑
双环[3.2.1]辛烷基]甲基]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸苄酯
[0857][0858]
将步骤2的产物(430mg,0.43mmol)用tfa(12ml)在0℃
°
处理1h。加入甲苯(10ml),并将所得溶液真空浓缩。将残余物在100ml的dcm与5%na2co3溶液(20ml)之间分配。有机相用盐水(20ml)洗涤,以无水硫酸钠干燥,并真空浓缩以得到标题化合物(385mg),为淡黄色固体。lcms(esi):[m h]
=896.6。
[0859]
步骤4.n
‑
[3
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
19
‑
[[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]甲]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]丙基氨基]丙基]氨基甲酸叔丁酯
[0860][0861]
将步骤3的产物(385mg,0.430mmol)和n
‑
(3
‑
氧代丙基)氨基甲酸叔丁酯(66.9mg,0.39mmol)在dcm(30ml)中在室温搅拌2h。将混合物冷却至0℃
°
并在该温度加入三乙酰氧基硼氢化钠(137mg,0.646mmol)。使反应混合物回温至室温并在室温搅拌3h。用5%nh4cl水溶液(30ml)猝灭反应并分离各相。水相用dcm(100ml)萃取。合并的有机层用盐水洗涤(30ml),经无水硫酸钠干燥并真空浓缩。将残余物通过反相快速色谱在c18柱上纯化,以得到标题化合物(194mg,42.9%收率),为白色固体。lcms(esi):[m h]
=1053.7。
[0862]
步骤5.n
‑
[3
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
19
‑
[[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]甲基]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]丙基氨基]丙基]氨基甲酸叔丁酯
[0863][0864]
向n
‑
[3
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
19
‑
[[(1s,5r)
‑3‑
双环[3.2.1]辛烷基]甲基]
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]丙基氨基]丙基]氨基甲酸酯(194mg,0.184mmol)在异丙醇(30ml)中的溶液中加入钯(碳载,10wt%,194mg)。在氢气氛下将混合物在室温搅拌2.5h。经由过滤移除催化剂并真空浓缩滤液,以得到标题化合物(159mg,93.9%收率),为白色固体,不经进一步纯化即用于下一步。lcms(esi):[m h]
=919.7。
[0865]
步骤6.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
((3
‑
((3
‑
氨基丙基)氨基)丙氧基)甲基)
‑
19
‑
((1r,3s,5s)
‑
双环[3.2.1]辛烷
‑3‑
基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0866]
将步骤5的产物(159mg,0.173mmol)用tfa(10ml)在0℃
°
处理1h。加入甲苯(10ml),并将混合物真空浓缩。残余物通过制备型hplc纯化。柱:xselect csh prep c18 obd色谱柱,5um,19*150mm;流动相a:水(0.05%tfa),流动相b:meoh
‑‑
制备型;流速:25ml/min;梯度:37%b至67%b在20min内;254/220nm;rt:17.09min,以得到作为其tfa盐的标题化合物(65mg),为白色固体。lcms(esi):[m h]
=819.6,r
t
=2.64min,方法=i。1h nmr(400mhz,dmso
‑
d6):δ8.72
‑
8.42(m,3h),8.20
‑
8.02(m,5h),8.00
‑
7.82(m,3h),7.56
‑
7.44(m,1h),4.58
‑
4.45(m,1h),4.38
‑
4.11(m,2h),3.89
‑
3.65(m,4h),3.38
‑
3.12(m,4h),3.09
‑
2.72(m,11h),2.22
‑
1.99(m,4h),1.89
‑
1.52(m,15h),1.49
‑
1.21(m,16h),1.09
‑
0.87(12h)。
[0867]
实例25.(3r,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
((3
‑
(氨基甲基)氮杂环丁烷
‑1‑
基)甲基)
‑
12
‑
环己基
‑
19
‑
已基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0868][0869]
步骤1. 2
‑
(((苄氧基)羰基)氨基)
‑3‑
(3
‑
(((叔丁氧羰基)氨基)甲基)氮杂环丁烷
‑1‑
基)丙酸甲酯
[0870][0871]
在0℃向(2s)
‑2‑
(苄氧羰基氨基)
‑3‑
羟基
‑
丙酸甲酯(2.5191g,9.9471mmol)和三乙胺(3.0ml,21mmol)在dcm(20ml)中的混合物中加入甲磺酰氯(0.90ml,12mmol)。将该混合物在室温搅拌2h,然后加入(氮杂环丁烷
‑3‑
基甲基)氨基甲酸叔丁酯盐酸盐(2.5107g,10.935mmol)和三乙胺(4.0ml,28mmol)。将反应混合物在室温搅拌16h。反应混合物用饱和nahco3水溶液洗涤,水层用另外的100ml dcm萃取,合并的有机萃取物用mgso4干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(120g二氧化硅,溶剂梯度:0
‑
10%meoh在dcm中)。将所得粘稠胶真空干燥过夜以产生3.4092g(81%)标题化合物。lcms(esi):[m h]
=422.25;1h nmr(400mhz,dmso
‑
d6)δ7.53(d,j=7.8hz,1h),7.40
‑
7.25(m,5h),6.84(d,j=6.3hz,1h),5.03(s,2h),4.02
‑
3.93(m,1h),3.62(s,3h),3.17(q,j=7.7hz,2h),3.03(t,j=6.3hz,2h),2.80(q,j=6.7hz,2h),2.70
‑
2.55(m,2h),2.42
‑
2.33(m,1h),1.37(s,9h)。
[0872]
步骤2. 2
‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑3‑
(3
‑
(((叔丁氧羰基)氨基)甲基)氮杂环丁烷
‑1‑
基)丙酸
[0873][0874]
向2
‑
(((苄氧基)羰基)氨基)
‑3‑
(3
‑
(((叔丁氧羰基)氨基)甲基)氮杂环丁烷
‑1‑
基)丙酸甲酯(3.4092g,8.088mmol)在1,4
‑
二氧六环(20ml)中的溶液中加入氢氧化锂(1.0 m水溶液,10.0ml,10mmol)。将反应混合物在室温搅拌6h。将反应混合物倒入100ml dcm中并用10%柠檬酸水溶液洗涤。水性部分用另外的3x100ml dcm萃取,合并的有机萃取物用硫酸
镁干燥,过滤,真空蒸发,并真空干燥过夜。将所得物质溶解在乙醇(20ml)中。在氮气氛下,加入钯(碳载,10wt.%)(271.7mg,0.2554mmol)。然后将反应混合物在氢气球下在室温搅拌4h,然后通过硅藻土过滤,用200ml乙醇冲洗并真空蒸发。将所得物质溶解在具有碳酸氢钠(2.066g,24.5mmol)和fmoc
‑
osu(3.274g,9.706mmol)的水(15ml)和1,4
‑
二氧六环(15ml)中。将反应混合物在室温搅拌2h,然后用100ml dcm稀释并用10%柠檬酸水溶液洗涤。水性部分用另外的100ml dcm萃取,合并的有机萃取物用硫酸镁干燥,过滤并真空蒸发。粗产物通过硅胶快速色谱纯化(120g二氧化硅,溶剂梯度:0
‑
15%甲醇在dcm中),得到2.3477g(59%)标题化合物。lcms(esi):[m h]
=496.25。
[0875]
步骤3.(3r,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
((3
‑
(氨基甲基)氮杂环丁烷
‑1‑
基)甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0876]
使用2
‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑3‑
(3
‑
(((叔丁氧羰基)氨基)甲基)氮杂环丁烷
‑1‑
基)丙酸并且按照类似于那些针对实例11描述的过程,制备标题化合物(2种非对映异构体的混合物)。lcms(esi):r
t
(min)=8.55和8.70,[m h]
=735.6,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.80
‑
8.51(m,1h),8.38
‑
8.17(m,1h),8.17
‑
7.91(m,3h),7.91
‑
7.56(m,4h),4.73
‑
3.64(m,8h),3.02(d,j=104.9hz,10h),2.73(s,1h),2.09
‑
1.45(m,10h),1.45
‑
0.73(m,33h)。
[0877]
实例26.(2s)
‑
2,3
‑
二氨基
‑
n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环已基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]丙酰胺(三氟乙酸盐)
[0878][0879]
步骤1.(2s)
‑2‑
[[(2s)
‑2‑
[[(2s)
‑2‑
[[(2s)
‑2‑
[[(2r,3r)
‑3‑
[(2r)
‑2‑
氨基丙氧基]
‑2‑
甲基
‑
壬酰基]
‑
甲基
‑
氨基]
‑4‑
甲基
‑
戊酰基]氨基]
‑2‑
环己基
‑
乙酰基]氨基]
‑3‑
(苄氧羰基氨基)丙酰基]氨基]
‑3‑
(叔丁氧羰基氨基)丙酸
[0880][0881]
使用适当受保护氨基酸,按照类似于那些针对实例11步骤1所述的过程制备标题化合物。lcms(esi):[m h]
=918.5。
[0882]
步骤2.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环己基
‑
19
‑
已基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]氨基甲酸叔丁酯
[0883][0884]
将(2s)
‑2‑
[[(2s)
‑2‑
[[(2s)
‑2‑
[[(2s)
‑2‑
[[(2r,3r)
‑3‑
[(2r)
‑2‑
氨基丙氧基]
‑2‑
甲基
‑
壬酰基]
‑
甲基
‑
氨基]
‑4‑
甲基
‑
戊酰基]氨基]
‑2‑
环己基
‑
乙酰基]氨基]
‑3‑
(苄氧羰基氨基)丙酰基]氨基]
‑3‑
(叔丁氧羰基氨基)丙酸(1.2g,1.3mmol)溶解在1.2l的thf中,并且充入dipea(0.69ml,4mmol)。将hatu(1.1g,2.6mmol)溶解在6ml dmf中,并加入前述混合物中。将混合物在室温搅拌过夜并真空蒸发。然后将残余物用乙酸乙酯和水稀释,剧烈振摇并分配。然后将有机部分真空中浓缩,并且将残余物通过硅胶快速色谱纯化(125g二氧化硅,0
‑
10%meoh在dcm中),以得到标题化合物(650mg,0.72mmol,55%收率)。lcms(esi):[m h]
=900.5。
[0885]
步骤3.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸苄酯
[0886][0887]
将n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]氨基甲酸叔丁酯(600mg,0.66mmol)溶解在4ml的dcm中,并且充入1.6ml的三异丙基硅烷和0.8ml的tfa。然后将混合物在室温搅拌1h,用甲苯稀释并真空浓缩。然后用etoac稀释残余物,加入0.10ml tea,并用水稀释。然后剧烈振摇混合物并分配。将有机相真空浓缩,以得到标题化合物(540mg,0.66mmol,100%收率)。lcms(esi):[m h]
=800.5。
[0888]
步骤4.n
‑
[(1s)
‑2‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基氨基]
‑1‑
[(叔丁氧羰基氨基)甲基]
‑2‑
氧代
‑
乙基]氨基甲酸叔丁酯
[0889][0890]
将(2s)
‑
2,3
‑
双(叔丁氧羰基氨基)丙酸(190mg,0.63mmol)溶解在2ml dmf中并充入hatu(243mg,0.63mmol)和tea(174ul,1.25mmol)。然后将混合物在室温下搅拌5min并加入溶解在0.30ml dmf中的n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸苄酯(290mg,0.36mmol)中。然后将混合物在室温搅拌30min。然后向混合物中充入1ml饱和nahco3水溶液并在室温搅拌5min。然后用etoac和水稀释混合物,剧烈振摇并分配。然后将有机部分用水洗涤两次并真空浓缩,以得到标题化合物(400mg,0.36mmol,100%收率)。lcms(esi):[m h]
=1086.7。
[0891]
步骤5.(2s)
‑
2,3
‑
二氨基
‑
n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]丙酰胺(三氟乙酸盐)
[0892]
将n
‑
[(1s)
‑2‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基氨基]
‑1‑
[(叔丁氧羰基氨基)甲基]
‑2‑
氧代
‑
乙基]氨基甲酸叔丁酯(400mg,0.36mmol)溶解在40ml的1∶1异丙醇:etoac中,并通过hcube设备(10%pd/c,50℃,75psi)进行氢化。然后将混合物真空浓缩。
[0893]
然后将残余物溶解在4ml dcm中并充入1ml三异丙基硅烷和0.80ml tfa。然后将混合物在室温搅拌1h,真空浓缩,并与二氧六环共沸两次。然后将残余物通过反相hplc纯化,以得到标题化合物(88mg,0.11mmol,36%收率)。lcms(esi):rt(min)=9.11,[m h]
=753.8,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.67(s,1h),8.44(s,1h),8.35(s,1h),8.22(d,j=5.7hz,1h),8.11(s,1h),8.02(s,1h),7.96(d,j=4.9hz,1h),7.69(d,j=8.3hz,1h),7.54(d,j=8.1hz,1h),4.66(t,j=7.7hz,1h),4.25(s,1h),4.05(s,1h),3.85(s,2h),3.72(s,1h),3.20(s,1h),3.11(d,j=6.2hz,1h),2.98
‑
2.70(m,5h),1.87(s,2h),1.65
‑
1.58(m,14h),1.27(s,13h),1.11(s,1h),1.06
‑
0.81(m,17h)。
[0894]
实例27. 3
‑
(氨基甲基)
‑
n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]氮杂环丁烷
‑1‑
甲酰胺(三氟乙酸盐)
[0895][0896]
步骤1.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
[[[3
‑
[(叔丁氧羰基氨基)甲基]氮杂环丁烷
‑1‑
羰基]氨基]甲基]
‑
12
‑
环庚基
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸叔丁酯
[0897][0898]
将n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸酯(70.1mg,0.0.090mmol,实例14步骤2)和1,1
′‑
羰基二咪唑(29.7mg,0.18mmol)在thf(5ml)中的溶液在室温搅拌2h。然后加入(氮杂环丁烷
‑3‑
基甲基)氨基甲酸叔丁酯(34.4mg,0.18mmol)。将所得混合物在室温搅拌2h,然后真空浓缩。将残余物通过反相快速色谱在c18柱上纯化,用0
‑
53%乙腈/0.1%tfa水溶液洗脱,以得到标题化合物(65.4mg,73.1%收率),为白色固体。lcms(esi):[m h]
=978.8。
[0899]
步骤2. 3
‑
(氨基甲基)
‑
n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]氮杂环丁烷
‑1‑
甲酰胺
[0900]
在搅拌下,将步骤1的产物(93.2mg,0.10mmol)用tfa(3ml)在0℃处理30min。将混合物在真空下浓缩。将残余物通过制备型lcms纯化,以得到作为其tfa盐的标题化合物(28.4mg),为白色固体。lcms(esi):[m h]
=778.6,r
t
=2.7min,方法=i。1h nmr(400mhz,dmso
‑
d6):δ8.75
‑
8.55(m,1h),8.21
‑
7.75(m,8h),7.45
‑
7.26(m,1h),4.65
‑
4.17(m,3h),3.93
‑
3.85(m,5h),3.61
‑
3.44(m,4h),3.39
‑
3.35(m,3h),3.15
‑
3.03(m,4h),2.93
‑
2.82(m,3h),2.19
‑
1.98(m,2h),1.86
‑
1.65(m,8h),1.55
‑
1.25(m,18h),1.00
‑
0.88(m,12h)。
[0901]
实例28.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
己基
‑3‑
(1
‑
羟基
‑2‑
(哌嗪
‑1‑
基)乙基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0902][0903]
步骤1.4
‑
(2
‑
((2s,8s,11s,14s,17s)
‑
17
‑
((((苄氧基)羰基)氨基)甲基)
‑
14
‑
环己基
‑7‑
己基
‑
11
‑
异丁基
‑
8,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)
‑2‑
((叔丁基二甲基甲硅烷基)氧基)乙基)哌嗪
‑1‑
甲酸苄酯
[0904][0905]
使用中间体9和中间体t6并且按照类似于那些针对实例22描述的过程,获得标题化合物。
[0906]
步骤2.(3s,6s,9s,12s,15s)
‑6‑
(氨基甲基)
‑9‑
环己基
‑
16
‑
己基
‑3‑
(1
‑
羟基
‑2‑
(哌嗪
‑1‑
基)乙基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0907]
向溶解在thf(1ml)中的来自步骤1的物质(50mg)中加入四丁基氟化铵(1ml,在thf中的1n溶液),并将混合物搅拌过夜。反应混合物用ipac稀释,用水和盐水洗涤,以硫酸钠干燥并浓缩,获得粗制4
‑
(2
‑
((2s,8s,11s,14s,17s)
‑
17
‑
((((苄氧基)羰基)氨基)甲基)
‑
14
‑
环己基
‑7‑
己基
‑
11
‑
异丁基
‑
8,10
‑
二甲基
‑
3,9,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)
‑2‑
羟基乙基)哌嗪
‑1‑
甲酸苄酯。使用类似于那些在实例22(步骤6)中描述的过程将该物质转化为标题化合物,然后通过反相hplc纯化以获得作为三氟乙酸盐的标题化合物。lcms(esi):r
t
(min)=5.95;[m h]
=736.5,方法=b。
[0908]
实例29.(3s,6s,9s,12s,15s,17s)
‑6‑
(氨基甲基)
‑3‑
((3
‑
氨基丙氧基)甲基)
‑9‑
环已基
‑
16
‑
(环戊基甲基)
‑
13,15,17
‑
三甲基
‑
12
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮(三氟乙酸盐)
[0909][0910]
步骤1.n
‑
me
‑
正缬氨酸
‑
环已基甘氨酸
‑
dap(boc)
‑3‑
((叔丁氧羰基)氨基)丙基)
‑
丝氨酸
‑
(2
‑
氯三苯甲基树脂)
[0911][0912]
按照类似于那些在实例7步骤1描述的方法制备树脂结合的四肽。
[0913]
步骤2.(3s,6s,9s,12s,15s,17s)
‑6‑
(氨基甲基)
‑3‑
((3
‑
氨基丙氧基)甲基)
‑9‑
环己基
‑
16
‑
(环戊基甲基)
‑
13,15,17
‑
三甲基
‑
12
‑
丙基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14五酮
[0914]
使用来自步骤1的树脂结合的四肽和(2s)
‑2‑
[环戊基甲基
‑
[(1s)
‑2‑
(9h
‑
芴
‑9‑
基甲氧羰基氨基)
‑1‑
甲基
‑
乙基]氨基]丙酸(中间体t21),并且按照类似于那些在实例12步骤2
‑
4中描述的方法,制备作为其tfa盐的标题化合物。lcms(esi):[m h]
=693.5,r
t
=1.9min,方法=j。1h nmr(400mhz,dmso
‑
d6):δ8.92
‑
8.75(m,1h),8.18
‑
7.92(m,4h),7.81
‑
7.70(m,4h),4.85
‑
4.72(m,1h),4.58
‑
4.50(m,1h),4.24
‑
4.07(m,3h),3.95
‑
3.77(m,2h),3.28
‑
3.09(m,8h),2.91
‑
2.82(m,4h),2.14
‑
1.92(m,5h),1.80
‑
1.71(m,5h),1.67
‑
1.58(m,6h),1.57
‑
1.50(m,5h),1.44
‑
1.40(m,2h),1.29
‑
1.02(m,10h),0.99
‑
0.88(m,4h)。
[0915]
实例30.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[[3
‑
氨基丙基(甲基)氨基]甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0916][0917]
步骤1.n
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
[(叔丁氧羰基氨基)甲基]
‑
12
‑
环庚基
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基
‑
甲基
‑
氨基]丙基]氨基甲酸叔丁酯
[0918][0919]
向n
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
[(叔丁氧羰基氨基)甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基氨基]丙基]氨基甲酸叔丁酯(实例14,步骤3)(201mg,0.218mmol)在乙醇(5ml)中的溶液中加入多聚甲醛(71.7mg,2.39mmol)和乙酸(26.1mg,0.435mmol)。将反应混合物在室温搅拌2h,然后加入氰基硼氢化钠(54.7mg,0.87mmol)。将反应混合物在室温搅拌3天,然后真空浓缩。将残余物通过反相色谱在c18柱上纯化(0
‑
50%乙腈/0.1%tfa水溶液),以得到标题化合物(119mg),为白色固体。lcms(esi):[m h]
=937.8。
[0920]
步骤2.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[[3
‑
氨基丙基(甲基)氨基]甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0921]
将步骤1的产物(119mg,0.126mmol)用tfa(5.0ml)在0℃
°
处理30min,然后真空浓缩。将残余物通过制备型lcms纯化,以得到作为其tfa盐的标题化合物(9.90mg),为白色固体。lcms(esi):[m h]
=737.6,r
t
=2.5min,方法=i。1h nmr(400mhz,dmso
‑
d6):δ9.62
‑
9.45(m,1h),8.62
‑
8.35(m,1h),8.18
‑
8.12(m,1h),8.02
‑
7.92(m,2h),7.88
‑
7.74(m,3h),7.52
‑
7.41(m,1h),7.29
‑
6.95(m,1h),4.62
‑
4.55(m,1h),4.30
‑
4.22(m,1h),4.09
‑
4.02(m,1h),3.92
‑
3.80(m,2h),3.30
‑
3.08(m,10h),2.97
‑
2.74(m,8h),2.15
‑
2.07(m,2h),1.98
‑
1.88(m,4h),1.75
‑
1.59(m,7h),1.45
‑
1.24(m,17h),1.05
‑
0.85(m,11h)。
[0922]
实例31. 1
‑
((1s,3r)
‑3‑
(((3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)甲氧基)环丁基)胍(三氟乙酸盐)
[0923][0924]
步骤1.n
‑
[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]环丁基]氨基甲酸叔丁酯
[0925][0926]
按照类似于那些在实例8步骤1
‑
3描述的方法制备标题化合物。lcms(esi):[m h]
=994.6。
[0927]
步骤2.(((3r,6s,9s,12s,15s,18r,19r)
‑6‑
(((1r,3s)
‑3‑
氨基环丁氧基)甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基)甲基)氨基甲酸苄酯
[0928]
[0929]
将步骤1的产物(577mg,0.580mmol)用tfa(10ml)在0℃
°
处理1h,然后真空浓缩。将残余物与甲苯(2x10ml)共蒸发以移除残余的tfa。将粗产物不经纯化即作为tfa盐直接用于下一步。lcms(esi):[m h]
=894.6。
[0930]
步骤3.(nz)
‑
n
‑
[[[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(苄氧羰基氨基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]环丁基]氨基]
‑
(叔丁氧羰基氨基)亚甲基]氨基甲酸叔丁酯
[0931][0932]
在室温向来自步骤2的粗产物(519mg)在乙醇(60ml)中的溶液中加入(z)(((叔丁氧羰基)氨基)(1h
‑
吡唑
‑1‑
基)亚甲基)氨基甲酸叔丁酯(360mg,1.16mmol)和dipea(0.40ml,2.3mmol)。将反应混合物在室温搅拌4h并真空浓缩。将残余物溶解在dcm(150ml)中并用10%氯化铵水溶液(2x150ml)洗涤。有机层经无水硫酸钠干燥并真空浓缩。将残余物通过反相色谱在c18柱上纯化(0
‑
100%乙腈/0.05%tfa水溶液),以得到标题化合物(281mg),为无色固体。lcms(esi):[m h]
=1136.7。
[0933]
步骤4.(nz)
‑
n
‑
[[[3
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲氧基]环丁基]氨基]
‑
(叔丁氧羰基氨基)亚甲基]氨基甲酸叔丁酯
[0934][0935]
将来自步骤3的产物(281mg,0.247mmol)与pd/c(280mg,碳上负载10%pd)在2
‑
丙醇(35ml)中合并,并在氢气氛下在室温搅拌6h。经由过滤移除催化剂,并将滤液减压浓缩,以得到标题化合物(220mg,88.8%收率),为无色固体。lcms(esi):[m h]
=1002.8。
[0936]
步骤5. 1
‑
((1s,3r)
‑3‑
(((3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑
19
‑
(螺[3.3]庚烷
‑2‑
基甲基)
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基)甲氧基)环丁基)胍
[0937]
将来自步骤4的产物(220mg,0.220mmol)用tfa(4.0ml)在0℃
°
处理4h,然后真空浓缩。将残余物与甲苯(2x10ml)共蒸发。粗产物通过制备型hplc纯化,以得到作为其tfa盐的标题化合物(45.3mg),白色固体。lcms(esi):[m h]
=802.6,r
t
=2.72min,方法=i。1h nmr(400mhz,dmso
‑
d6):δ8.56
‑
8.31(m,1h),8.18
‑
8.12(m,2h),8.08
‑
7.99(m,4h),7.92
‑
7.85(m,1h),7.34
‑
7.06(m,4h),4.52
‑
4.35(m,2h),4.20
‑
4.08(m,2h),4.02
‑
3.92(m,2h),3.88
‑
3.80(m,1h),3.68
‑
3.60(m,1h),3.52
‑
3.42(m,1h),3.41
‑
3.34(m,1h),3.32
‑
3.12(m,6h),3.15(s,1h),2.73(s,2h),2.31
‑
2.23(m,3h),2.16
‑
1.98(m,8h),1.88
‑
1.81(m,2h),1.78
‑
1.72(m,3h),1.69
‑
1.55(m,8h),1.53
‑
1.45(m,6h),1.32
‑
1.21(m,3h),0.99
‑
0.89(m,6h),0.88
‑
0.83(m,3h)。
[0938]
实例32.(3s,6s,9s,12s,15s,18s)
‑6‑
(氨基甲基)
‑
16
‑
丁基
‑9‑
环己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
18
‑
(羟基甲基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0939][0940]
步骤1:n
‑
(叔丁氧羰基)
‑
o
‑
(叔丁基二苯基甲硅烷基)
‑
d
‑
丝氨酸甲酯
[0941][0942]
向(2r)
‑2‑
氨基
‑3‑
羟基
‑
丙酸甲酯盐酸盐(2.8g,18mmol)在thf/水混合物中的悬浮液中加入碳酸二叔丁酯(3.8g,22mmol)和碳酸氢钠(1.5g,18mmol),并将混合物搅拌过夜。将反应混合物用ipac稀释,用盐水洗涤,经硫酸钠干燥并浓缩。将残余物溶解在dcm(30ml)中并加入叔丁基氯二苯基硅烷(5.9g,22mmol)和咪唑(2.5g,36mmol),并将混合物搅拌过夜。将反应混合物用ipac稀释,用水和盐水洗涤,经硫酸钠干燥并浓缩。将残余物通过色谱纯化(硅胶,0
‑
50%ipac/庚烷),得到标题化合物(8.1g,98%)。1h nmr(400mhz,氯仿
‑
d)δ7.61
‑
7.53(m,3h),7.48
‑
7.34(m,7h),5.41(d,j=8.8hz,1h),4.50
‑
4.30(m,1h),4.06(dd,j=10.1,3.0hz,1h),3.89(dd,j=10.1,3.1hz,1h),3.74(s,3h),1.46(s,9h),1.03(s,9h)。
[0943]
步骤2:n
‑
((s)
‑2‑
((叔丁氧羰基)氨基)
‑3‑
((叔丁基二苯基甲硅烷基)氧基)丙基)
‑
n
‑
己基
‑
l
‑
丙氨酸苄酯
[0944][0945]
将来自步骤1的残余物溶解在干燥thf(30ml)中并在冰浴中冷却。加入氢化铝锂(8.85ml,在thf中的2m溶液),并历时2h使反应混合物回温至室温。用饱和氯化铵水溶液猝灭反应混合物,通过硅藻土过滤移除固体。滤液用硫酸钠干燥并浓缩,以得到粗制(s)
‑
(1
‑
((叔丁基二苯基甲硅烷基)氧基)
‑3‑
羟基丙烷
‑2‑
基)氨基甲酸叔丁酯。
[0946]
如实例20的步骤4中所述,将该物质转化为醛,获得n
‑
[(1r)
‑1‑
[[叔丁基(二苯基)甲硅烷基]氧基甲基]
‑2‑
氧代
‑
乙基]氨基甲酸叔丁酯(3.5g,54%)。1h nmr(400mhz,氯仿
‑
d)δ9.66(s,1h),7.69
‑
7.56(m,4h),7.51
‑
7.35(m,6h),5.40(d,j=7.6hz,1h),4.42
‑
3.88(m,3h),1.45(d,j=9.5hz,9h),1.03(s,9h)。
[0947]
按照实例20的步骤5中所述,将该物质转化为标题化合物(1.75g,55%)。1h nmr(400mhz,chlorofom
‑
d)δ7.70
‑
7.52(m,4h),7.48
‑
7.30(m,11h),5.10(s,2h),4.73(s,1h),3.79
‑
3.50(m,4h),2.88
‑
2.67(m,2h),2.63
‑
2.40(m,2h),1.43(s,9h),1.38
‑
1.18(m,11h),1.05(s,9h),0.94
‑
0.81(m,3h).
[0948]
步骤3.n
‑
((s)
‑2‑
((((9h
‑
芴
‑9‑
基)甲氧基)羰基)氨基)
‑3‑
((叔丁基二苯基甲硅烷基)氧基)丙基)
‑
n
‑
己基
‑
l
‑
丙氨酸
[0949][0950]
如实例20的步骤6和7中所述,将n
‑
((s)
‑2‑
((叔丁氧羰基)氨基)
‑3‑
((叔丁基二苯基甲硅烷基)氧基)丙基)
‑
n
‑
己基
‑
l
‑
丙氨酸苄酯(1.75g)转化为标题化合物(1.0g,54%)。1h nmr(400mhz,氯仿
‑
d)δ7.74(d,j=7.6hz,2h),7.60(dd,j=18.3,7.4hz,6h),7.46
‑
7.34(m,8h),7.32
‑
7.19(m,2h),5.98(d,j=7.4hz,1h),4.33(d,j=8.5hz,2h),4.18(t,j=7.2hz,1h),3.97
‑
3.59(m,4h),3.07
‑
2.93(m,2h),2.73(d,j=56.2hz,2h),1.51(s,2h),1.42
‑
1.35(m,3h),1.22(d,j=11.7hz,6h),1.08(s,9h),0.88
‑
0.78(m,3h)。
[0951]
步骤4.(((2s,5s,8s,11s,14s,17s)
‑5‑
((s)
‑1‑
(苄氧基)乙基)
‑
17
‑
环己基
‑8‑
((((2,2
‑
二甲基
‑
1,1
‑
二苯基丙基)二甲基甲硅烷基)氧基)甲基)
‑
10
‑
己基
‑
14
‑
异丁基
‑
11,13
‑
二甲基
‑
3,6,12,15,18
‑
五氧代
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑2‑
基)甲基)氨基甲酸苄酯
[0952][0953]
使用来自步骤2的中间体并且按照类似于那些在实例20的步骤8和9中描述的过程,制备标题化合物(0.30g)。lcms(esi):[m h]
=1145.8。
[0954]
步骤5.(3s,6s,9s,12s,15s,18s)
‑6‑
(氨基甲基)
‑
16
‑
丁基
‑9‑
环己基
‑3‑
((s)
‑1‑
羟基乙基)
‑
18
‑
(羟基甲基)
‑
12
‑
异丁基
‑
13,15
‑
二甲基
‑
1,4,7,10,13,16
‑
六氮杂环十八烷
‑
2,5,8,11,14
‑
五酮
[0955]
使用来自步骤4的中间体并且按照类似于那些在实例28步骤2中描述的过程,将标题化合物制备为其tfa盐。lcms(esi):r
t
(min)=7.807,[m h]
=682.5,方法b(纯度:82%)。
[0956]
实例33.n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基]甲磺酰胺(三氟乙酸盐)
[0957][0958]
将n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环己基
‑
19
‑
己基
‑
15
‑
异丁基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸叔丁酯(实例26,步骤3)(60mg,0.68mmol)溶解在0.20ml的dcm中,并且充入dipea(26ul,0.15mmol)和甲磺酰氯(6ul,0.075mmol)。然后将混合物在室温搅拌一小时。然后将混合物用etoac和水稀释,剧烈振摇并分配。然后将有机部分用水洗涤一次并真空浓缩。
[0959]
然后将残余物溶解在5ml 1∶1异丙醇:etoac中并充入0.1ml tfa。然后使用hcube设备(10%pd/c,50℃,75psi)将混合物氢化。然后将混合物真空浓缩并通过反相hplc纯化,得到标题化合物(21mg,0.028mmol,41%收率)。lcms(esi):r
t
(min)=12.17,[m h]
=744.5,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.33
‑
8.07(m,2h),7.86(d,j=7.1hz,3h),7.46(t,j=8.8hz,1h),7.14
‑
7.00(m,1h),6.51(s,1h),4.61(dd,j=10.3,5.1hz,1h),4.19(d,j=8.1hz,1h),3.99(s,1h),3.93(s,1h),3.83(s,1h),3.73(s,1h),3.10(s,1h),2.90(dd,j=10.0,8.1hz,4h),2.81(d,j=6.9hz,1h),2.07(s,1h),1.75(d,j=12.9hz,1h),1.65(s,8h),1.53(s,1h),1.26(s,15h),1.10(d,j=12.2hz,1h),1.09
‑
0.93(m,4h),0.97
‑
0.81(m,13h)。
[0960]
实例34.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑6‑
(((1,5
‑
二氨基戊烷
‑3‑
基)氧基)甲基)
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0961][0962]
步骤1. 3
‑
(苄氧基)戊二酸二甲酯
[0963][0964]
向圆底烧瓶中加入3
‑
羟基戊二酸二甲酯(10ml,67.7mmol)、三甲基氯硅烷(12.9ml,135mmol)和dcm(135ml)。将反应烧瓶在冰水浴中冷却至0℃。一旦冷却后,就分两部分加入咪唑(9.2g,135mmol)。10min后,移除冷却浴并将反应在室温搅拌过夜。将反应过滤移除过量的咪唑,用dcm冲洗,并浓缩至~20%体积。加入乙醚,并将有机层用水洗涤两次。有机层经硫酸镁干燥,过滤并浓缩。将粗制残余物吸收在乙腈(150ml)中,并用三乙基硅烷(7.2ml,45.0mmol)和氯化铁(1.2g,7.5mmol)处理。将反应在室温搅拌25min,然后加入苯甲醛(4.6ml,45.0mol)并搅拌过夜。第二天将反应浓缩,用水处理,用dcm萃取(三次)。将合并的有机层用盐水洗涤,经硫酸镁干燥,过滤并浓缩。将残余物通过硅胶快速色谱纯化(0
‑
100%乙酸乙酯/庚烷),得到标题化合物(6.71g,67%收率),为透明无色油状物。1h nmr(400mhz,dmso
‑
d6)δ7.33(m,2h),7.26(m,3h),4.5(s,2h),4.18(m,1h),3.60(s,6h),2.65(d,j=6.3hz,4h)。
[0965]
步骤2. 3
‑
(苄氧基)戊烷
‑
1,5
‑
二醇
[0966][0967]
向圆底烧瓶中加入3
‑
苄氧基戊二酸二甲酯(1.05g,19.7mmol)和thf(8ml)。将溶液冷却至0℃,然后历时10min将氢化铝锂(在乙醚中的2m溶液)(9.8ml,19.7mmol)逐步加入反应中。在0℃搅拌20min后,使反应回温至室温并搅拌3h。将反应冷却至0℃,并且用0.75ml水、然后用0.75ml naoh(15%w/w水溶液)、最后用2.24ml水猝灭反应。10min后,将烧瓶回温至室温,用乙酸乙酯稀释,再搅拌45min。过滤移除所得悬浮液,滤液用水稀释,并且用乙酸乙酯萃取三次。合并的有机层经硫酸镁干燥,过滤并浓缩。将残余物通过硅胶快速色谱纯化(0
‑
15%甲醇/dcm),以得到标题化合物(0.702g,85%收率),为透明无色油状物。1h nmr(400mhz,dmso
‑
d6)δ7.38
‑
7.22(m,5h),4.46(s,2h),4.37(t,j=5.1hz,2h),3.64(tt,j=6.9,5.3hz,1h),3.49(td,j=6.7,5.1hz,4h),1.74
‑
1.56(m,4h)。
[0968]
步骤3.(((1,5
‑
二叠氮基戊烷
‑3‑
基)氧基)甲基)苯
[0969][0970]
向圆底烧瓶中加入3
‑
苄氧基戊烷
‑
1,5
‑
二醇(0.49g,2.33mmol)和dcm(11.6ml)。将溶液冷却至0℃,然后用三乙胺(0.97ml,6.99mmol)和甲磺酸酐(1.05g,5.82mmol)处理。在0
℃搅拌5min后,使反应烧瓶回温至室温。4h后,真空蒸发反应。将粗制残余物吸收在dmf(10.5ml)中并用叠氮化钠(0.76g,11.6mmol)处理,然后加热至50℃过夜。第二天,将反应用水猝灭并用dcm萃取三次。合并的有机层用硫酸镁干燥并减压浓缩,得到0.588g粗制物质,将其原样用于下一步。1h nmr(400mhz,dmso
‑
d6)δ7.40
‑
7.24(m,5h),4.48(s,2h),3.59(quin,j=5.9hz,1h),3.48
‑
3.33(m,4h),1.78(td,j=7.0,6.0hz,4h)。
[0971]
步骤4.(3
‑
羟基戊烷
‑
1,5
‑
二基)二氨基甲酸二叔丁酯
[0972][0973]
将来自步骤3的粗制残余物与乙醇(111ml)和二碳酸二叔丁酯(1.5g,6.68mmol)合并,并用鼓泡氮气吹扫5min。加入钯(碳载,10wt%)(0.950g,0.891mmol)并将反应烧瓶吹扫空气,用氢气回填(重复三次),然后在氢气球下搅拌过夜。通过硅藻土垫过滤反应物,用乙醇洗涤。收集滤液并浓缩。将残余物通过硅胶快速色谱纯化(0
‑
15%甲醇/dcm),得到标题化合物(0.502g,70%收率),为白色固体。lcms(esi):[m h]
=319;1h nmr(400mhz,dmso
‑
d6)δ6.67(t,j=5.7hz,2h),4.42(d,j=5.4hz,1h),3.49
‑
3.39(m,1h),3.06
‑
2.88(m,4h),1.59
‑
1.27(m,22h)。
[0974]
步骤5.n
‑
((苄氧基)羰基)
‑
o
‑
(2,2,14,14
‑
四甲基
‑
4,12
‑
二氧代
‑
3,13
‑
二氧杂
‑
5,11
‑
二氮杂十五烷
‑8‑
基)丝氨酸甲酯
[0975][0976]
向烘干的圆底烧瓶中加入(3
‑
羟基戊烷
‑
1,5
‑
二基)二氨基甲酸二叔丁酯(0.55g,1.73mmol)、氮杂环丙烷
‑
1,2
‑
二甲酸1
‑
苄酯2
‑
甲酯(0.37g,1.57mmol)、氯仿(4ml),然后冷却至0℃。逐步加入三氟化硼醚合物(0.019ml,0.157mmol),产生黄色溶液。在0℃搅拌30min后,使反应回温至室温并搅拌过夜。加入甲醇(~5ml)并将反应浓缩。将残余物通过硅胶快速色谱纯化(0
‑
100%乙酸乙酯/庚烷),得到标题化合物(0.30g,35%收率)。lcms(esi):[m h]
=554.2;1h nmr(400mhz,dmso
‑
d6)δ7.42
‑
7.28(m,5h),6.67(t,j=5.7hz,2h),5.11
‑
5.00(m,2h),4.43(d,j=5.4hz,1h),3.68
‑
3.57(m,4h),2.97(tt,j=13.8,6.0hz,5h),1.53
‑
1.42(m,4h),1.37(s,20h)。
[0977]
步骤6.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2,2,14,14
‑
四甲基
‑
4,12
‑
二氧代
‑
3,13
‑
二氧杂
‑
5,11
‑
二氮杂十五烷
‑8‑
基)丝氨酸
[0978][0979]
向小瓶中加入2
‑
(苄氧羰基氨基)
‑3‑
[3
‑
(叔丁氧羰基氨基)
‑1‑
[2
‑
(叔丁氧羰基氨基)乙基]丙氧基]丙酸甲酯(0.50g,0.90mmol)、thf(1.8ml)和氢氧化锂(1m水溶液)(1.3ml,1.3mmol)。将小瓶用氮气吹扫,盖上盖子,然后放在振荡器上1h。然后将反应浓缩,用dcm稀释。有机层用1m hcl水溶液洗涤。有机层经硫酸镁干燥,过滤并浓缩。将粗制残余物吸收在1,4
‑
二氧六环(2.1ml)和水(0.1ml)中,并用碳酸氢钠(0.184g,2.19mmol)处理。将混合物冷却至0℃,然后加入n
‑
琥珀酰亚胺碳酸9
‑
芴基甲酯(0.40g,1.20mmol)。将反应回温至室温并搅拌过夜,然后将反应过滤并浓缩。将残余物通过硅胶快速色谱纯化(0
‑
100%乙酸乙酯/庚烷),得到标题化合物(0.268g,39%收率),为白色固体。lcms(esi):[m h]
=628.3;1h nmr(400mhz,dmso
‑
d6)δ12.75(s,1h),7.89(d,j=7.5hz,2h),7.73(d,j=7.3hz,2h),7.41(t,j=7.5hz,2h),7.32(t,j=7.4hz,2h),6.74(s,2h),4.38
‑
4.05(m,4h),3.78
‑
3.52(m,1h),3.07
‑
2.84(m,5h),2.62
‑
2.57(m,2h),1.56
‑
1.51(m,4h),1.40
‑
1.32(m,18h)。
[0980]
步骤7.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑6‑
(((1,5
‑
二氨基戊烷
‑3‑
基)氧基)甲基)
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0981]
使用n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2,2,14,14
‑
四甲基
‑
4,12
‑
二氧代
‑
3,13
‑
二氧杂
‑
5,11
‑
二氮杂十五烷
‑8‑
基)丝氨酸苄酯并且按照类似于那些针对实例11所述的过程,将标题化合物制备为三氟乙酸盐。lcms(esi):r
t
(min)=8.53,[m h]
=768.6,方法=b。
[0982]
实例35.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑6‑
(((1,3
‑
二氨基丙烷
‑2‑
基)氧基)甲基)
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0983][0984]
步骤1.n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2,2,12,12
‑
四甲基
‑
4,10
‑
二氧代
‑
3,
11
‑
二氧杂
‑
5,9
‑
二氮杂十三烷
‑7‑
基)丝氨酸
[0985][0986]
使用(2
‑
羟基丙烷
‑
1,3
‑
二基)二氨基甲酸二叔丁酯并且按照类似于那些针对实例34步骤5
‑
6所述的过程,制备标题化合物。lcms(esi):[m h]
=600.2;1h nmr(400mhz,dmso
‑
d6)δ12.76(s,1h),7.89(d,j=7.6hz,2h),7.73(d,j=7.5hz,2h),7.42(t,j=7.5hz,2h),7.33(t,j=7.5hz,2h),6.80(s,1h),6.66(s,1h),6.59(s,1h),4.28(dt,j=16.6,7.4hz,3h),4.16(s,1h),3.78(t,j=7.9hz,1h),3.72
‑
3.60(m,1h),2.94(dd,j=19.4,6.7hz,4h),1.37(s,18h)。
[0987]
步骤2:(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑
12
‑
环庚基
‑6‑
(((1,3
‑
二氨基丙烷
‑2‑
基)氧基)甲基)
‑
19
‑
已基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0988]
使用n
‑
(((9h
‑
芴
‑9‑
基)甲氧基)羰基)
‑
o
‑
(2,2,12,12
‑
四甲基
‑
4,10
‑
二氧代
‑
3,11
‑
二氧杂
‑
5,9
‑
二氮杂十三烷
‑7‑
基)丝氨酸并且按照类似于那些针对实例11所述的过程,将标题化合物制备为三氟乙酸盐。lcms(esi):r
t
(min)=8.53,[m h]
=739.6,方法=b;1h nmr(400mhz,dmso
‑
d6)δ8.77
‑
8.07(m,2h),7.96(dd,j=30.6,17.0hz,9h),7.67(dd,j=14.1,8.2hz,1h),4.68
‑
4.37(m,2h),4.31
‑
4.06(m,1h),3.92(s,1h),3.83(d,j=10.6hz,3h),3.34(t,j=11.1hz,3h),3.16(d,j=16.2hz,7h),2.96(d,j=6.5hz,2h),2.74(s,1h),2.17
‑
1.95(m,2h),1.91
‑
1.24(m,24h),1.12
‑
0.69(m,12h)。
[0989]
实例36.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[[[6
‑
(氨基甲基)嘧啶
‑4‑
基]氨基]甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮(三氟乙酸盐)
[0990][0991]
步骤1.n
‑
[[6
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑9‑
[(叔丁氧羰基氨基)甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑6‑
基]甲基氨基]嘧啶
‑4‑
基]甲基]氨基甲酸叔丁酯
[0992][0993]
向n
‑
[[(3r,6s,9s,12s,15s,18r,19r)
‑6‑
(氨基甲基)
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
5,8,11,14,17
‑
五氧代
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑9‑
基]甲基]氨基甲酸酯(200mg,0.26mmol,实例14步骤2)在dmf(2.5ml)中的溶液中加入n
‑
[(6
‑
氯嘧啶
‑4‑
基)甲基]氨基甲酸叔丁酯(95.6mg,0.39mmol)和dipea(0.09ml,0.52mmol)。将混合物在氮气下在100℃
°
搅拌过夜,然后冷却至室温。将反应混合物直接加载到c18柱上并通过反相色谱纯化(0
‑
100%乙腈/0.01%tfa水溶液),以得到标题化合物(211mg,83%收率),为固体。lcms(esi):[m h]
973.7。
[0994]
步骤2.(3r,6s,9s,12s,15s,18r,19r)
‑9‑
(氨基甲基)
‑6‑
[[[6
‑
(氨基甲基)嘧啶
‑4‑
基]氨基]甲基]
‑
12
‑
环庚基
‑
19
‑
己基
‑
3,16,18
‑
三甲基
‑
15
‑
丙基
‑1‑
氧杂
‑
4,7,10,13,16
‑
五氮杂环十九烷
‑
5,8,11,14,17
‑
五酮
[0995]
在搅拌下将来自步骤1的产物(210mg,0.22mmol)用tfa(3.0ml)在0℃
°
处理1h。加入甲苯(10ml)并将混合物真空浓缩。将残余物通过prep
‑
hplc纯化,以得到作为其tfa盐的标题化合物(56.5mg),为黄色固体。lcms(esi):[m h]
=773.7,r
t
=2.57min,方法=i。1h nmr(400mhz,dmso
‑
d6):δ8.94
‑
8.56(m,3h),8.52
‑
8.31(m,3h),8.30
‑
8.02(m,5h),7.52
‑
7.36(m,1h),6.58
‑
6.48(m,1h),4.62
‑
4.58(m,2h),4.43
‑
4.12(m,2h),4.05
‑
3.99(m,2h),3.86
‑
3.81(m,3h),3.50
‑
3.25(m,4h),3.15
‑
2.84(m,6h),2.15
‑
2.05(m,2h),1.89
‑
1.80(m,1h),1.74
‑
1.66(m,6h),1.62
‑
1.25(m,17h),1.01
‑
0.83(m,12h)。
[0996]
按照类似于那些在上述实例中描述的过程,利用所指示的合成方法,替换合适的fmoc保护的氨基酸,并从市售材料和/或中间体1
‑
15、中间体p2c
‑
p2k和中间体t1
‑
t24(结构见表1)中选择,将表4中所列的实例各自制备为三氟乙酸盐。
[0997]
表4
[0998]
[0999]
[1000]
[1001][1002]
生物测定
[1003]
实例b1:最小抑制浓度的确定
[1004]
每种化合物的体外抗微生物活性通过使用经临床和实验室标准研究所(clsi)批准的肉汤微量稀释技术测量最小抑制剂浓度(mic)来确定(methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically;approved standard
‑
第八版。clsi文件m07
‑
a8。wayne,pa:clinical and laboratroy standards;2009)。抗菌活性是针对大肠杆菌(escherichia coli(e..coli))菌株cft073 atcc 700928进行测量的,该菌株是临床相关的革兰氏阴性菌株。将细胞接种到mueller hinton琼脂平板上并在37℃生长16
‑
18小时。通过将细胞刮入1ml测试培养基(mueller hinton ii阳离子调节肉汤,补充有0.002%v/v tween
‑
80)并稀释至最终od
600nm
为0.01,制备接种物悬浮液。
[1005]
测试化合物在dmso中以100um的浓度制备。化合物在几种不同的稀释形式下进行测试,数据报告在表6、7和8中。在方案1中,将化合物原液以64μg/ml的浓度稀释到测试培养基中,并在相同的培养基中连续2倍稀释在96孔u底微量滴定盘中,总共10个化合物浓度。在方案2中,将化合物原液以4μg/ml的浓度稀释到测试培养基中,并在相同的培养基中连续2倍稀释在96孔u底微量滴定盘中,总共10个化合物浓度。在方案3中,将化合物原液以0.5μg/ml的浓度稀释到测试培养基中,并按上述方法进行连续2倍稀释。在方案4中,将化合物原液以0.13μg/ml的浓度稀释到测试培养基中,并按上述方法进行连续2倍稀释。将接种物悬浮液加入测试化合物的2倍系列稀释液中,使od od
600nm
的最终密度为0.0005,并在35℃孵育22小时。培养后,肉眼检查平板,将完全阻止细菌生长的测试化合物的最低浓度记录为mic。结果如表5中所列。
[1006]
表5
[1007]
[1008][1009]
药物组合物
[1010]
实例c1:肠胃外组合物
[1011]
为了制备适合通过注射施用的肠胃外药物组合物,将100mg本文公开的化合物溶解在dmso中,然后与10ml的0.9%无菌盐水混合。将混合物掺入适合注射施用的剂量单位形式中。
[1012]
在另一实施例中,将以下成分混合以形成可注射制剂:
[1013][1014]
将除水以外的所有上述成分混合并搅拌,并且,如果需要,可以稍微加热。然后加入足量的水。
[1015]
实例c2:口服组合物
[1016]
为了制备用于口服递送的药物组合物,将100mg本文公开的化合物与750mg淀粉混合。将混合物掺入适合口服施用的口服剂量单位,诸如硬明胶胶囊。
[1017]
在另一实施例中,将以下成分紧密混合并压制成单个刻痕片剂。
[1018][1019]
在又一实施例中,将以下成分紧密混合并装入硬壳明胶胶囊中。
[1020][1021]
在又一实施例中,将以下成分混合以形成用于口服施用的溶液/悬浮液:
[1022][1023]
实例c3:外用凝胶组合物
[1024]
为了制备药物局部凝胶组合物,将100mg本文公开的化合物与1.75g羟丙基纤维素、10ml丙二醇、10ml肉豆蔻酸异丙酯和100ml纯化醇usp混合。然后将所得凝胶混合物加入适合局部施用的容器诸如管中。
[1025]
虽然本文已经示出和描述了本公开的优选实施例,但是对于本领域技术人员来说,这些实施例仅作为示例提供是显而易见的。在不脱离本公开的情况下,本领域技术人员现在将想到许多变更、变化和替换。应当理解,在实践本发明时可以采用本文描述的实施例的各种替代方案。以下权利要求旨在限定本公开的范围,并且由此涵盖在这些权利要求及其等同物的范围内的方法和结构。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。