1.本发明属于医药技术领域,涉及一种核苷类化合物的晶型及其制备方法。
背景技术:
2.2019新型冠状病毒(严重急性呼吸综合征冠状病毒2,sars
‑
cov
‑
2)是一种具有包膜的单链rna 病毒,为β属冠状病毒。与sars和mers类似,sars
‑
cov
‑
2基因组编码非结构蛋白:3c样蛋白酶(3
‑
chymotrypsin
‑
like protease,3clpro)、木瓜蛋白酶样蛋白酶(papain
‑
likeprotease,plpro)、解旋酶(helicase)和rna依赖rna聚合酶(rna
‑
dependent rna polymerase,rdrp);结构蛋白:如棘突糖蛋白(spike glycoprotein)和附属蛋白(accessory proteins)。新冠病毒的表面棘突糖蛋白与人体细胞表面血管紧张素转换酶(ace2)受体的结合从而感染人的呼吸道上皮细胞。病毒在进入宿主细胞后解体,将核衣壳和病毒rna释放到细胞质中,病毒rna 5
ʹ
末端开放阅读框(orf1a/b)将编码多聚蛋白质(pp1a和pp1ab),它们对病毒复制所需酶的加工、成熟起重要作用。pp1a和pp1ab可被木瓜蛋白酶样蛋白酶(plpro)和3c样蛋白酶(3clpro)裂解,产生非结构蛋白,包括rna依赖性rna聚合酶和解螺旋酶等,它们对于新冠病毒的转录和复制的起着关键的作用。目前,冠状病毒识别受体的表面棘突糖蛋白、参与复制及转录过程的重要蛋白3clpro、plpro与rdrp是四个十分具有抗病毒药物研发吸引力的靶点。
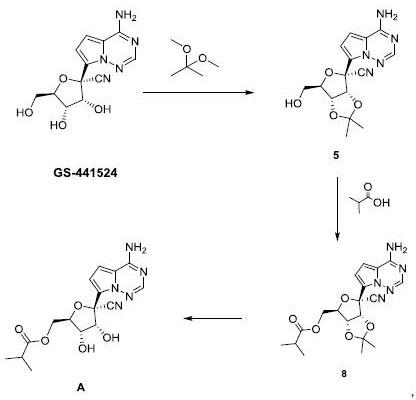
3.式a所示化合物是作用于rdrp的具有良好口服生物利用度和良好体内外抗sars
‑
cov
‑
2活性的核苷类似物,申请人已经申请了描述该化合物治疗病毒感染的应用专利和合成工艺专利(专利申请号:cn202011000517.2;专利申请号:cn202110562244.9)药物多晶型是药品研发中的常见现象,是影响药品质量的重要因素。同一药物的不同晶型在外观、溶解度、熔点、溶出度、生物有效性等方面可能会有显著不同,也会对药物的稳定性、生物利用度及疗效产生不同的影响。因此,在药品研发中,应全面考虑药品的多晶型问题。
4.发明人对式a所示化合物的晶型进行了相关研究,发现了制备简单、稳定性好和溶解度高的晶型。
技术实现要素:
5.本发明主要涉及式a所示化合物的晶型。本发明提供的部分晶型具有制备简单、制
备重现性高、物理和化学稳定性好、有利于实施应用的特性。
6.另一方面,本发明还提供了新晶型的制备方法、组合物和用途等。
7.第一方面,本发明提供一种式a所示化合物的晶型。
8.一种式a所示化合物的晶型,,包括:晶型i或晶型。
9.所述晶型i为无水物。
10.所述晶型i的x射线粉末衍射图谱在衍射角2θ为9.72
±
0.2
°
、10.13
±
0.2
°
、15.99
±
0.2
°
、19.05
±
0.2
°
和19.70
±
0.2
°
处有特征峰。在一些实施例中,所述晶型的x射线粉末衍射图谱在衍射角2θ为9.72
±
0.2
°
、10.13
±
0.2
°
、10.37
±
0.2
°
、13.12
±
0.2
°
、13.31
±
0.2
°
、15.99
±
0.2
°
、17.47
±
0.2
°
、19.05
±
0.2
°
、19.70
±
0.2
°
、20.69
±
0.2
°
、24.81
±
0.2
°
、25.32
±
0.2
°
和26.90
±
0.2
°
处有特征峰。在一些实施例中,所述晶型的x射线粉末衍射图谱在衍射角2θ为9.72
±
0.2
°
、10.13
±
0.2
°
、10.37
±
0.2
°
、12.29
±
0.2
°
、13.12
±
0.2
°
、13.31
±
0.2
°
、13.60
±
0.2
°
、15.99
±
0.2
°
、17.47
±
0.2
°
、17.77
±
0.2
°
、19.05
±
0.2
°
、19.70
±
0.2
°
、20.69
±
0.2
°
、21.63
±
0.2
°
、22.73
±
0.2
°
、24.81
±
0.2
°
、25.32
±
0.2
°
、26.90
±
0.2
°
、27.23
±
0.2
°
、28.78
±
0.2
°
、31.01
±
0.2
°
、31.30
±
0.2
°
、32.70
±
0.2
°
、33.97
±
0.2
°
和37.60
±
0.2
°
处有特征峰。在一些实施例中,所述晶型i的x射线粉末衍射图谱基本如图2所示。
11.所述晶型i的差示扫描量热曲线中在165℃
ꢀ‑
195℃处具有吸热峰。在一些实施例中,所述晶型i的差示扫描量热曲线基本如图3所示。
12.所述晶型i的热重分析曲线中在50℃
‑
150℃的温度范围内有0.2%以下的失重。在一些实施例中,所述晶型i的热重分析曲线中在50℃
‑
150℃的温度范围内有0.1%以下的失重。在一些实施例中,所述晶型i的热重分析曲线图谱基本如图3所示。
13.所述晶型为半水合物。
14.晶型的x射线粉末衍射图谱在衍射角2θ为5.85
±
0.2
°
、7.82
±
0.2
°
、10.34
±
0.2
°
、11.92
±
0.2
°
和18.02
±
0.2
°
处有特征峰。在一些实施例中,所述晶型的x射线粉末衍射图谱在衍射角2θ为5.85
±
0.2
°
、7.82
±
0.2
°
、10.34
±
0.2
°
、11.92
±
0.2
°
、14.74
±
0.2
°
、15.75
±
0.2
°
、17.30
±
0.2
°
、17.76
±
0.2
°
、18.02
±
0.2
°
、25.16
±
0.2
°
、25.86
±
0.2
°
、26.00
±
0.2
°
和26.41
±
0.2
°
处有特征峰。在一些实施例中,所述晶型的x射线粉末衍射图谱在衍射角2θ为5.85
±
0.2
°
、7.82
±
0.2
°
、10.34
±
0.2
°
、11.92
±
0.2
°
、14.74
±
0.2
°
、15.75
±
0.2
°
、17.30
±
0.2
°
、17.76
±
0.2
°
、18.02
±
0.2
°
、25.16
±
0.2
°
、25.86
±
0.2
°
、26.00
±
0.2
°
和26.41
±
0.2
°
处有特征峰。在一些实施例中,所述晶型的x射线粉末衍射图谱在衍射角2θ
为5.85
±
0.2
°
、7.82
±
0.2
°
、10.34
±
0.2
°
、11.92
±
0.2
°
、12.89
±
0.2
°
、13.82
±
0.2
°
、14.74
±
0.2
°
、15.75
±
0.2
°
、16.66
±
0.2
°
、17.30
±
0.2
°
、17.76
±
0.2
°
、18.02
±
0.2
°
、19.62
±
0.2
°
、20.16
±
0.2
°
、22.43
±
0.2
°
、25.16
±
0.2
°
、25.86
±
0.2
°
、26.00
±
0.2
°
、26.41
±
0.2
°
和30.36
±
0.2
°
处有特征峰。在一些实施例中,所述晶型的x射线粉末衍射图谱基本如图7所示。
15.所述晶型的差示扫描量热曲线中在50℃
‑
110℃和165℃
ꢀ‑
195℃处具有吸热峰。在一些实施例中,所述晶型的差示扫描量热曲线基本如图8所示。
16.所述晶型的热重分析曲线中在50
‑
100℃的温度范围内有3%以下的失重。在一些实施例中,所述晶型的热重分析曲线图谱基本如图8所示。
17.第二方面,本发明提供一种第一方面所述晶型的方法。
18.一种制备第一方面所述晶型的方法,其包括:取式a所示化合物,溶解于溶剂1中,过滤,挥发滤液,得到所述晶型;或者取式a所示化合物,与溶剂2混合,混悬打浆,过滤,滤饼挥发溶剂,得到所述晶型;或者取式a所示化合物溶于50℃
‑
60℃的溶剂3中,冷却,析出固体,过滤,滤饼挥干溶剂,得到所述晶型;或者取式a所示化合物溶于溶剂4中,再加入反溶剂,析出固体,过滤,滤饼挥干溶剂,得到所述晶型。
19.所述溶剂1可以包括选自丁酮、丙酮、甲醇、乙醇和乙腈中的至少一种。在一些实施例中,所述溶剂1包括选自丁酮和丙酮中的至少一种,得到晶型。在一些实施例中,所述溶剂1包括选自甲醇、乙醇和乙腈中的至少一种,得到晶型。
20.每一克所述式a所示化合物,所述溶剂1的用量可以为20ml
‑
1000ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为25ml
‑
1000ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为20ml
‑
50ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为25ml
‑
50ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为20ml
‑
100ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为25ml
‑
100ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为20ml
‑
500ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂1的用量为25ml
‑
50ml。
21.所述溶剂2可以包括选自水、甲苯、甲基叔丁基醚、异丙醇、乙酸异丙酯、正庚烷、乙酸乙酯和正丁醇中的至少一种,得到晶型。
22.每一克所述式a所示化合物,所述溶剂2的用量可以为1ml
‑
10ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂2的用量为2
‑
7ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂2的用量为2
‑
5ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂2的用量为3
‑‑
5ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂2的用量为4ml
‑
5ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂2的用量为5ml。
23.所述混悬打浆的温度可以为20℃
‑
60℃。在一些实施例中,所述混悬打浆的温度为20℃
‑
35℃。在一些实施例中,所述混悬打浆的温度为35℃
‑
60℃,所述溶剂2不为异丙醇或乙腈。
24.所述混悬打浆的时间可以为1天
‑
5天。在一些实施例中,所述所述混悬打浆的时间为3天
‑
4天。在一些实施例中,所述所述混悬打浆的时间为3天。
25.所述溶剂3包括选自乙腈、异丙醇和乙醇中的至少一种,得到晶型。
26.每一克所述式a所示化合物,所述溶剂3的用量可以为5ml
‑
20ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂3的用量为5ml
‑
15ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂3的用量为10ml
‑
15ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂3的用量为7ml
‑
12ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂3的用量为10ml
‑
12ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂3的用量为10ml。
27.所述冷却可以为冷却至10℃
‑
35℃。在一些实施例中,所述冷却为冷却至15℃
‑
30℃。在一些实施例中,所述冷却为冷却至15℃
‑
25℃。
28.所述溶剂4包括选自丙酮、乙醇、四氢呋喃和二甲基亚砜中的至少一种,所述反溶剂包括选自甲苯、正庚烷、甲苯、甲基叔丁基醚、甲苯、正庚烷和水中的至少一种。在一些实施例中,所述溶剂4为丙酮,所述反溶剂包括选自甲苯和正庚烷中的至少一种,得到晶型。在一些实施例中,溶剂4为乙醇,所述反溶剂包括或为甲苯,得到晶型。在一些实施例中,溶剂4为四氢呋喃,所述反溶剂包括选自甲基叔丁基醚、甲苯和正庚烷中的至少一种,得到晶型。在一些实施例中,溶剂4为二甲基亚砜,所述反溶剂包括或为水,得到晶型。
29.每一克所述式a所示化合物,所述溶剂4的用量可以为5ml
‑
200ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂4的用量为10ml
‑
100ml。在一些实施例中,每一克所述式a所示化合物,所述溶剂4的用量为10ml
‑
50ml。
30.所述溶剂4与反溶剂的体积比可以为1:5
‑
1:30。在一些实施例中,所述溶剂4与反溶剂的体积比为1:10
‑
1:30。在一些实施例中,所述溶剂4与反溶剂的体积比为1:15
‑
1:30。在一些实施例中,所述溶剂4与反溶剂的体积比为1:15
‑
1:25。在一些实施例中,所述溶剂4与反溶剂的体积比为1:15
‑
1:25。在一些实施例中,所述溶剂4与反溶剂的体积比为1:15
‑
1:20。在一些实施例中,所述溶剂4与反溶剂的体积比为1:20
‑
1:25。在一些实施例中,所述溶剂4与反溶剂的体积比为1:20。
31.第三方面,本发明提供一种组合物。
32.一种组合物,其包含第一方面所述的晶型和药学上可接受的辅料。
33.按照质量比计算,所述晶型为组合物总质量的至少0.05%
‑
95%。
34.第四方面,本发明提供一种前述晶型或前述组合物的用途。
35.所述药物组合物包含中药成分和/或西药成分。所述西药成分包括:阿匹莫德、r 82913、ds
‑
6930、ono 5334、瑞德西韦、磷酸奥司他韦、汉防己甲素、氯法齐明、阿司咪唑、重组人源血管紧缩素转换酶2或法匹拉韦和/或它们的药学上可接受的盐中的至少一种。
36.第一方面所述的晶型或第三方面所述的组合物在制备预防、缓解和/或治疗病毒感染,或病毒的复制或繁殖及其所产生的细胞病变效应的产品中的用途。
37.第一方面所述的晶型或第三方面所述的组合物在制备预防、缓解和/或治疗冠状病毒感染,或其同源变异病毒的复制或繁殖及其所产生的细胞病变效应的产品中的用途。
38.所述冠状病毒包括:mhv
‑
a59、hcov
‑
229e、hcov
‑
oc43、hcov
‑
nl63、hcov
‑
hku1、sars
‑
cov,mers
‑
cov、sars
‑
cov
‑
2、小鼠肝炎病毒、猫传染性腹膜炎病毒、犬冠状病毒、牛冠状病毒、禽传染性支气管炎病毒或猪冠状病毒。
39.第五方面,本发明提供一种制备第二方面所述方法中所述式a所示化合物的方法。
40.一种制备第二方面所述方法中所述式a所示化合物的方法,其包括:,取化合物gs
‑
441524溶于溶剂5中,在第一种酸存在的情况下,与2,2
‑
二甲氧基丙烷进行反应,经过第一后处理,得到化合物5;取化合物5溶于溶剂6中,在第二种酸存在的情况下,与4
‑
二甲氨基吡啶和二环己基碳二亚胺进行反应,经过第二后处理,得到化合物8;取化合物8溶于溶剂7中,搅拌,加入碳酸钠调节ph至7
‑
9,旋转蒸发除去有机溶剂,经过第三后处理,得到式a所示化合物。
41.所述溶剂5包括或为丙酮。
42.所述第一种酸包括或为浓硫酸。
43.所述溶剂6包括或为二氯甲烷.第二种酸包括或为异丁酸。
44.所述溶剂7可以包括或为盐酸水溶液和四氢呋喃的混合溶剂。
45.以盐酸水溶液的总质量计,所述盐酸的含量为35wt%
‑
38wt%。
46.所述搅拌的时间可以为1小时
‑
20小时。
47.所述第一后处理包括:旋转蒸发除去有机溶剂。用乙酸乙酯和饱和碳酸氢钠水溶液萃取,重复萃取三次,合并乙酸乙酯层,加入无水硫酸钠干燥,过滤除去硫酸钠,旋转蒸发除去有机溶剂,经过柱层析分离(洗脱液为:石油醚/乙酸乙酯(v/v)= 1/2)。
48.所述第二后处理包括:柱层析分离(洗脱液为:石油醚/乙酸乙酯(v/v)= 1/1)。
49.所述第三后处理包括:柱层析分离(洗脱液为:石油醚/乙酸乙酯(v/v)= 1/3)。
50.有益效果相比现有技术,本发明具有以下技术效果:(1)本发明所提供的式a所示化合物的晶型i在60 ℃(开口)和40 ℃/75%rh(开口)
条件下放置一周,晶型和化学纯度没有发生明显改变,物理和化学稳定性良好。
51.(2)本发明所提供的式a所示化合物的晶型i在研磨后晶型没有发生改变,其物理稳定性良好。
52.(3)本发明所提供的式a所示化合物的晶型i的粒径小,结晶度高,无吸湿性。
53.术语定义除非另外说明,否则如本文使用的以下术语和短语意图具有以下含义:“本发明所述化合物”意指式i所示化合物或其药学上可接受的盐、互变异构体、多晶型物、异构体和溶剂合物。同样地,短语“式i所示化合物”意指该式的化合物和其药学上可接受的盐、互变异构体、多晶型物、异构体和溶剂合物。
54.本发明中,如“化合物i”和“式i所示化合物”的表述,表示的是同一个化合物。
[0055]“v/v”表示体积比。ic
50
表示半数抑制浓度。
[0056]“wt%”表示该组分占混合物总质量的质量百分比。
[0057]
本发明中“室温”指的是环境温度,温度由大约10℃到大约40℃。在一些实施例中,“室温”指的是温度由大约20℃到大约30℃;在另一些实施例中,“室温”指的是温度由大约25℃到大约30℃;在又一些实施例中,“室温”指的是10℃、15℃、20℃、25℃、30℃、35℃、40℃等。
[0058]
本文使用的术语“治疗”,除非另外表明,否则意指逆转、减轻该术语所适用的病症或疾患或这样的病症或疾患的一个或多个症状、抑制所述病症或疾患或其一个或多个症状的进展或防止所述病症或疾患或其一个或多个症状。如本文使用的术语“治疗”是指治疗行为,如“治疗”在上文刚定义的。
[0059]
仪器参数除非参数中另行规定,以下所有分析都在室温下进行。
[0060]
射线粉末衍射研究在装配有自动化3*15零背景样品架的透射反射样品台的荷兰panalyticalempyrean x
‑
射线衍射仪上收集x
‑
射线粉末衍射图谱。所用辐射源为(cu,kα,kα1(
å
):1.540598;kα2(
å
):1.544426;kα2/kα1强度比例:0.50),其中电压设定在45kv,电流设定在40ma.x
‑
射线的束发散度,即样品上x
‑
射线约束的有效尺寸,为6.6mm.采用θ
‑
θ连续扫描模式,得到3
°‑
40
°
的有效2θ范围。取适量样品在环境条件(约18℃
‑
32℃)下于零背景样品架圆形凹槽处,用洁净的载玻片轻压,得到一个平整的平面,并将零背景样品架固定。将样品以0.013
°
的扫描步长在3
‑
40
°
2θ
±
0.2
°
范围内产生传统的x射线粉末衍射图谱。用于数据收集的软件为data collector ,数据用data viewer 和 highscore plus分析和展示。
[0061]
差示扫描量热法(dsc)dsc测量在tainstrumentstm型号q2000中用密封盘装置进行。将样品(约2
‑
3mg)在铝盘中称量,用tzero压盖,精密记录到百分之一毫克,并将样品转移至仪器中进行测量。仪器用氮气以50ml/min吹扫。在30℃到300℃之间以10℃/min的加热速率收集数据。以吸热峰向下进行绘图,数据用ta universal analysis分析和展示。
[0062]
热重分析法(tga)在ta instruments q500上采集tga数据。使用认证的镍校准仪器的温度。通常将8
‑
12mg样品加载到预称重的铂金坩埚上,并以10℃/min从30℃加热至300℃。在样品上方保
持60ml/min的氮气清扫。在tga图中,横坐标表示温度(temperature,℃),纵坐标表示失重的百分含量(weight(%))。
[0063]
动态水蒸气吸附(dynamic vapor sorption, dvs)样品的水蒸气吸附/脱附数据是在水蒸气吸附仪(proumid gmbh & co. kg,德国)上收集完成的。将样品置于已去皮的样品盘中,自动称重。仪器参数设置如下:偏折光显微镜(plm)plm所用到的仪器为polarizing microscope eclipse lv100pol(nikon, jpn)。
[0064]
氢核磁共振(1h
‑
nmr) 1h
‑
nmr是在配备有samplexpress 60自动进样器的avance iii hd 300或者400上完成的。
[0065]
晶型的高效液相色谱(hplc)检测液相色谱分析所用到的仪器为安捷伦hplc 1260系列,用于样品固态稳定性研究的分析方法如表1所示。
[0066]
上述各种剂型的药物均可以按照药学领域的常规方法制备。
[0067]
本发明中,一些化合物的缩写所表示的化合物结构:在描述实验细节时,使用了某些缩写和缩略词。尽管它们中的大多数能被本领域技术人员所理解,但下表包含了这些缩写和缩略词的列表。
附图说明
[0068]
图1示晶型i的偏折光显微镜检测图。
[0069]
图2示晶型i的x射线粉末衍射检测谱图。
[0070]
图3示晶型i的差示扫描量热检测谱图和热重分析谱图。
[0071]
图4示实施例9中晶型i的动态水蒸气吸附结果。
[0072]
图5示实施例9中晶型i的动态水蒸气吸附前后的x射线粉末衍射检测谱图。
[0073]
图6示晶型ii的偏折光显微镜检测图。
[0074]
图7示晶型ii的x射线粉末衍射检测谱图。
[0075]
图8示晶型ii的差示扫描量热检测谱图和热重分析谱图。
[0076]
图9示实施例12中晶型i的影响因素稳定性x射线粉末衍射检测结果。
[0077]
图10示实施例10中晶型i的研磨实验结果。
[0078]
图11示实施例11中晶型i的水活度实验x射线粉末衍射检测结果。
具体实施方式
[0079]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例以对本发明作进一步的详细说明。
[0080]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0081]
本发明中,μm表示微摩尔每升;mmol表示毫摩尔;equiv表示当量;aw表示水分活度。
[0082]
实施例1:(3ar,4r,6r,6ar)
‑4‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4] 三嗪
‑7‑
基)
‑6‑
(羟甲基
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3] 间二氧杂环戊烷基
‑4‑
甲腈(化合物5)的合成将5.62g的化合物gs
‑
441524溶于30ml丙酮中,再加入11.50ml的2,2
‑
二甲氧基丙烷和1.34ml硫酸,45℃搅拌半小时,冷却至25℃,旋转蒸发除去有机溶剂。用100ml的乙酸乙酯和100ml的饱和碳酸氢钠水溶液萃取,重复萃取三次,合并乙酸乙酯层,加入无水硫酸钠干燥,过滤除去硫酸钠。旋转蒸发除去有机溶剂,经过柱层析分离(洗脱液为:石油醚/乙酸乙酯(v/v)=1/2)得到6.20g化合物5(白色固体,产率97%)。取所得化合物5检测氢谱,结果如下:氢谱:1h nmr (400 mhz, chloroform
‑
d) δ 7.95 (s, 1h), 7.11 (d, j = 4.7 hz, 1h), 6.69 (dd, j = 4.8, 2.4 hz, 1h), 5.77 (s, 2h), 5.42 (d, j = 6.6 hz, 1h), 5.24 (dd, j = 6.6, 2.4 hz, 1h), 4.67 (q, j = 1.9 hz, 1h), 3.99 (dd, j = 12.5, 1.9 hz, 1h), 3.84 (dd, j = 12.5, 1.7 hz, 1h), 1.81 (s, 3h), 1.40 (s, 3h)。
[0083]
实施例2:((3ar,4r,6r,6ar)
‑6‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑6‑
氰基
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]间二氧杂环戊
‑4‑
基)甲基异丁酸酯(化合物8)的合成
将1.50g的化合物5溶于15ml的二氯甲烷中,再加入0.42ml异丁酸和55.40mg的4
‑
二甲氨基吡啶,搅拌10min后,加入1.02g的二环己基碳二亚胺,25℃搅拌24h。经过柱层析分离(洗脱液为:石油醚/乙酸乙酯(v/v)=1/1),得到1.71g化合物8(白色固体,产率94%)。取得到的化合物8检测氢谱和碳谱,结果如下:氢谱:1h nmr (400 mhz, cdcl3, zqf
‑
rd01
‑
2) δ (ppm): 7.99 (s, 1h), 6.99 (d, j=4.6 hz, 1h), 6.62 (d, j=4.6 hz, 1h), 5.72 (br, 2h), 5.49 (d, j=6.8 hz, 1h), 4.93
‑
4.90 (dd, j=6.8 hz, 4.3 hz, 1h), 4.61
‑
4.58 (q, j=4.4 hz, 1h), 4.44
‑
4.26 (m, 2h), 2.61
‑
2.50 (m, 1h), 1.77 (s, 3h), 1.42 (s, 3h), 1.17
‑
1.14 (q, j=3.8 hz, 6h)。
[0084]
碳谱:
13
c nmr (100 mhz, cdcl3, zqf
‑
rd01
‑
2) δ (ppm): 176.7, 155.2, 147.3, 123.5, 117.2, 116.7, 115.6, 112.6, 100.0, 83.8, 83.0, 82.0, 81.4, 63.1, 33.8, 26.4, 25.6, 18.9。
[0085]
实施例3:((2r,3s,4r,5r)
‑5‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4] 三嗪
‑7‑
基)
‑5‑
氰基
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基) 甲基异丁酸酯(化合物a)的合成将1.50g的化合物8溶于3 ml质量百分比为37%的盐酸水溶液和15ml的四氢呋喃中,搅拌6小时后,加入碳酸钠调节ph至8,旋转蒸发除去有机溶剂,经过柱层析分离(洗脱液为:石油醚/乙酸乙酯(v/v)=1/3),得到0.66g式a所示化合物(白色固体,产率49%)。取得到的式a所示化合物检测氢谱、碳谱和高效液相色谱,结果如下:氢谱:1h nmr (400 mhz, methanol
‑
d4) δ 7.76 (s, 1h), 6.78 (s, 2h), 4.78 (d, j = 5.3 hz, 1h), 4.40
‑
4.24 (m, 2h), 4.24
‑
4.11 (m, 1h), 4.10
‑
4.01 (m, 1h), 2.42 (p, j = 7.0 hz, 1h), 0.99 (dd, j = 7.0, 4.1 hz, 6h)。
[0086]
碳谱:
13
c nmr (101 mhz, methanol
‑
d4) δ 176.96, 155.82, 146.92, 124.25, 116.54, 116.29, 110.75, 101.20, 82.04, 80.00, 74.27, 70.68, 62.93, 33.58, 25.00, 17.95, 17.87。
[0087]
高效液相色谱:流动相为水/乙腈(v/v) = 10/90, 流速为0.8ml/min,检测波长为254 nm, 式a所示化合物的保留时间为2.036min。
[0088]
实施例4:式a所示化合物的固体形式的制备(蒸发结晶法)取200mg实施例3所得式a所示化合物,分别溶于5ml表2所示溶剂中,过滤,所得滤液于30℃挥发,挥干溶剂,得到所述式a所示化合物的固体形式。取所得固体形式进行氢谱检测有机溶剂和x射线粉末衍射检测,其结果如表2所示。
[0089]
实施例5:式a所示化合物的固体形式的制备(混悬打浆法)取约1g实施例3所得式a所示化合物 ,分别与5ml表3所述溶剂混合,得混悬液,再分别于21℃和50℃打浆3天,过滤,滤饼挥干溶剂,得到式a所示化合物的固体形式。取所得固体形式进行x射线粉末衍射检测,其结果如表3所示。
[0090]
实施例6:式a所示化合物的固体形式的制备(冷却结晶法)取500mg实施例3所得式a所示化合物 ,分别溶解于5ml 50℃的表4所述溶剂,过滤,冷却至室温,搅拌,若有析出固体,过滤,滤饼挥干溶剂,得到式a所示化合物的固体形式。取所得固体形式进行x射线粉末衍射检测,其结果如表4所示。
[0091]
实施例7:式a所示化合物的固体形式的制备(反溶剂沉淀法)取100mg实施例3所得式a所示化合物 ,分别溶解于1ml表5所述溶剂,得溶液a,再
分别取0.2ml溶液a加入4ml表5所述反溶剂,若有固体析出,则过滤,滤饼挥干,得到式a所示化合物的固体形式。取所得固体形式进行x射线粉末衍射检测,其结果如表5所示。
[0092]
实施例8:晶型表征检测分别取实施例4实施例7所得晶型i和晶型ii,分别进行偏折光显微镜检测、x射线粉末衍射检测、差示扫描量热检测和热重分析检测,结果分别见图1
‑
图3、图5
‑
图8。
[0093]
结论:(1)晶型i呈现不规则形状,粒径小于20μm, 具有较高结晶度,由于晶型i的氢谱显示无有机溶剂残留,其差示扫描量热图和热重分析图显示其在熔溶前又无失重,表明晶型i为无水晶型。
[0094]
(2)晶型ii呈现棒状晶体,较高结晶度,其差示扫描量热图和热重分析图显示样品在100 ℃之前约有2.3%的失重,是由脱水造成的,氢谱没有检测到明显的有机溶剂残留,说明晶型ii为半水合物。
[0095]
实施例9:晶型i的动态水蒸气吸附检测取晶型i进行动态水蒸气吸附检测,结果如图4所示。再取动态水蒸气吸附前和动态水蒸气吸附后的晶型i分别进行x射线粉末衍射检测,结果如图5所示。
[0096]
结论:动态水蒸气吸附检测结果显示晶型i不具有吸湿性,在80%rh,25℃的条件下,增重<0.2%,且测试后晶型未发生改变,证明晶型i的晶型稳定性好。
[0097]
实施例10:晶型i的研磨实验
取晶型i用研钵和研杵手动研磨5分钟,并对研磨后固体进行xrpd测试。研磨五分钟后,晶型没有改变,结果见图10。
[0098]
结论:晶型i的晶型物理稳定性好。
[0099]
实施例11:晶型i的水活度实验取50mg晶型i,加入1.0 ml不同水活度的乙醇水溶液(水活度用dynochem计算得到),混合,得到混悬液,混悬液分别在21℃和50℃打浆7天,过滤,得到固体产品,取所得固体产品进行x射线粉末衍射检测,结果如表6和图11所示。
[0100]
备注:在50℃条件下,起始物料在etoh/water中具有较高的溶解度,因此只进行了aw=0.1的实验。
[0101]
结论:晶型i在不同水活度中的晶型稳定性好。
[0102]
实施例12:影响因素稳定性研究取晶型i,分别于40℃/75%rh(开口)和60℃(开口)条件下放置7天,分别于0天和7天进行高效液相色谱检测和x射线粉末衍射检测,结果如表7和图9所示。
[0103]
结论:结果显示,晶型i在实验条件(40℃/75%rh/开口,60℃/开口)下放置7天,式a所示化合物纯度不变,晶型不变,其物理和化学稳定性良好。
[0104]
本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。