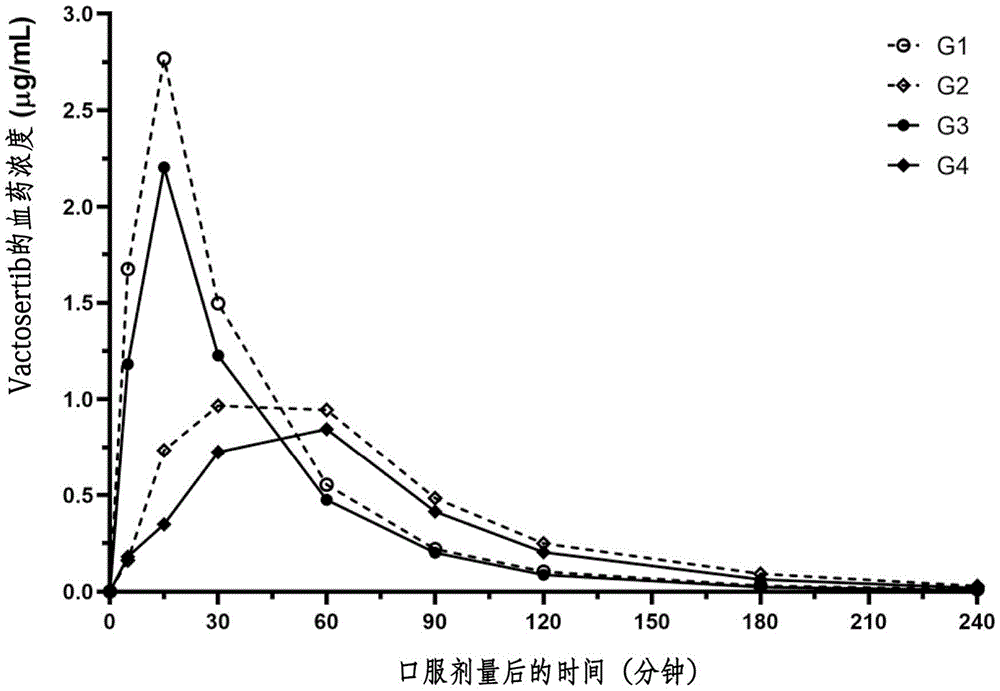
本发明涉及包含粘土矿物复合物的控释的用于口服给药的组合物、其制备方法及控释方法。
背景技术:
蒙皂石为以2个由Al、Mg、Fe组成的八面体薄片位于由Si、Al、Fe组成的四面体薄片之间来以三明治形状结合,由此组成一个单元层(2:1层)的叶状硅酸盐矿物质。蒙皂石的单元层具有负电荷,这是由具有4价正电荷的四面体Si被具有3价正电荷的Al或者Fe同晶置换(isomophicsubstitution)或者具有3价正电荷的八面体Al或者Fe3 被具有2价正电荷的Mg或者Fe2 同晶置换而产生的。通过单元层中产生的负电荷来在单元层与单元层之间诱导阳离子,这意味着蒙皂石可以用作药物载体(carrier)。下述化学式1的化合物为韩国授权专利第10-1500665号记载的N-((4-([1,2,4]三唑并[1,5-a]吡啶-6-基)-5-(6-甲基吡啶-2-基)-1H-咪唑-2-基)甲基)-2-氟苯胺(N-((4-([1,2,4]triazolo[1,5-a]pyridine-6-yl)-5-(6-methylpyridin-2-yl)-1H-imidazol-2-yl)methyl)-2-fluoroaniline),已知具有现有的抗癌剂的活性等。本发明人在研究上述化合物的药理作用时确认到上述化合物在体内的吸收不规律的问题。于是,在为了改善化学式1的化合物在体内作用的研究中确认到在特定条件下与粘土矿物形成复合物时,不仅可以改善向结肠传递药物,还可以改善在结肠中的药物作用,从而完成本发明。[化学式1]技术实现要素:技术问题本发明的目的在于,提供药物向结肠的传递及药物在结肠中的作用得到改善的组合物。技术方案为了实现上述目的,本发明提供一种控释的用于口服给药的组合物,其包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物。[化学式1]并且,本发明提供控释的用于口服给药的组合物的制备方法,包括混合包含化学式1的化合物或者其药学上可接受的盐的溶液、酸性反应溶液以及粘土矿物悬浮液并将上述化合物吸附于粘土矿物来制备复合物的步骤,上述控释的用于口服给药的组合物包含化学式1的化合物及粘土矿物的复合物。并且,本发明提供控释方法,其特征在于,向对象口服给药组合物,上述药物组合物包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物。[化学式1]有益效果本发明的组合物可以控制化学式1的化合物的释放,不仅可以有效地向结肠传递,而且还可以使上述化合物在结肠中长时间缓慢地起作用。附图说明图1为使用铂涂敷vactosertib粉末表面后通过扫描电子显微镜SEM拍摄的照片。图2为使用铂涂敷膨润土/vactosertib复合物后进行观察的扫描电子显微镜SEM照片。图3为以vactosertib、膨润土、膨润土/vactosertib复合物以及将vactosertib与膨润土以与上述复合物相同的比例简单混合的混合物为对象进行X射线衍射分析(XRD)后示出XRD图谱的曲线图。图4示出将包含0.1%的吐温80(Tween80)的0.1N的HCl溶液(pH1.2)设定为溶出液后进行的vactosertib粉末(●)及膨润土/vactosertib复合物(□)的药物释放模式。图5示出将包含0.1%的吐温(Tween)80的50mM的醋酸盐缓冲液(pH4.5)设定为溶出液后进行的vactosertib粉末(●)及膨润土/vactosertib复合物(□)的药物释放模式。图6示出将包含0.1%的吐温80的50mM的磷酸盐缓冲液(pH7.4)设定为溶出液后进行的vactosertib粉末(●)及膨润土/vactosertib复合物(□)的药物释放模式。图7示出在以vactosertib的量为基准设定剂量为10mg/kg并向大鼠口服给药vactosertib粉末(●)、vactosertib水溶液(△)以及膨润土/vactosertib复合物悬浮液(□)后的随时间的血药浓度曲线。图8示出利用由DSS诱导的溃疡性结肠炎大鼠模型的药物给药方案。图9示出在由DSS诱导的溃疡性结肠炎大鼠模型中的根据时间的vactosertib的血药浓度变化。连接曲线的点表示属于各组的动物在不同时间段的血药浓度数据的平均值,○表示G1组,◇表示G2组,●表示G3组,◆表示G4组。图10示出在由DSS诱导的溃疡性结肠炎大鼠模型中口服给药试验物质后vactosertib的血药浓度达峰时间(tmax),○表示G1组,◇表示G2组,●表示G3组,◆表示G4组。图11示出在由DSS诱导的溃疡性结肠炎大鼠模型中口服给药试验物质后的vactosertib的药峰浓度(Cmax),○表示G1组,◇表示G2组,●表示G3组,◆表示G4组。图12示出在由DSS诱导的溃疡性结肠炎大鼠模型中口服给药试验物质后的vactosertib的血药浓度-时间曲线下面积(AUC),○表示G1组,◇表示G2组,●表示G3组,◆表示G4组。图13示出在由DSS诱导的溃疡性结肠炎大鼠模型中口服给药试验物质后的vactosertib在体内的药物清除半衰期(t1/2),○表示G1组,◇表示G2组,●表示G3组,◆表示G4组。图14示出用于进行动物实验的溃疡性结肠炎诱导方法及试验物质给药方案。图15示出在由DSS诱导的溃疡性结肠炎小鼠模型中通过组织病理学评估的炎症的严重程度。以中间值、25-75%值(方框内)、最小值-最大值(两个极端)示出数据,利用内置于GraphpadPrism8.01版本的作为非参数检验法的邓恩检验法(Dunn'stest)来检验阳性对照组(▲G2组)与试验物质给药组(◇G3组、●G4组、◆G5组)之间的统计学显著性。膨润土给药组(G3组)和vactosertib给药组(G4组)的炎症指数中间值略低于阳性对照组(G2组),vactosertib/膨润土复合物给药组(G5组)的中间值虽然更低,但没有统计学显著性。图16示出在由DSS诱导的溃疡性结肠炎小鼠模型中通过组织病理学评估的隐窝细胞损伤严重程度。以中间值、25-75%值(方框内)、最小值-最大值(两个极端)示出数据,利用内置于GraphpadPrism8.01版本的作为非参数检验法的邓恩检验法来检验阳性对照组(▲G2组)与试验物质给药组(◇G3组、●G4组、◆G5组)之间的统计学显著性。膨润土给药组(G3组)和vactosertib给药组(G4组)的隐窝细胞损伤指数中间值低于阳性对照组(G2组),但没有统计学显著性,而vactosertib/膨润土复合物给药组(G5组)的中间值在统计学上显著减少。图17示出在由DSS诱导的溃疡性结肠炎小鼠模型中通过组织病理学评估的溃疡的严重程度。以中间值、25-75%值(方框内)、最小值-最大值(两个极端)示出数据,利用内置于GraphpadPrism8.01版本的作为非参数检验法的邓恩检验法来检验阳性对照组(▲G2组)与试验物质给药组(◇G3组、●G4组、◆G5组)之间的统计学显著性。膨润土给药组(G3组)和vactosertib给药组(G4组)的溃疡指数中间值低于阳性对照组(G2组),但没有统计学显著性,而vactosertib/膨润土复合物给药组(G5组)的中间值在统计学上显著减少。图18示出在由DSS诱导的溃疡性结肠炎小鼠模型中通过组织病理学评估的浮肿的严重程度。以中间值、25-75%值(方框内)、最小值-最大值(两个极端)示出数据,利用内置于GraphpadPrism8.01版本的作为非参数检验法的邓恩检验法来检验阳性对照组(▲G2组)与试验物质给药组(◇G3组、●G4组、◆G5组)之间的统计学显著性。与阳性对照组(G2组)相比,无法在各试验物质给药组中观察到显著变化。图19示出在由DSS诱导的溃疡性结肠炎小鼠模型中通过组织病理学评估的纤维化的严重程度。以中间值、25-75%值(方框内)、最小值-最大值(两个极端)示出数据,利用内置于GraphpadPrism8.01版本的作为非参数检验法的邓恩检验法来检验阳性对照组(▲G2组)与试验物质给药组(◇G3组、●G4组、◆G5组)之间的统计学显著性。膨润土给药组(G3组)与vactosertib给药组(G4组)的纤维化指数中间值略低于阳性对照组(G2组),vactosertib/膨润土复合物给药组(G5组)的中间值更低,但没有统计学显著性。图20示出比较在将由DSS诱导的溃疡性结肠炎小鼠模型中通过组织病理学评估的炎症、隐窝细胞损伤、溃疡及浮肿的严重程度分数总合的图(纤维化分数除外)。以中间值、25-75%值(方框内)、最小值-最大值(两个极端)示出数据,利用内置于GraphpadPrism8.01版本的作为非参数检验法的邓恩检验法来检验阳性对照组(▲G2组)与试验物质给药组(◇G3组、●G4组、◆G5组)之间的统计学显著性。膨润土给药组(G3组)与vactosertib给药组(G4组)的溃疡指数中间值低于阳性对照组(G2组),但没有统计学显著性,而vactosertib/膨润土复合物给药组(G5组)的中间值在统计学上显著减少。图21为示出在由DSS诱导的溃疡性结肠炎小鼠模型的结肠中试验物质对于白细胞介素6(IL-6)的影响的免疫组织化学染色的结果。图22为在由DSS诱导的溃疡性结肠炎小鼠模型的结肠中测量IL-6染色的发色面积的结果。具体实施方式本发明涉及控释的用于口服给药的组合物,上述组合物包含化学式1的化合物(以下称“vactosertib”)或其药学上可接受的盐以及粘土矿物的复合物。[化学式1]并且,本发明涉及控释的用于口服给药的组合物的制备方法,包括混合包含化学式1的化合物或者其药学上可接受的盐的溶液、酸性反应溶液以及粘土矿物悬浮液使上述化合物吸附于粘土矿物来制备复合物的步骤。上述控释的用于口服给药的组合物包含化学式1的化合物及粘土矿物的复合物。并且,本发明涉及控释方法,其特征在于,向对象口服给药组合物,上述组合物包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物。[化学式1]以下,详细说明本发明。化学式1的化合物本发明的化学式1的化合物可以利用公知的方法制备。例如,化学式1的化合物为N-((4-([1,2,4]三唑并[1,5-a]吡啶-6-基)-5-(6-甲基吡啶-2-基)-1H-咪唑-2-基)甲基)-2-氟苯胺。上述化合物的分子式为C22H18FN7,在25±3℃的温度下的饱和水溶液的pH值为pH7.2,溶解度(aqueoussolubility)<0.02mg/ml。在低的pH值范围内,溶解度更高(即,在pH2.81的酸碱度下,溶解度为25.05mg/ml),在pH4.9以上的酸碱度下,溶解度为0.02mg/ml以下。分配系数(辛醇/水)LogDo/w为3.31。化学式1的化合物可以利用韩国专利公开公报第10-2013-0028749号公开的方法制备。[化学式1]粘土矿物通常,粘土矿物具有由硅片(silicasheet)与铝片(aluminasheet)结合形成的结晶单元层叠的层状结构,即,板状结构,在如上所述的粘土矿物中的具有层间膨胀性的粘土矿物中,在结晶单元之间不存在氢键,结晶单元之间的结合力弱,因此当在这些结晶单元之间吸入水分时,会发生膨胀。因此,即使具有较大尺寸的离子也可以轻松进入具有层间膨胀性的粘土矿物的结晶单元之间。另一方面,在具有层间膨胀性的粘土矿物中,具有4价正电荷的四面体Si被具有3价正电荷的Al或者Fe同晶置换或者具有3价正电荷的八面体Al或者Fe3 被具有2价正电荷的Mg或者Fe2 同晶置换而产生层的负层电荷,但在层与层之间或者在表面结合有钙离子(Ca2 )、镁离子(Mg2 )、钠离子(Na )、钾离子(K )等阳离子,因此整体上呈电中性。本发明的粘土矿物对阳离子物质的吸附优秀,上述吸附的模式根据周围pH值而变化。本发明的粘土矿物具有板状结构,具体地,具有层间膨胀性,是可以在内部插入抗生素来用作传递体的粘土矿物。本发明的粘土矿物可以为蒙皂石类矿物,例如,可以为蒙脱石(montmorillonite)或者膨润土(bentonite)、蒙皂石(smectite)、蛭石(vermiculite)、贝德石(beidellite)、绿脱石(nontronite)、皂石(saponite)、水辉石(hectorite)等。例如,本发明的粘土矿物可以为包含50%重量以上的蒙脱石的膨润土。优选地,本发明的粘土矿物为膨润土。粘土矿物/vactosertib复合物在本发明的粘土矿物/vactosertib复合物中,将粘土矿物用作使vactosertib经过胃(pH1.5至2.0)和小肠(pH7.2至7.5)后传递至结肠(pH7.9至8.5)来在结肠中起作用的传递体,即,载体。即,本发明的粘土矿物/vactosertib复合物抑制vactosertib在上消化道中被吸收,并将vactosertib传递到结肠部位。本发明的粘土矿物为层状形粘土矿物,层状表面带有负电荷,vactosertib以非结晶状态吸附于粘土矿物。上述复合物能够以1:0.02至1:0.26的重量比包含粘土矿物与化学式1的化合物或者其药学上可接受的盐。本发明的粘土矿物优选为膨润土。在低pH值的条件下,具有胺基的弱碱性药物分子带有正电荷,可以与膨润土结合,但在高pH值的条件下,可以失去电荷而从膨润土中释放出来。考虑到体内上消化道的pH值低、下消化道的pH值高,本发明的膨润土/vactosertib复合物具有pH值依赖性释放调节能力。即,将本发明的复合物放入pH1.2的溶出液时的溶出速度慢于将化学式1的化合物粉末放入pH1.2的溶出液时的溶出速度(图4)。但是,将上述复合物放入pH7.4的溶出液时的溶出速度快于将化学式1的化合物粉末放入pH7.4的溶出液时的溶出速度(图6)。vactosertib在酸性环境下容易溶解,但pH7的酸碱度下呈中性电荷,其在中性中的溶解度低,因此其吸收集中出现在上消化道中,具有无法在作为目标器官的下消化道中充分吸收的吸收性问题。即,当口服给药vactosertib时,通过胃酸使其带有正电荷而增加溶解度后,在通过小肠时由于pH值再次变回中性而使一部分中性分子在小肠上部被吸收。因此在药物动态学上具有在口服给药vactosertib的初期血药浓度剧增的问题。并且,vactosertib在小肠中被中性分子化后,溶解度剧减而析出,之后无法在小肠中被吸收。从而在药物动态学上出现血药浓度剧减。即,vactosertib的电荷与溶解度随着pH值的变化而变化,从而发生其在小肠中可被吸收的区域非常有限的缺点。并且,vactosertib在溶解和析出时严重受到胃液和肠液的影响,由于每个人分泌胃液和肠液的程度不同,因此很难在患者中期待规定的效果。并且,vactosertib以分子结团的非晶体型固体剂的形式摄取,因此需要通过胃酸将其溶解为单个分子的过程。并且,由于vactosertib的药物本身的半衰期非常短,因此,即使在上消化道快速吸收药物,也很难期待药理效果的长期作用。然而,在粘土矿物/vactosertib复合物的情况下,优选地,在vactosertib吸附于膨润土的膨润土/vactosertib复合物的情况下,vactosertib以阳离子形态的分子插入膨润土的层间,没有在胃酸中溶解的过程。并且,膨润土具有涂敷能力,吸附于小肠上皮细胞后,作为药物的vactosertib脱离并中性化后被吸收,因此可以被小肠的全部区域吸收。而且,移动到结肠的粘土矿物/vactosertib复合物吸附与患处直接释放vactosertib,从而具有以双重方式向患处传递药物的效果。包含粘土矿物/vactosertib复合物的组合物本发明涉及控释的用于口服给药的组合物,上述组合物包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物。vactosertib在酸性环境下容易溶解,而在中性环境中的溶解度低,因此具有在体内的吸收不确定的问题。即,在包括胃在内的上消化道的情况下,pH值为非常低的1至3左右,从十二指肠通过小肠时,pH值增加至5至8的水平。因此,vactosertib只在上消化道集中被吸收,而在下消化道中经常出现无法充分被吸收的情况。并且,药物本身的半衰期非常短,因此,即使在上消化道快速吸收药物,也很难期待药理效果的长期作用。相反,在包含粘土矿物/vactosertib复合物的组合物的情况下,vactosertib在酸性pH值环境下被离子化,因此,在低的pH值下吸附于粘土矿物,而在高pH值下被释放出来。因此,vactosertib以吸附于粘土矿物的状态稳定地传递到pH值高的下消化道,例如结肠,因此不仅延迟药物的释放,还帮助vactosertib在结肠中持续起作用。并且,在酸性条件下,本发明的粘土矿物/vactosertib复合物的vactosertib与粘土矿物之间保持很强的吸附,因此防止给药后药物的急剧吸收。并且,本身可以很好地分散于水中,因此比片剂更易于吸收,并且能够以具有再现性的方式实施。并且,本发明的粘土矿物/vactosertib复合物与利用醋酸邻苯二甲酸纤维素、邻苯二甲酸羟丙基甲基纤维素醋酸酯、聚醋酸乙烯邻苯二甲酸酯、聚丙烯酸酯L及聚丙烯酸酯S系列的高分子等现有的肠溶性高分子的情况不同,不需要用于防止药物在初期释放的涂敷工序。向大鼠口服给药上述组合物时的血药浓度达峰时间(tmax)可以为30分钟至60分钟,向人口服给药时的血药浓度达峰时间(tmax)可以为3小时至10小时。并且,口服给药上述组合物时的血药浓度达峰时间(tmax)长于口服给药化学式1的化合物水溶液时的血药浓度达峰时间(tmax)。优选地,若化学式1的化合物的剂量相同,则口服给药上述组合物时的血药浓度达峰时间(tmax)比口服给药化学式1的化合物水溶液时的血药浓度达峰时间(tmax)长2倍至10倍,更优选地,长2.3倍至8倍,更加优选地,长2.4倍至6倍,进而优选地,长2.6倍至5倍,进而更优选地,长2.7倍至4倍。口服给药上述组合物时的药峰浓度(Cmax)可以为0.7μg/ml至1.9μg/ml。并且,口服给药上述组合物时的药峰浓度(Cmax)低于口服给药化学式1的化合物水溶液时的药峰浓度(Cmax)。优选地,若化学式1的化合物的剂量相同,则口服给药上述组合物时的药峰浓度(Cmax)为口服给药化学式1的化合物水溶液时药峰浓度(Cmax)的20%至80%,更优选为25%至70%,更加优选为30%至60%,进而优选为35%至50%,进而更优选为37%至45%。将本发明的复合物放入pH1.2的溶出液时的溶出速度慢于将化学式1的化合物粉末放入pH1.2的溶出液时的溶出速度。但是,将上述复合物放入pH7.4的溶出液时的溶出速度快于将化学式1的化合物粉末放入pH7.4的溶出液时的溶出速度。并且,口服给药上述组合物时化学式1的化合物在小肠中的吸收率低于口服给药化学式1的化合物水溶液时在小肠中的吸收率。上述吸收率是指相对于单位时间内的给药的化学式1的化合物的量而被吸收的化合物的量。口服给药上述组合物时减少的化学式1的化合物在小肠中的吸收量意味着可以相对增加在结肠中起作用的化学式1的化合物的量。并且,口服给药本发明的组合物时化学式1的化合物在小肠中的吸收速度(rate)慢于口服给药化学式1的化合物水溶液时在小肠中的吸收速度。在此情况下,在小肠中的吸收速度的变化可以利用血药浓度达峰时间(tmax)及药峰浓度(Cmax)得知。例如,若血药浓度达峰时间(tmax)增加、药峰浓度(Cmax)降低,则意味着吸收速度减少,若血药浓度达峰时间(tmax)减少、药峰浓度(Cmax)增加,则意味着吸收速度增加。因此,本发明的组合物适合在结肠中释放化学式1的化合物。口服给药本发明的组合物时化学式1的化合物在结肠中对选自由炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化组成的组中的症状的药物改善效果优于口服给药化学式1的化合物水溶液时在结肠中对选自由炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化组成的组中的症状的药物改善效果。优选地,口服给药本发明的组合物时化学式1的化合物在结肠中的记载于表8的炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化的组织病理学总分所示的药物改善效果优于口服给药化学式1的化合物水溶液时在结肠中的记载于表8的炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化的组织病理学总分所示的药物改善效果。尤其,本发明的组合物可以将以结肠为目标器官的药物组合物传递到结肠来增加结肠中的药物作用的药物组合物。本发明的组合物为控释组合物。控释(缓释或控释(Modified-orControlled-release))是指给药药物后药物的血药浓度迅速上升到有效浓度,只在所希望的时间内保持规定的药物血药浓度,相比于其他普通制剂,给药频率低,活体反应均匀,副作用少。本发明的组合物可以为药物组合物。本发明的药物组合物可以包含0.01重量百分比至80重量百分比的包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物,优选地,可以包含0.02重量百分比至65重量百分比。但是,上述含量可以根据给药人员的需要增减,可根据年龄、饮食、营养状态、疾病的进程等状况适当地增减。本发明所属
技术领域:
的普通技术人员可以适当地决定本发明的药物组合物中所含的包含化学式1的化合物或者其药学上可接受的盐以及粘土矿物的复合物的含量。本发明的药物组合物可以口服给药,可以用作通常的药品制剂的形态,例如,本发明的药物组合物可以用做如片剂、颗粒剂、胶囊剂、悬浮剂等的用于口服给药的制剂的形态,上述制剂可以使用可接受的通常的载体来制备,例如,在用于口服给药的制剂的情况下,可以使用赋形剂、结合剂、崩解剂、润滑剂、溶解剂、着色剂、涂敷剂、悬浮化剂、保存剂或者增量剂来制备。上述仅为制备本发明的药物组合物的可采用的制剂形态及添加剂的例示,本发明的药物组合物不限定于此。本发明的药物组合物的剂量可以根据患者的状态、年龄、性别及并发症等各种因素由专家来决定,但通常能够以成人每公斤体重为0.1mg至10g的剂量来给药,优选地,能够以每公斤体重10mg至5g的剂量来给药。并且,单位剂型可以包含上述药物组合物的1天剂量或其的1/2、1/3或者1/4的剂量,可以一日给药1次至6次,但不限定于此,主治医师可以适当调节上述剂量。本发明的组合物的制备方法本发明涉及控释的用于口服给药的组合物的制备方法,其包括混合包含化学式1的化合物或者其药学上可接受的盐的溶液、酸性反应溶液以及粘土矿物悬浮液并将上述化合物吸附于粘土矿物来制备复合物的步骤,上述控释的用于口服给药的组合物包含化学式1的化合物及粘土矿物的复合物。在此情况下,为了使化学式1的化合物以非结晶的状态吸附于层状形的粘土矿物,可以使用将上述化合物与粘土矿物溶解并分散于相同的水溶液中来混合的方法。在此情况下,由于粘土矿物(例如膨润土)的层状表面带有负电荷,因此,若溶解于水溶液的药物带有阳离子,则可以更为有效地进行吸附。由于vactosertib在中性pH值环境下的溶解度非常低,很难在相应pH值条件下被离子化来吸附于粘土矿物。但在反应溶剂的pH值呈酸性的情况下,vactosertib的溶解度增加,可以使大量的药物与膨润土反应,由于vactosertib的结构特性在低的pH值下带正电荷,通过更强的离子结合反应以高吸附率结合于粘土矿物。因此,优选地,上述反应溶液的pH值为pH0.5至pH3.5。在此情况下,为了向反应溶液赋予酸性条件,可以选择盐酸、磷酸、硫酸、醋酸、甲酸中的一种。而且,vactosertib在pH3的酸碱度下的溶解度约为1mg/mL,考虑pH值越低溶解度越增加的情况,更优选地,反应pH值在1至3的范围内选择。在此情况下,为了更充分的吸附,优选地,上述复合物以0.1:5至1.3:5的重量比包含化学式1的化合物或者其药学上可接受的盐及粘土矿物。若在粘土矿物中反应过量的药物(化学式1的化合物),则并不附于粘土矿物而损失的药物增多。相反,若粘土矿物的量比药物量高,则虽可提高整体的吸附率,但最终获得的复合物中药物含量低且服用量增加,由此会为给药带来负担。为了获得在规定条件下形成本发明的复合物后干燥形态的复合物,可以使用多种方法。并且,将vactosertib吸附于粘土矿物后,可以通过离心分离、去除上清液等方法去除未结合的vactosertib。并且,在将沉淀物冷冻干燥的情况下,可以获得性状与现有的粘土矿物相似的粉末形态的复合物。另一方面,为了分离反应液与复合物,可以包括通过过滤器仅使溶剂通过,使用蒸馏水反复洗涤未通过的复合物沉淀物的过程,通过上述过程可以在不使用离心分离的情况下去除未吸附的药物和酸性溶液。或者,也可以利用在吸附反应结束后缓解溶液的强酸性条件并冷冻干燥的方法。具体地,在能够使药物有效吸附的低的pH值下进行反应后,提高反应溶液的pH值后进行冷冻干燥,从而可以降低仪器被腐蚀的可能性,使冷冻干燥机顺畅地运行。本发明的组合物可以应用于vactosertib具有预防、改善或者治疗效果而周知的多种疾病中。例如,如韩国授权专利第10-1500665号所记载的,本发明的组合物对由间变性淋巴瘤激酶5(ALK5)和/或间变性淋巴瘤激酶4(ALK4)介导的多种疾病状态,例如肾小球肾炎、糖尿病性视网膜病变、狼疮性肾炎、高血压诱发的肾脏病变、肾间质纤维化、药源性并发症引起的肾纤维化、与免疫缺陷综合征(HIV)相关的肾脏病变、移植性肾脏病变、任意病变引起的肝纤维化、肝炎引起的肝功能障碍、酒精性肝炎、胆道疾病、囊性纤维化、肺纤维化、间质性肺疾病、急性肺损伤、成人呼吸窘迫综合征、特发性肺纤维化、慢性阻塞性肺疾病、感染因子或者毒性因子引起的肺疾病、心肌梗死后心脏纤维化、充血性心力衰竭、扩张型心肌病、心肌炎、血管内膜增厚、血管狭窄、高血压诱发的血管再生、肺动脉高压、冠状动脉再狭窄、末梢血管再狭窄、颈动脉再狭窄、血管支架诱发的再狭窄、动脉粥样硬化症、眼部瘢痕、角膜瘢痕、增生性玻璃体视网膜病变、青光眼、眼内压、因外伤或者手术生成的上后在治愈过程中发生的皮肤中形成的过度的或者肥厚性的瘢痕或者瘢痕疙瘩、覆膜及皮下狭窄、硬皮症、纤维硬化症、进行性全身硬化病、皮肌炎、多发性肌炎、关节炎、骨质疏松症、溃疡、神经功能损伤、男性阳痿、佩罗尼氏病、杜普伊特伦挛缩、阿尔茨海默病、雷诺综合征、放射线诱发的纤维化、血栓病、肿瘤转移性生长、多发性骨髓瘤、黑色素瘤、神经胶质瘤、胶质母细胞瘤、白血病、肉瘤、平滑肌瘤、间皮瘤以及肺、乳腺、结肠、肾脏、卵巢、颈部、肝、胆道、胃肠道、胰脏、前列腺、头颈部的肿瘤的预防、治疗或者改善效果,同时,对骨髓增生异常综合征(myelodysplasticsyndrome)或者淋巴瘤(lymphoma)具有预防、治疗或者改善效果。并且,本发明的组合物可以为以下消化道,例如结肠为目标器官的药物组合物。例如,本发明的组合物可以为用于预防、改善或者治疗炎症性肠疾病的组合物。例如,本发明的组合物可以为用于预防、改善或者治疗克罗恩氏病(Crohn'sdisease)或者溃疡性结肠炎(ulcerativecolitis)的组合物。<材料及方法>从韩国(株)MedPacto公司获得化学式1的化合物(以下称“vactosertib”)。从韩国地质资源研究院获得膨润土来使用。<实验例1><1-1>评估vactosertib与膨润土根据溶剂pH值的吸附率。在此情况下,考虑到vactosertib在中性环境下不易溶解,而从pH3的条件下以约1mg/mL的溶解度开始溶解,因此,将反应pH值选定为pH1、pH2以及pH3进行试验。首先,将vactosertib以10mg/mL的浓度溶解于0.1N的盐酸水溶液,将其稀释于与各pH值相对应的盐酸水溶液或者磷酸盐缓冲液(pH1:0.1N的盐酸水溶液,pH2、pH3:磷酸盐缓冲液)来将浓度配制为1mg/mL。将1mL的相关药物溶液、8mL的与各pH值相对应的反应溶液(即,0.1N的盐酸水溶液或者磷酸盐缓冲液)以及1mL的将膨润土悬浮于蒸馏水中配制为5mg/mL的悬浮液混合后放置30分钟来使药物吸附于膨润土。然后,对通过离心分离(3,000rpm,5分钟)获得的上清液的药物浓度进行分析。通过此种方式,在使用的总药物(vactosertib)中减去留在上清液中的药物量,通过数学式1间接地计算吸附于膨润土的药物量来计算吸附率。[数学式1]其结果,组间的差异虽不显著,但确认随着pH值的降低,吸附率有些许的增加,在pH1的情况下,vactosertib的吸附效率最高(表1)。因此,在以下试验中,制备vactosertib及膨润土的复合物时,将溶剂的pH值设定为pH1来进行试验。[表1]上述浓度以最终浓度为基准。<1-2>评估根据vactosertib与膨润土的比例的吸附率。这是为了寻找在使vactosertib的损失最小化的情况下最大限度地吸附于膨润土的组成。为此,通过与实验例1相同的方式准备试样并制备,将膨润土的反应浓度与反应pH值分别固定在0.5mg/mL、pH1,而使用的vactosertib的浓度配制为从0.01mg/mL到0.5mg/mL的不同浓度来制备复合物。然后检测吸附率根据vactosertib浓度的差异。其结果,在vactosertib的浓度低的情况下,虽然大部分药物都保持了结合在膨润土的状态,但吸附率从0.1mg/mL的vactosertib的浓度开始减少,在这个浓度以上的浓度中,药物吸附率为60%以下,非常低(表2)。因此,将在保证足够高的吸附率的同时保证复合物中vactosertib的含量也高的组成,即,相对于0.5mg/ml的膨润土浓度,vactosertib的浓度为0.025mg/ml至0.1mg/ml的比例选定为最佳的膨润土/vactosertib的比例。[表2]<1-3>在反应pH值及膨润土/vactosertib的比例固定的状态下,评估吸附率根据使用的vactosertib及膨润土的绝对浓度变化的变化。为此,通过与实验例1相同的方式准备试样并制备,利用pH1的溶剂,将vactosertib及膨润土的浓度比配制为1:5,绝对浓度(即,实际浓度)配制为不同来制备复合物。并检测吸附率根据绝对浓度变化的差异。其结果,随着复合物中使用的vactosertib及膨润土的绝对浓度增加到10倍,药物吸附率增加了,据此确认可以减少药物的损失(表3)。[表3]<实验例2><2-1>利用pH1的溶剂,在1mg/ml的vactosertib的浓度5mg/ml的膨润土的浓度的条件下,使用实验例1的方法制备膨润土/vactosertib复合物。然后,将其冷冻干燥后,为了检测vactosertib的实际含量,进行了vactosertib的提取及分析。将上述冷冻干燥的复合物分散于包含0.5%的吐温80的磷酸盐缓冲生理盐水(phosphatebufferedsaline,pH7.4)来提取vactosertib的结果,计算的膨润土/vactosertib复合物的反应重量百分比为与现有的vactosertib及膨润土的反应重量百分比近似的约16.9±0.2%。反应重量百分比通过下述数学式2计算。[数学式2]<2-2>评估上述<2-1>中制备的膨润土/vactosertib复合物的形态学特性。为此,将在<2-1>中制备的膨润土/vactosertib复合物与vactosertib分别粘附于铜带来在表面涂敷铂后,使用扫描电子显微镜(scanningelectronmicroscopy,SEM)拍照。其结果,观察到vactosertib粉末具有1μm以上大小的特有的结晶形状(图1)。然而,膨润土/vactosertib复合物确没有表现出这样的结晶形状,而是表现出膨润土的层状结构不规则地聚集在一起的形态(图2)。<2-3>通过XRD分析了膨润土/vactosertib复合物的结晶学特性。通过分析、比较vactosertib、膨润土、vactosertib与膨润土的物理混合物以及膨润土/vactosertib复合物的XRD图案来确认根据药物吸附现象的结晶结构变化。其结果,在vactosertib的情况下,具有固有的XRD曲线,这在物理混合物中也有相似的表现。然而,膨润土/vactosertib复合物的情况下,未表现出固有的波峰,确认到表现出与现有的膨润土的图案非常相似的图案(图3)。图3以曲线图示出各物质的XRD图案。在vactosertib的情况下,具有固有的XRD波峰图案,这在物理混合物中也有相似的表现。然而,膨润土/vactosertib复合物的情况下,未表现出固有的波峰,确认到表现出与现有的膨润土的图案非常相似的图案。<实验例3><3-1>为了确认vactosertib吸附于膨润土后释放的变化,进行了vactosertib的溶出试验。在此情况下,为了预测在上消化道及下消化道中的药物释放,在不同的pH值条件下进行溶出。为此,分别将0.1N的HCl溶液(pH1.2)、50mM的醋酸盐缓冲液(pH4.5)、50mM的磷酸盐缓冲液(pH7.4)用作溶出液来进行试验。在此情况下,为了赋予充分溶出vactosertib的溶出环境,在所有溶出液中以0.1%的浓度溶解吐温80并使用。以使vactosertib浓度为基准配制为1mg/mL的方式将vactosertib粉末及膨润土/vactosertib复合物制备为分散液,将其中的0.3mL密封于半透膜后浸在30mL的各溶出液中进行溶出。然后,在各规定的不同时间收集0.2mL的溶出液,通过分析药物浓度来比较溶出速度。其结果,vactosertib粉末在pH1.2下迅速溶出,而在更高的pH值中,溶出速度显著变慢。相反,膨润土/vactosertib复合物在pH1.2及pH4.5下都表现出比现有的vactosertib粉末更低的溶出速度,但在pH7.4下表现出更高的溶出速度(图4:pH1.2,图5:pH4.5,图6:pH7.4)。因此,确认到在将能够在上消化道中迅速溶出的vactosertib吸附于膨润土的情况下,可以降低vactosertib的溶出及吸收来使vactosertib在具有更高pH值的下消化道中溶出及吸收。<3-2>为了确认实际口服给药膨润土/vactosertib复合物时血药浓度的变化,利用约7周龄的SD大鼠进行了药物动态分析试验。将现有的vactosertib粉末以及将其溶解于pH3的缓冲液中的水溶液,还有将膨润土/vactosertib复合物用作试样来进行试验,以药物(即,vactosertib)量为基准将剂量设定为10mg/kg。向大鼠口服给药各试样后在不同时间点采血,对其进行前处理来分析血药浓度。其结果,与现有的vactosertib粉末相比,在以水溶液的形态采用时,作为药物的vactosertib的吸收变高,在15分钟的时候就到达药峰浓度。另一方面,在将vactosertib吸附于膨润土的膨润土/vactosertib复合物的情况下,与vactosertib水溶液相比,不但药峰浓度显著降低,而且到达药峰浓度的时间更慢,后半部的血药浓度比水溶液更高。并且,从整体上来看,与vactosertib粉末组相比,膨润土/vactosertib复合物组的血药浓度曲线下面积(AUC)更大。由此确认,与现有的vactosertib粉末相比,膨润土/vactosertib复合物表现出更为优秀的吸收,与急速吸收的vactosertib的水溶液形态相比,可以减慢药物(即,vactosertib)的吸收(图7)。基于上述图7的结果分析了各组的体内动态学特性。其结果,将vactosertib以粉末形态采用的组与其他组相比,血药浓度曲线下面积显著低,与各参数的平均值相比,偏差值比其他组高。在将vactosertib以水溶液的形态采用的情况下,表现出最高的Cmax(药峰浓度)和最快的Tmax(血药浓度达峰时间),这表示在溶液的状态下,药物的吸收很快。另一方面,在采用膨润土/vactosertib复合物的情况下,与vactosertib水溶液相比,虽然Cmax更低,但Tmax更慢。即,口服给药膨润土/vactosertib复合物时,血药浓度达峰时间(Tmax)为45±18.37分钟,比口服给药vactosertib水溶液时的血药浓度达峰时间(Tmax)15±0分钟长约3倍。并且,口服给药膨润土/vactosertib复合物时,药峰浓度(Cmax)为1.12±0.37μg/mL,仅为口服给药vactosertib水溶液时药峰浓度(Cmax)2.77±0.55μg/mL的约40.4%。由此确认可以通过将vactosertib吸附于膨润土来延缓vactosertib的吸收。与vactosertib粉末相比,膨润土/vactosertib复合物的药物动态血压参数的偏差值也低,可知膨润土/vactosertib复合物与vactosertib相比更为均匀地吸收(表4)。[表4]<实验例4>以10mg/mL的浓度将vactosertib溶于0.1N的盐酸水溶液,使用pH1的0.1N的盐酸水溶液将其稀释至浓度为1mg/mL。将1mL的相应药物(即,vactosertib)溶液、8mL的0.1N的盐酸水溶液以及1mL的将膨润土悬浮于蒸馏水中配制为5mg/mL的悬浮液混合后放置30分钟来使药物吸附于膨润土。然后,进行离心分离(3,000rpm,5分钟)获得上清液,将其冷冻干燥来制备膨润土/vactosertib复合物。<实验例5><5-1>通过掌握在由DSS(硫酸葡聚糖钠(dextransulfatesodium))诱导溃疡性结肠炎的大鼠模型中的vactosertib的药物动态来评估vactosertib剂型根据是否与膨润土结合的生物药剂学差异。首先,将vactosertib溶于0.1N的盐酸水溶液使浓度为10mg/mL,然后再使用pH3.0的磷酸盐缓冲液稀释,配制成浓度为1mg/mL的溶液来使用。将vactosertib-膨润土复合物以5.92mg/mL悬浮于蒸馏水,使其药物浓度与vactosertib水溶液相同来使用。若使用Bathsonicator对所有制剂进行超声波处理,则更好地分散,分散后易于沉淀,因此要在给药前充分摇晃混匀后给药。利用高效液相色谱HPLC对于制备的膨润土/vactosertib复合物的吸附量、药物释放等体外(Invitro)实验试样进行分析。而对相当于低浓度的血浆分析等活体试样的分析则利用了液相色谱质谱联用测定(LC/MS/MS)。将各血浆试样与乙腈按照一定比例混合来脱蛋白,离心分离后只取上清液进行分析。将SD大鼠用作试验动物。上述SD大鼠是从韩国(株)OrientBio公司的加平中心(韩国京畿道加平郡北面牧童里699-13)获得的,购入时约6周龄。购入后进行约3天的检疫及驯化后,只将健康的动物用于试验。给药试验物质时约为8周龄,使用了40只。试验动物是在仁荷大学医学研究所生命科学研究室二楼的清洁动物室中饲育的,以最少10次/小时的频率换气,在温度为22±4℃、相对湿度为50±20%的条件及12小时点灯(照明:08:00~20:00)、照度为150Lux~300Lux的条件下饲育。使用啮齿类用放射线照射固体饲料(PMI5053,美国),利用饲喂器使实验动物自由摄取,将自来水进行逆渗透压处理后,使用紫外线水消毒器及高压蒸汽灭菌后,利用聚砜材质的饮水瓶(500ml)供实验动物自由摄取。为了诱导溃疡性结肠炎,将5.5%的硫酸葡聚糖钠掺入大鼠的饮水中持续供应6天。试验物质是在过夜(Overnight)(12小时)禁食后口服给药1次。采血期间也是禁食的。试验组如下述表5所示。在此情况下,G1是从第一天(DAY1)到第六天(DAY6)供应普通饮食,过夜禁食后在第七天(DAY7)单次口服给药vactosertib。G2是从第一天到第六天供应普通饮食,过夜禁食后在第七天单次口服给药膨润土/vactosertib复合物。G3是从第一天到第六天使用5.5%硫酸葡聚糖钠诱导炎症后,过夜禁食后在第七天单次口服给药vactosertib。G4是从第一天到第六天使用5.5%硫酸葡聚糖钠诱导炎症后,过夜禁食后在第七天单次口服给药膨润土/vactosertib复合物。上述给药是将试验物质放入注射器后连接大鼠用饲管注入进行口服给药。然后按照组别给药vactosertib或者膨润土/vactosertib复合物1次(图8)。[表5]()内表示可以最终评估的动物数量临床症状观察是在试验期间对不同个体一天一次进行一般症状(是否有血便、腹泻等)以及是否死亡/濒死的观察。在此情况下,只以出现特异症状的个体为限保持记录(正常的情况下不记录)。并且,试验动物的购入、分组以及给药试验物质之前每天都测量体重。试验动物的购入、分组以及给药试验物质之前测量各组的平均体重的结果,未在各组间观察到统计学上的显著差异。而在解剖检查日(第七天)测量的G3组及G4组(硫酸葡聚糖钠诱发组)的平均体重显出与G1组及G2组(蒸馏水诱发组)相比在统计学上的显著减少。(p<0.05,表6)。[表6]试验期间的体重值数据以平均±标准偏差来表示,利用SPSSstatistic19(曼-惠特尼检验)进行统计分析。*p<0.05,与G1组相比具有显著差异#p<0.05,与G2组相比具有显著差异G1组:n=10,G2组:n=9,G3组:n=9,G4组:n=10。<5-2>检测vactosertib的血药浓度随时间的变化后评估了药物在体内的动态。如图9所示(○表示G1组,◇表示G2组,●表示G3组,◆表示G4组),评估了作为药物成分在体内吸收量指标的血药浓度的曲线下面积(AUC,浓度-时间曲线下面积(areaundertheconcentration-timecurve))、作为表示吸收速度有多快的指标的血药浓度达峰时间(tmax)以及药峰浓度(Cmax,最大血药浓度(maximumplasmaconcentration))。其结果确认到与G3组相比,G4组的血药浓度达峰时间tmax增加,药峰浓度Cmax减少。并且,确认到与G3组相比,G4组到达药峰浓度Cmax后,时间-浓度曲线后半部分长时间保持更高的药物(即,vactosertib)的血药浓度(图9)。具体地,在血药浓度达峰时间(tmax)的情况下,在给药膨润土/vactosertib复合物的大部分动物中,血药浓度达峰时间延长约2倍至4倍(图10,○表示G1组,◇表示G2组,●表示G3组,◆表示G4组)。这意味着在胃肠道中实现药物缓慢释放来延长药物的吸收时间,从而增大药物效果的作用时间。另一方面,在药峰浓度(Cmax)的情况下,在给药膨润土/vactosertib复合物的组中确认到药峰浓度降低至一半以下(图11,○表示G1组,◇表示G2组,●表示G3组,◆表示G4组)。因此,通过使vactosertib与膨润土结合来防止急剧的血药浓度的增加并减少因给药药物引起危害反应的危险。并且,在膨润土/vactosertib复合物给药组中,血药浓度达峰时间(tmax)变长,血药浓度缓慢减少,确认可以减少药物给药频率的优点。在血药浓度-时间曲线下面积(AUC)的情况下,vactosertib与膨润土的结合未给vactosertib的血药浓度-时间曲线下面积带来显著影响(图12,○表示G1组,◇表示G2组,●表示G3组,◆表示G4组)。因此,与膨润土的结合保持血液内总药物流入度(exposure)并延长药效的作用时间,确认即使vactosertib与膨润土结合,流入体内的vactosertib的量也未表现出显著差异。在体内药物清除半衰期(t1/2)的情况下,vactosertib与膨润土的结合未给半衰期带来显著影响(图13,○表示G1组,◇表示G2组,●表示G3组,◆表示G4组)。因此,确认这与药物的缓慢吸收无关,仍可保持相似的药物在体内消失的速度。<实验例6>利用作为炎症性肠疾病之一的溃疡性结肠炎的小鼠模型进行vactosertib/膨润土复合物的抗炎症、抗隐窝细胞损伤、抗溃疡、抗浮肿、抗纤维化效果的动物试验。用于进行动物实验的炎症诱导方法及药物给药方案如图14所示。选定C57BL/6小鼠作为试验动物,为了诱导溃疡性结肠炎,反复如下周期:将作为炎症诱导物质的硫酸葡聚糖钠掺入食物中后供应7天,然后在供应14天的正常饮食。反复3次这样的炎症诱导周期后,诱导结肠内的慢性炎症、溃疡以及纤维化状态。从第一个炎症诱导周期结束后过了3周开始,以一天两次(上午10点、下午4点)、每周5天共给药6周的方式向除对照组以外的试验动物组给药膨润土、vactosertib或者vactosertib/膨润土复合物。换算的剂量以每公斤小鼠体重为基准,膨润土为106.8mg,vactosertib为20.0mg,复合物为126.8mg。在此情况下,将实验例5中制备的复合物用作vactosertib/膨润土复合物。在溃疡性结肠炎动物试验中使用的不同试验组的炎症诱导物质、试验物质、试验物质剂量及试验动物数量如下述表7所示。[表7]()内表示可以最终评估的动物数量试验中使用的C57BL/6小鼠是从韩国(株)OrientBio公司的加平中心(韩国京畿道加平郡北面牧童里699-13)获得的,购入时约6周龄。购入后进行约7天的检疫及驯化后,只将健康的动物用于试验。给药诱导物质时试验动物约为8周龄,使用了52只。试验动物是在仁荷大学医学研究所生命科学研究室二楼的清洁动物室中饲育的,以最少10次/小时的频率换气,在温度为22±4℃、相对湿度为50±20%的条件及12小时点灯(照明:08:00~20:00)、照度为150Lux~300Lux的条件下饲育。使用啮齿类用放射线照射固体饲料(FeedLabGMOFreeRodentDietGF2005),利用饲喂器使实验动物自由摄取,将自来水进行逆渗透压处理后,使用紫外线水消毒器及高压蒸汽灭菌后,利用聚砜材质的饮水瓶(300ml)供实验动物自由摄取。临床症状观察是在试验期间对不同个体一天一次进行一般症状(是否有血便、腹泻等)以及是否死亡/濒死的观察。在此情况下,只以出现特异症状的个体为限保持记录(正常的情况下不记录)。并且,试验动物的购入、分组以及给药试验物质之前每天都测量体重。为了观察结肠组织的免疫组织化学变化,将在10%的中性缓冲福尔马林溶液中固定的组织制备为薄切片后,附着于涂片(Coatingslide)并脱石蜡后进行染色,将抗体白细胞介素6(IL-6,Interleukin-6)、α-平滑肌肌动蛋白(α-SMA,α-smoothmuscleactin)、转化生长因子-β(TGF-β,transforminggrowthfactorbeta)分别在各切片上反应的免疫组织化学(IHC)则委托外部机构(Knotus公司)进行。不同个体的组织切片指定规定的三个区域并放大100倍来拍照,测量相比于总面积的染色部分后计算平均值(Zen2.3blueedtion,CarlZeiss公司,德国)。为了诱导溃疡性结肠炎,将2%的硫酸葡聚糖钠掺入小鼠的饮水中在每个实验周期(3周)的前7天持续供应。试验物质则同第二个硫酸葡聚糖钠诱发一同,以一天2次、每周5天的方式共口服给药6周(图14)。试验物质的剂量根据体重,在不同个体中进行换算,以每公斤体重20mL的药量给药。上述给药是将试验物质放入注射器后连接大鼠用饲管注入进行口服给药。在试验期间,所有硫酸葡聚糖钠DSS摄取组在7天后都出现以显著的体重减少及腹泻为特征的溃疡性结肠炎疾病活动指数(diseaseactivityindex,DAI)明显增加。14天的恢复期后,再开始下一个硫酸葡聚糖钠诱导周期之前,疾病活动指数恢复至接近正常。试验物质给药组之间未观察到显著的疾病活动指数的显著差异。在结肠重量、长度及结肠比例的变化中,在所有的硫酸葡聚糖钠摄取组中均观察到结肠重量增加、结肠长度缩短及结肠比例的增加。在试验物质给药组中未观察到因摄取硫酸葡聚糖钠引起的上述症状的显著差异。为了评估试验物质对溃疡性结肠炎的治疗效果,在试验结束后,将动物麻醉后并开腹放血后,对不同个体进行组织病理检查。组织病理学所见如下述表8所示,细分为炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化的严重程度变化并评分。并且,将除纤维化以外的上述四种组织病理学严重程度分数相加后,将阳性对照组(G2)与试验物质给药组(G3、G4、G4)进行比较。[表8]利用GraphpadPrism8.1版本进行统计学分析,以p<0.05为显著性的最小单位。测定的所有组织病理指数为非参数数据,在曲线图中以中间值、25-75%值(方框内)、最小值-最大值(两个极端)来表示(图15至图20)。利用作为非参数检验法的克鲁斯卡尔-沃利斯检验进行方差分析(ANOVA),利用邓恩检验法检验阳性对照组(▲表示G2组)与试验物质给药组(◇表示G3组,●表示G4组,◆表示G5组)之间的统计学显著性。并且,其平均值如表9所示。[表9]测量IL-6染色的发色面积并计算平均值及标准差的结果,相比于G1组,G2组观察到增加15.8倍的面积,示出统计学显著性(p<0.01)。相比于G2组,在试验物质给药组(G3组至G5组)中分别表现出分别减少8.7%、23.0%以及56.3%的面积,尤其,G5组与G2组相比表现出显著的减少(表10、图21、图22)。[表10]试验组白细胞介素6(面积%)G10.8±0.3G212.6±4.2**G311.5±1.9G49.7±2.9G55.5±1.5##。权利要求书(按照条约第19条的修改)1.(修改后)一种控释的用于口服给药的组合物,其特征在于,包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物,调节上述组合物的吸收速度,[化学式1]2.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,口服给药上述组合物时的血药浓度达峰时间(tmax)比口服给药化学式1的化合物水溶液时的血药浓度达峰时间(tmax)长2倍至10倍。3.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,口服给药上述组合物时的血药浓度达峰时间(tmax)长于口服给药化学式1的化合物水溶液时的血药浓度达峰时间(tmax)。4.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,口服给药上述组合物时的药峰浓度(Cmax)为口服给药化学式1的化合物水溶液时的药峰浓度(Cmax)的20%至80%。5.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,口服给药上述组合物时的药峰浓度(Cmax)低于口服给药化学式1的化合物水溶液时的药峰浓度(Cmax)。6.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,口服给药上述组合物时的化学式1的化合物在结肠中对选自由炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化组成的组中的症状的药物改善效果优于口服给药化学式1的化合物水溶液时在结肠中的药物改善效果。7.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,口服给药上述组合物时的化学式1的化合物在小肠中的吸收速度慢于口服给药化学式1的化合物水溶液时在小肠中的吸收速度。8.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,将上述复合物放入pH1.2的溶出液中时的溶出速度慢于将化学式1的化合物放入pH1.2的溶出液时的溶出速度。9.根据权利要求1所述的控释的用于口服给药的组合物,其特征在于,将上述复合物放入pH7.4的溶出液中时的溶出速度快于将化学式1的化合物放入pH7.4的溶出液时的溶出速度。10.(修改后)一种控释的用于口服给药的组合物的制备方法,其特征在于,包括混合包含化学式1的化合物或者其药学上可接受的盐的溶液、酸性反应溶液以及粘土矿物悬浮液并将上述化合物吸附于粘土矿物来制备复合物的步骤,上述控释的用于口服给药的组合物包含化学式1的化合物及粘土矿物的复合物,调节上述组合物的吸收速度,[化学式1]11.根据权利要求10所述的控释的用于口服给药的组合物的制备方法,其特征在于,上述反应溶液的pH值为pH0.5至pH3.5。12.(修改后)一种控释方法,其特征在于,向对象口服给药组合物,上述组合物包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物,调节上述组合物的吸收速度,[化学式1]13.根据权利要求12所述的控释方法,其特征在于,口服给药上述组合物时的血药浓度达峰时间(tmax)比口服给药化学式1的化合物水溶液时的血药浓度达峰时间(tmax)长2倍至10倍。14.根据权利要求12所述的控释方法,其特征在于,口服给药上述组合物时的血药浓度达峰时间(tmax)长于口服给药化学式1的化合物水溶液时的血药浓度达峰时间(tmax)。15.根据权利要求12所述的控释方法,其特征在于,口服给药上述组合物时的药峰浓度(Cmax)为口服给药化学式1的化合物水溶液时的药峰浓度(Cmax)的20%至80%。16.根据权利要求12所述的控释方法,其特征在于,口服给药上述组合物时的药峰浓度(Cmax)低于口服给药化学式1的化合物水溶液时的药峰浓度(Cmax)。17.根据权利要求12所述的控释方法,其特征在于,口服给药上述组合物时的化学式1的化合物在结肠中对选自由炎症、隐窝细胞损伤、溃疡、浮肿以及纤维化组成的组中的症状的药物改善效果优于口服给药化学式1的化合物水溶液时在结肠中的药物改善效果。18.根据权利要求12所述的控释方法,其特征在于,口服给药上述组合物时的化学式1的化合物在小肠中的吸收速度慢于口服给药化学式1的化合物水溶液时在小肠中的吸收速度。19.根据权利要求12所述的控释方法,其特征在于,将上述复合物放入pH1.2的溶出液中时的溶出速度慢于将化学式1的化合物放入pH1.2的溶出液时的溶出速度。20.根据权利要求12所述的控释方法,其特征在于,将上述复合物放入pH7.4的溶出液中时的溶出速度快于将化学式1的化合物放入pH7.4的溶出液时的溶出速度。说明或声明(按照条约第19条的修改)条约第19条(1)规定的说明1.修改的权利要求修改权利要求1、10、12,使其更加明确。2.修改内容将权利要求1中记载的“一种控释的用于口服给药的组合物,其特征在于,包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物”修改为“一种控释的用于口服给药的组合物,其特征在于,包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物,调节上述组合物的吸收速度”,将权利要求10中记载的“一种控释的用于口服给药的组合物的制备方法,其特征在于,包括混合包含化学式1的化合物或者其药学上可接受的盐的溶液、酸性反应溶液以及粘土矿物悬浮液并将上述化合物吸附于粘土矿物来制备复合物的步骤”修改为“一种控释的用于口服给药的组合物的制备方法,其特征在于,包括混合包含化学式1的化合物或者其药学上可接受的盐的溶液、酸性反应溶液以及粘土矿物悬浮液并将上述化合物吸附于粘土矿物来制备复合物的步骤,调节上述组合物的吸收速度”,将权利要求12中记载的“一种控释方法,其特征在于,向对象口服给药组合物,上述组合物包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物”修改为“一种控释方法,其特征在于,向对象口服给药组合物,上述组合物包含化学式1的化合物或其药学上可接受的盐以及粘土矿物的复合物,调节上述组合物的吸收速度”,使上述权利要求1、10、12更加明确。3.权利要求的修改与说明书和附图的关系上述权利要求的修改没有增加新的内容,只是使表达更加清楚,不影响说明书和附图。当前第1页12
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。