1.本发明涉及病毒学和生物技术领域,涉及一种利用反向遗传学技术的圆圈病毒3型双拷贝全长基因感染性克隆及其构建方法和应用。
背景技术:
2.圆圈病毒3型(gyrovirus 3,gyv3)是继鸡传染性贫血病毒(chicken infectious anaemiavirus,cav或ciav)之后的第三个圆圈病毒属成员,分类于编码复制相关蛋白的单链环状dna病毒门(circular replication
‑
associated protein
‑
encoding single
‑
stranded dnaviruses,cress dna viruses)的指环病毒科(anelloviridae)。gyv3自2012年在智利儿童未知病因急性肠胃炎腹泻粪便中通过宏病毒组学鉴定命名以来,先后在人类不同年龄腹泻粪便、雪貂腹泻粪便和市场鸡肉中被发现。由于其发现样品的广泛性和复杂性,gyv3与疾病的相关性和宿主范围引起医学和兽医学界的共同关注。2018年,发明人在临床患有未知病因腺胃炎的白羽肉鸡腺胃中鉴定到gyv3,并在回顾性流行病学调查中发现其在2013
‑
2017年收集的肉鸡腺胃炎病料中有12.5%(42/336)的感染率,且在之后的致病性实验中明确其可感染鸡和小鼠,同时实现两群体间的水平传播。由此可见gyv3与肉鸡腺胃炎有很大的相关性,而且其跨物种传播现象提示潜在的公共卫生意义,其深入研究亟待进行,但由于缺乏成熟的分离纯化技术和有效的体外培养体系,gyv3确切的致病性和研究受到严重阻碍。
3.gyv3基因结构为单链环状的负义dna,大小为2.3kb,包含3个同向重叠的开放阅读框 (orf)和一个非编码区(utr)。3个orfs分别编码病毒的3个蛋白,即衣壳蛋白vp1,支架蛋白vp2和凋亡蛋白vp3,这些蛋白质通过交替使用起始密码子从单个多顺反子mrna翻译而来,但是密码子交替使用的真实机制还没有被阐明。圆圈病毒的复制方式尚未得到所有科学家的公认,至今缺乏明确结论。目前主流观点认为其dna采取滚环复制(rcr)的方式进行复制,但是已经明确的是病毒缺乏复制自身dna的机制,依赖于分裂宿主细胞进行dna复制,在此过程中经历一个双链dna复制形式(dsrf),这为gyv3全基因组的普通pcr扩增和感染性克隆构建奠定分子基础。
4.感染性克隆作为病毒反向遗传学研究中一种重要的研究工具,广泛应用于rna病毒及其他基因类型病毒的拯救以及病毒基因结构与功能、病毒致病性、病毒宿主相互作用和疫苗研制等基础和应用研究中。感染性克隆是指人工构建的可实现体外和体内感染并装配出感染性病毒粒子的遗传物质。gyv3感染性克隆构建面临的最大难点是全基因组的扩增,因为病毒utr 区5
′
端含有一段gc高含量区,给病毒全基因的测序和扩增造成很大困难,至今圆圈病毒属的13个成员只有首位成员鸡传染性贫血病毒(ciav)完成了感染性克隆的构建,并实现体内外的病毒拯救,而其他12种病毒均没有构建感染性克隆,也没有明确的致病性研究。如何填补这一空白成为本领域亟待解决的问题。
技术实现要素:
5.本发明的发明人针对现有技术存在的空白之处,首次利用具体利用gyv3的环状基因组结构特点和自带的酶切位点,使用高保真酶扩增到两段不同起止点的病毒全基因组,通过两次连接转化连接于真核表达pcdna3.1( )构建gyv3双拷贝全长感染性克隆pcdna3.1
‑
2gyv3,并且实现spf鸡体内的病毒拯救和致病性研究;最终提供了一种简单、快捷的圆圈病毒反向遗传学操作系统,为gyv3致病性及基因结构与功能研究等方面提供平台,也为研究针对gyv3 的基因工程疫苗奠定了基础。
6.1991年,noteborn和claessens等人先后分别在ciav感染的lscc
‑
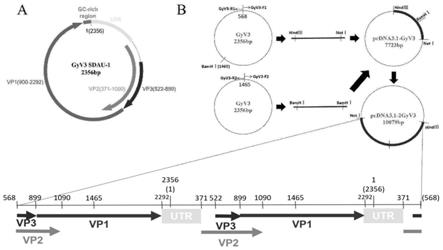
1104
‑
x
‑
5和 mdcc
‑
msb1中分离到病毒的双链复制形式(dsrf)dna并克隆到真核表达载体pic20h和 pgem
‑
7zf( )上,然后转染mdcc
‑
msb1细胞,观察到细胞病变,传代之后,利用病毒中和实验检测到上清中存在传染性病毒粒子。noteborn等人继续用转染mdcc
‑
msb1细胞上清肌肉接种1日龄spf鸡,复制出与野毒株相同的临床症状和组织病变。2000年,brown等人利用重叠pcr扩增得到一株ciav澳大利亚分离株的全基因组并连接到载体pgem
‑
4z中,后将重组的环形载体以及酶切线性化的全基因组转染mdcc
‑
msb1细胞,通过间接免疫荧光技术(ifa)和转染细胞上清接种新细胞观察细胞病变证明感性克隆构建成功。2011年,李秀梅利用普通pcr 扩增ciav全基因组,经过摸索最佳不完全酶切条件通过两次连接转化将2个全长基因组顺向连接到pbluescriptπsk( )质粒载体中,最后通过ifa体外验证和感染性克隆质粒肌肉接种1日龄spf鸡复制疾病验证感染性克隆构建成功。2017年,kaffashi等人利用普通pcr扩增得到一株ciav以色列分离株的全基因组并连接到载体ptz57r/t中,后将酶切线性化的全基因组转染mdcc
‑
msb1细胞,通过观察细胞病变和细胞上清接种1日龄spf鸡复制疾病证明成功构建ciav感性克隆。此外猪圆环病毒(pcv)也有双拷贝感染性克隆的构建先例,但由于pcv无gc高含量区,pcv全基因组的获取与双拷贝连接较为简单。
7.由此可见,圆圈病毒本身基因结构小型、环状的特点以及复制形成dsrf的特点有利于感染性克隆的构建。本发明双拷贝感染性克隆的构建利用了病毒自带的酶切位点,避免了不完全酶切条件的摸索,提高了构建效率。
8.本发明的具体技术方案是:
9.发明人首先提供了一株gyv3病毒毒株,命名为gyv3 sdau
‑
2株,发明人对其进行了生物保藏,生物保藏编号为cctcc no:v202061;
10.进一步的,发明人提供了一种gyv3双拷贝全基因组感染性克隆质粒,该克隆质粒由真核表达载体pcdna3.1( )和gyv3双拷贝全基因组组成,命名为pcdna3.1
‑
2gyv3,其中双拷贝全基因组感染性克隆的核苷酸序列如seq id no.1所示;
11.发明人进一步提供了上述gyv3双拷贝全基因组感染性克隆质粒的构建方法,具体步骤如下:
12.利用病毒环状基因组特点,人工从第568个核苷酸设计引物,上下游引物5
′
端分别添加hindⅲ和notⅰ酶切位点和对应保护碱基,将gc高含量区放在扩增片段中间区域,利用高保真酶扩增出线性全基因组hind
ⅲ‑
gyv3
‑
notⅰ,连接于pcdna3.1( )构建gyv3单拷贝重组质粒pcdna3.1
‑
gyv3;从病毒基因组自带bamhⅰ酶切位点处(第1465个核苷酸)人工设计第二对引物,上游引物直接在自带的bamhⅰ酶切位点5
′
端添加保护碱基,下游引物人工添加 bamhⅰ酶切位点和保护碱基,扩增出线性全基因组bamh
ⅰ‑
gyv3
‑
bamhⅰ,通过第二次连接转
化,连接于上述获得的pcdna3.1
‑
gyv3,最终合成gyv3双拷贝全长基因感染性克隆 pcdna3.1
‑
2gyv3;上述方案根据gyv3的特性重新选取了酶切位点和引物的位置,并重新设计了引物序列,与现有常规技术具有显著不同;
13.除此之外,发明人还提供了上述gyv3双拷贝全基因组感染性克隆质粒的应用,具体提供了一种体外验证感染性克隆的方法,包括以下步骤:mdcc
‑
msb1细胞的培养和原代鸡胚肾小管上皮细胞的制备;感染性克隆的转染;间接免疫荧光检测感染性克隆转录表达情况;更加具体的是利用上述构建的gyv3双拷贝全长基因感染性克隆,对6胚龄卵黄囊、1日龄肌肉和腹腔接种spf鸡,进行体内病毒拯救,7、14、21、28dpi检测接种鸡病毒血症和组织病毒载量,验证是否有感染性病毒拯救成功,进而建立gyv3单纯感染模型。同时以同样方式接种gyv3组织分离毒(gyv3 sdau
‑
2株,保藏号为cctcc no:v202061)作为阳性对照和pbs作为阴性对照。
14.上述获得的质粒可用于gyv3体内的病毒拯救及致病性研究,且所涉及的质粒载体等原材料均廉价易得,大大缩短感染性克隆构建成本和时间。同时本技术的技术方案填补了本领域的空白,对于其他圆圈病毒均可使用本发明的构建策略进行感染性克隆的构建和病毒拯救。
15.保藏信息
16.保藏时间:2020年10月1日
17.保藏单位名称:中国典型培养物保藏中心
18.保藏编号:cctcc no:v202061
19.保藏单位地址:中国武汉武汉大学
20.分类命名:圆圈病毒3型sdau
‑
2株(gyrovirus 3 sdau
‑
2)
附图说明
21.图1为gyv3双拷贝感染性克隆构建原理图;
22.图中a:gyv3 sdau
‑
1基因组结构示意图;b:gyv3双拷贝感染性克隆构建原理图;
23.图2为gyv3全基因组pcr扩增结果示意图;
24.图中phanta max master mix和vazyme lamp master mix为两种高保真酶;f1/r1为第一对全长基因扩增引物,f2/r2为第二对全长基因扩增引物;阴:正常spf鸡血液dna;
25.图3为pcdna3.1
‑
gyv3阳性克隆的筛选结果示意图;
26.图中m:dna marker;1
‑
10:菌落编号;阳:gyv3阳性鸡血液dna;阴:pcdna3.1空载体菌液;
27.图4为pcdna3.1
‑
gyv3质粒双酶切鉴定结果示意图;
28.图中m:dna marker;2/5/9:阳性克隆质粒编号;
29.图5为pcdna3.1
‑
2gyv3阳性克隆的筛选结果示意图;
30.图中m:dna marker;1
‑
8:菌落编号;
31.图6为pcdna3.1
‑
2gyv3质粒单酶切鉴定结果示意图;
32.图中m:dna marker;5/8:阳性克隆质粒编号。
33.图7为mdcc
‑
msb1/鸡原代肾小管上皮细胞转染pcdna3.1
‑
2gyv3细胞病变观察与ifa检测结果示意灰度图;
34.图8为gyv3绝对荧光定量pcr标准曲线;
35.图中a.溶解曲线;b.扩增曲线;c.标准曲线;
36.图9为实验鸡血液病毒血症检测曲线图;
37.图中nc:正常组;gyv3
‑
ic:gyv3感染性克隆组;gyv3:gyv3阳性对照组;
38.图10为实验鸡各器官和粪便病毒载量检测结果柱状图。
具体实施方式
39.下面结合附图和实施例对本发明做进一步详细说明。所述实施例用于解释本发明,而非对本发明的限定。本实施例中除特殊说明外,其他均采用现有技术完成。
40.实施例1 gyv3全基因组的pcr扩增
41.1.引物设计与合成
42.依据构建策略(附图1),根据ncbi genbank数据库的gyv3 sdau
‑
1核酸序列,使用 snapgene viewer软件查询gyv3自带酶切位点,人工设计两对含酶切位点的gyv3全长基因扩增引物。下划线部分为酶切位点,5’端为保护碱基。
43.gyv3全基因扩增引物
[0044][0045][0046]
2.gyv3模板dna的制备
[0047]
使用gyv3
‑
sdau
‑
2株第2次回归spf鸡组织进行gyv3模板dna的制备。
[0048]
使用细胞/组织/血液dna提取试剂盒(天根,北京)提取gyv3感染鸡肾脏dna;
[0049]
3.gyv3全基因组的pcr扩增与纯化回收
[0050]
为了准确扩增gy v3全基因组,尝试两种高保真酶2
×
phanta max master mix和2
×
vazyme lamp master mix(vazyme,南京),以上步所得含gyv3 dna样品为模板进行pcr反应。上样体系和pcr反应程序见下表:
[0051]
pcr加样体系
[0052][0053]
pcr反应程序
[0054][0055]
反应结果经过琼脂糖凝胶电泳分离纯化(附图2)和胶回收(天根,北京)备用。结果显示phanta max master mix成功扩增出病毒的两种全基因组:hind
ⅲ‑
gyv3
‑
notⅰ(seq idno.6)和bamh
ⅰ‑
gyv3
‑
bamhⅰ(seq id no.7)。
[0056]
实施例2单拷贝全长基因重组质粒pcdna3.1
‑
gyv3的构建与鉴定
[0057]
1.pcdna3.1( )和hind
ⅲ‑
gyv3
‑
notⅰ双酶切和连接
[0058]
使用quickcut hindⅲ和quickcut notⅰ(takara)分别对空载体pcdna3.1( )和 hind
ⅲ‑
gyv3
‑
notⅰ进行双酶切,下表双酶切体系加样(空载体和hind
ⅲ‑
gyv3
‑
notⅰ的量不超过1μg),在金属浴锅中37℃反应10min,然后琼脂糖凝胶电泳纯化酶切产物并胶回收待用。在双酶切同一天进行pcdna3.1( )与hind
ⅲ‑
gyv3
‑
notⅰ的连接,下表连接体系加样, 16℃反应30min。
[0059]
双酶切体系
[0060][0061][0062]
连接体系
[0063][0064]
2.pcdna3.1( )和hind
ⅲ‑
gyv3
‑
notⅰ连接产物的转化
[0065]
①
将感受态细胞dh5α(100μl)从-80℃冰箱取出直接置于冰浴中;
[0066]
②
在超净台中向感受态细胞悬液中加入10μl连接产物(注意连接产物体积不要超过感受态细胞悬液体积的十分之一),轻弹混匀,在冰浴中静置30min;
[0067]
③
将离心管置于42℃水浴中放置90s,然后快速将管转移到冰浴中,使细胞冷却2.5min,该过程要避免离心管的移动,不要在有离心机、旋涡震荡工作的台面上进行;
[0068]
④
向离心管中加入900μl无菌的lb液体培养基(不含氨苄青霉素),混匀后置于37℃摇床150rpm振荡培养45min,使pcdna3.1( )质粒上氨苄抗性标记基因表达,使菌体复苏;
[0069]
⑤
在超净台中将离心管内容物混匀,吸取100μl已转化的感受态细胞滴加到含氨苄青霉素的lb固体琼脂培养基平板上,酒精灯火焰灼烧三角玻璃棒灭菌,用三角棒轻轻的将细胞均匀涂开,直到液体渗透,重复此操作直至涂满10个平板。将全部平板倒置于37℃恒温恒湿培养箱培养12
‑
16h,随时观察菌落状态。
[0070]
3.pcdna3.1
‑
gyv3阳性克隆的筛选与酶切鉴定
[0071]
每个平板用小白枪头挑取一个乳白色、孤立和生长较大的菌落,置于10ml含氨苄青霉素的lb液体培养基中,试管置于37℃摇床220rpm振荡培养12
‑
16h,至菌液od
600
达到2.0
‑
3.0 之间,停止培养。直接以各管菌液为模板,以实施例1所述上样体系和pcr反应程序进行pcdna3.1
‑
gyv3阳性克隆的筛选。结果显示在挑取的10个单克隆中,有3个包含gyv3的全基因(附图3)。
[0072]
使用质粒小提试剂盒(天根,北京)提取pcr鉴定阳性的菌液质粒,具体步骤见产品说明书。
[0073]
取提取的质粒pcdna3.1
‑
gyv3进行双酶切鉴定。按实施例2双酶切体系加样(空载体和 hind
ⅲ‑
gyv3
‑
notⅰ的量不超过1μg),在金属浴锅中37℃反应10min,之后琼脂糖凝胶电泳鉴定。琼脂糖凝胶电泳结果显示两条目的大小的片段:pcdna3.1载体和gyv3全长基因 (去保护碱基),证明pcdna3.1
‑
gyv3构建成功(附图4)。
[0074]
实施例3双拷贝全长基因重组质粒pcdna3.1
‑
2gyv3的构建
[0075]
1.pcdna3.1
‑
gyv3和bamh
ⅰ‑
gyv3
‑
bamhⅰ酶切和连接
[0076]
使用quickcut bamhⅰ分别对gyv3单拷贝克隆质粒pcdna3.1
‑
gyv3和 bamh
ⅰ‑
gyv3
‑
bamhⅰ进行酶切,按下表酶切体系加样(pcdna3.1
‑
gyv3和bamh
ⅰ‑
gyv3
‑
bamhⅰ的量不超过1μg),在金属浴锅中37℃反应10min,然后琼脂糖凝胶电泳纯化酶切产物并胶回收待用。在酶切同一天进行pcdna3.1
‑
gyv3与bamh
ⅰ‑
gyv3
‑
bamhⅰ的连接,按下表连接体系加样,16℃反应30min。连接产物-20℃保存备用。
[0077]
酶切体系
[0078][0079]
连接体系
[0080][0081]
2.pcdna3.1
‑
gyv3和bamh
ⅰ‑
gyv3
‑
bamhⅰ连接产物的转化
[0082]
同实施例2步骤2进行pcdna3.1
‑
gyv3和bamh
ⅰ‑
gyv3
‑
bamhⅰ连接产物的转化
[0083]
3.pcdna3.1
‑
2gyv3阳性克隆的筛选与酶切鉴定
[0084]
同实施例步骤3进行pcdna3.1
‑
2gyv3阳性克隆的菌液pcr筛选,使用pcdna3.1( )通用引物(上游)t7
‑
taatacgactcactataggg/(下游)bgh
‑
tagaaggcacagtcgagg,(seq id no.8/9 所示)按下表所示体系和程序进行pcr反应,结果经琼脂糖凝胶电泳鉴定。结果显示在挑取的10个单克隆中(其中2个菌落摇菌扩增没有成功),有2个包含双拷贝gyv3全基因(附图5);
[0085]
菌液pcr加样体系
[0086][0087]
菌液pcr反应程序
[0088][0089]
菌液pcr鉴定阳性的菌液,转移至200ml含氨苄青霉素的新鲜lb液体培养基中,锥形瓶置于37℃摇床220rpm振荡培养12
‑
16h,至菌液od600达到2.0
‑
3.0之间,停止培养,使用无内毒素质粒大提试剂盒(天根,北京)提取菌液质粒,具体步骤见产品说明书。
[0090]
取提取的质粒pcdna3.1
‑
2gyv3进行酶切鉴定。按表8酶切体系加样,在金属浴锅中37℃反应10min,之后琼脂糖凝胶电泳鉴定。琼脂糖凝胶电泳结果显示两条预期目的大小的片段,证明pcdna3.1
‑
2gyv3构建成功(附图6)。
[0091]
实施例4双拷贝全长基因感染性克隆pcdna3.1
‑
2gyv3体外转染验证
[0092]
1.鸡原代肾小管上皮细胞的制备与培养
[0093]
发明人研究显示,gyv3在鸡肾脏肾小管上皮细胞中有强烈的特异性信号定位,因此发明人利用现有技术制作了鸡原代肾小管上皮细胞,对pcdna3.1
‑
2gyv3进行体外转染验证,制作及培养过程如下:
[0094]
(1)在超净台中无菌取18胚龄小鸡,放入一次性无菌平皿中,用无菌眼科剪和镊子摘取小鸡肾脏,在含1%双抗的d’hanks液中尽量剔除肾脏血管、神经等结缔组织膜并剪碎;
[0095]
(2)用d’hanks液冲洗2遍;
[0096]
(3)用1%i型胶原酶(用无血清opti
‑
mem培养基配置)轻轻清洗1遍;转入灭菌 50ml离心管;
[0097]
(4)加入组织约5倍体积量的1%i型胶原酶,37℃水浴锅恒温孵育40min,孵育过程中轻轻震荡锥形瓶;
[0098]
(5)孵育结束后,用5ml的移液枪轻轻吹打30次,吸弃胶原酶;
[0099]
(6)加入一定量的d’hanks液,继续吹打30次;
[0100]
(7)用40μm孔径的一次性细胞滤器过滤,同时用d’hanks液充分冲洗滤器并收集滤液;
[0101]
(8)将滤液悬浮于d’hanks液中,以800rpm离心8min,如此清洗2遍;
[0102]
(9)弃上清,加入配置好的50%percoll细胞分离液,吹打混匀,12000rpm,4℃离心30min;
[0103]
(10)吸取最底层的高密度条带悬浮于d’hanks液中(此时可能看到红色的红细胞沉淀,注意不要吸到红细胞),吹打混匀,800rpm离心10min,反复清洗3遍;
[0104]
(11)用不含血清的dmem清洗1遍,800rpm离心10min;
[0105]
(12)将收集的细胞悬浮于鸡上皮细胞完全培养基,细胞计数,以2.5
×
106个/孔接种到6孔细胞培养板,另接种一个t25细胞瓶用于传代,置37℃含5%co2细胞培养箱中培养备用。
[0106]
2.mdcc
‑
msb1细胞的传代培养
[0107]
鉴于大多数ciav毒株可在mdcc
‑
msb1中稳定繁殖,发明人使用的mdcc
‑
msb1(商品
化的细胞系,购自bncc(北京))进行pcdna3.1
‑
2gyv3的体外转染验证,细胞传代和铺板步骤如下所示:
[0108]
(1)当原有细胞培养液颜色发黄变淡,或显微镜下观察细胞出现重叠现象时,便可进行传代。pbs、1640培养基和胎牛血清在37℃水浴锅中温育30min;
[0109]
(2)在超净台内,将t25细胞瓶内细胞悬液液转移到灭菌10ml离心管内,800rpm离心10min;
[0110]
(3)加入3ml pbs,轻柔吹打混悬细胞,800rpm离心10min,重复清洗2遍;
[0111]
(4)加入适量4ml含10%胎牛血清的1640培养基,轻柔吹打混悬,平均分装至2个 t25细胞瓶,每瓶2ml,再每瓶补足培养基至5ml;将细胞瓶置于含5%co2的37℃细胞培养箱中培养;
[0112]
(5)在合适时间,将处于对数生长期、活性良好的细胞铺到六孔板中,每孔加含10%胎牛血清的1640培养基2ml,细胞培养箱培养备用。
[0113]
3.感染性克隆pcdna3.1
‑
2gyv3的转染
[0114]
(1)当鸡原代肾小管细胞和mdcc
‑
msb1细胞汇合度分别达到70%左右时进行转染;
[0115]
(2)将罗氏脂质体转染试剂x
‑
tremegene hp dna transfection reagent、质粒(空载体pcdna3.1( )、感染性克隆质粒pcdna3.1
‑
2gyv3)与opti
‑
mem从冰箱取出置于室温,平衡至 15至 25℃。短暂漩涡混匀x
‑
tremegene hp dna transfection reagent;
[0116]
(3)取3个无菌的1.5ml离心管,在其中两个离心管中使用opti
‑
dmem稀释质粒,使空质粒和感染性克隆质粒的终浓度为0.01μg/μl(各总体积不能小于100μl),总量为各 400μl,轻柔混匀;第3个离心管加400μl opti
‑
dmem;
[0117]
(4)取x
‑
tremegene hp dna transfection reagent 3μl直接添加至3个离心管内,直接打入液面之下,切勿使转染试剂接触到塑料管壁,轻柔混合;
[0118]
(5)室温下孵育转染试剂:dna复合物15min;
[0119]
(6)从细胞培养箱中取出6孔细胞培养板,不必换培养基,将转染复合物直接逐滴加至细胞中,每孔100μl;
[0120]
(7)用手轻柔振荡或旋转培养板,使转染复合物均匀分布于培养板表面,然后放回细胞培养箱继续培养72h。
[0121]
4.间接免疫荧光检测蛋白表达
[0122]
取上一步孵育的6孔板细胞,用兔源抗gyv3 vp1多抗血清(参考袁世玉.圆圈病毒3型 (gyv3)对鸡和小鼠致病特性研究[d].山东农业大学,2020.公开的方法制备获得)对 pcdna3.1
‑
2gyv3在体外细胞内的转录表达进行间接免疫荧光检测。由于两种细胞生长特性的不同,前期的细胞固定操作有所差异:
[0123]
mdcc
‑
msb1属于完全悬浮细胞,固定时取每孔的细胞悬液于2ml灭菌离心管内,1000 r/min离心10min沉淀细胞,无菌收取上清,沉淀细胞用pbs清洗两遍,再用少量pbs将细胞悬浮,然后分别涂布于新的6孔板中央,待细胞干燥后,用-20℃预冷的丙酮乙醇3∶ 2混合固定液室温固定10min。
[0124]
鸡原代肾小管上皮细胞属于贴壁细胞,无菌收取上清后,贴壁细胞用pbs清洗两遍,每次每孔加2ml pbs在摇床上清洗5分钟,之后加入相同丙酮乙醇预冷固定液固定10min。之后两种细胞的抗体孵育操作相同:吸弃每孔固定液,用pbs清洗每孔细胞3遍,每遍
3min;每孔加入1∶100稀释的兔源抗gyv3 vp1多抗血清500μl,37℃孵育1小时;用pbs清洗 3遍,每遍3分钟;每孔加入1∶300稀释的fitc标记山羊抗兔igg 500μl,用锡箔纸包裹透明细胞板,避光放入37℃恒温恒湿培养箱孵育1小时;用pbs避光清洗3遍,每遍3分钟;用倒置荧光显微镜观察拍摄荧光情况。
[0125]
结果显示,可在两种转染细胞中检测到pcdna3.1
‑
2gyv3 vp1的表达,且可观察到细胞肿胀、破裂的细胞病变(附图7)。
[0126]
实施例5 gyv3绝对荧光定量标准曲线的构建
[0127]
为了检测感染性克隆pcdna3.1
‑
2gyv3感染模型中鸡的病毒血症水平和组织病毒载量,以验证病毒是否拯救成功,需构建针对gyv3基因的绝对荧光定量检测方法。
[0128]
以gyv3 sdau
‑
1株vp1基因为模板,使用primer 6.0设计绝对荧光定量pcr引物,引物序列见下表。
[0129]
gyv3绝对荧光定量引物序列
[0130][0131]
以实施例2中构建提取的质粒pcdna3.1
‑
gyv3为标准品,测量标准品浓度,按照质粒浓度与拷贝数换算公式:
[0132]
质粒拷贝数(copies/μl)=阿伏伽德罗常数6.02
×
10
23
(copies/mol)
×
质粒浓度(ng/μl)
ꢀ×
10
‑9/[(载体分子量 插入片段分子量)
×
660](g/mol),计算标准品拷贝数。
[0133]
用双蒸水将质粒标准品倍比稀释为1
×
109‑1×
103,每一梯度设置3个平行重复,每个重复设置3个技术学重复,荧光定量pcr反应体系和二步法扩增程序分别见表12和13,在罗氏荧光定量pcr仪上进行反应,仪器自带分析软件自动生成标准曲线。结果成功构建gyv3绝对荧光定量标准曲线。
[0134]
附图8a显示溶解曲线峰值单一,说明本发明所用qpcr引物灵敏性和特异性良好,可用于gyv3标准曲线建立;附图8b显示,扩增曲线起峰明显,间距均已,说明标准品稀释准确,重复性较好;从图8c获得该绝对荧光定量的标准曲线方程为:y=
‑
2.7465x 34.54(y:ct值, x:模板起始拷贝数),回归系数r2=0.9951,表明该曲线具有良好的相关性,可用于本发明 gyv3的绝对定量分析。
[0135]
实施例6 gyv3体内病毒拯救和spf鸡感染模型的建立
[0136]
采用6胚龄卵黄囊接种、1日龄腹腔和肌肉接种进行感染性克隆pcdna3.1
‑
2gyv3感染模型的建立(采用毒株为gyv3 sdau
‑
2),以确保感染性克隆质粒与体内细胞的充分作用。肌肉腹腔接种后7、14、21、28天,每组随机选取3只鸡采血、采泄殖腔拭子剖杀,进行拯救病毒检测。分组情况和接种方式总结如下表
[0137]
gyv3感染性克隆体内病毒拯救实验分组与接种方式
[0138][0139]
病毒血症检测是检验病毒感染性克隆是否具有感染性的重要指标,本发明监测了7dpi 各组所有鸡只,以及14/21/28dpi每组随机3只鸡的血液病毒载量水平。结果发现,在实验期内,gyv3感染性克隆组和阳性对照组均存在持续的病毒血症,其中gyv3
‑
ic组血液病毒载量在14dpi达到峰值,之后出现下降,gyv3组血液病毒载量的升高持续到21dpi,之后也呈现下降趋势(附图9)。由此表明双拷贝全长基因感染性克隆pcdna3.1
‑
2gyv3实现体内病毒拯救,并对spf鸡产生有效的感染。
[0140]
使用实施例5中成功构建的绝对荧光定量标准曲线,提取各组心脏、肝脏、脾脏、肺脏、肾脏、大脑、腺胃、十二指肠、骨髓、胸腺、法氏囊和泄殖腔拭子(粪便)12个组织样品dna,对7dpi、14dpi、21dpi、28dpi四个时间点进行定量pcr检测。
[0141]
结果显示(附图10):四个时间点gyv3
‑
ic和gyv3组各组织的病毒载量动态变化趋势基本一致。其中7dpi骨髓病毒载量最高,其次为肾脏、法氏囊、胸腺和脾脏,心脏、肝脏、肺脏、腺胃、十二指肠和粪便载量较低;14dpi肾脏病毒载量最高,腺胃和肝脏的病毒载量最低,粪便病毒载量较7dpi升高;21dpi和28dpi肾脏病毒载量依然最高,其次为法氏囊,胸腺和骨髓,其他组织和粪便病毒载量依然很低。从四个时间点的整体病毒载量来看,病毒载量在7和14dpi最高,载量最高可达7.92
×
108copies/g,而21和28dpi病毒载量呈整体下降趋势;从泄殖腔拭子的病毒载量来看,gyv3
‑
ic和gyv3组均可通过泄殖腔排泄病毒,其中在14dpi病毒载量最高,同时也直接表明本发明的感染性克隆可在体内合成完整的病毒粒子。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。