1.本发明属于咖啡因的检测技术领域,具体涉及一种茶叶中咖啡因的检测方法。
背景技术:
2.咖啡因是一种生物碱化合物,属于中枢神经兴奋剂,临床上用于治疗神经衰弱和 昏迷复苏。随着工作和生活节凑的加快,人们在日常生活中对于咖啡因的需求也越来 越大,用于暂时驱走睡意并恢复精力,含有咖啡因成分的产品比较丰富,例如咖啡、 茶、软饮料或能量饮料等。世界卫生组织国际癌症研究机构公布咖啡因属于“尚不能 分类”的致癌物。因此,人们对于咖啡因的摄入应该适量,
3.茶叶作为中国传统饮品,有着几千年的历史,在日常生活中消耗量较大,因此, 对于茶叶中的咖啡因的含量的测定,具有重要意义,能够指导人们科学合理的规划饮 茶量。
4.咖啡因能溶于一些极性或非极性溶剂,而茶叶中的色素成分或显色物质在溶剂中 会产生强烈的色素干扰,严重影响咖啡因的检测。在传统方法中,茶叶中咖啡因的检 测是采用分离的方法,分离丹宁、色素和蛋白,最后通过生化结晶,获得咖啡因晶体, 然而,该方法的咖啡因损失较大。目前,本领域内多采用液相色谱法测量咖啡因,虽 然准确性较之前有了提升,但茶叶中含有大量天然成分,且较为复杂,本领域技术人 员在研究液相色谱中茶叶的各种化合物的检测峰分离时投入了时间,耗时耗力。而且 对于不同种类的茶叶,由于其中化合物种类和含量不同,本领域技术人员还要花费大 量时间研究不同种类的茶叶。因此,利用溶解显色法测量咖啡因的方法方便快捷,成 本低廉,但需要首先解决色素干扰的问题。
技术实现要素:
5.针对上述问题,本发明提供了一种茶叶中咖啡因的检测方法,使用高精度光热电 位分析仪分析茶叶中的咖啡因含量,包括以下步骤:
6.s100:将茶叶研磨后,取样称重并记录茶叶样品的重量;
7.s200:在茶叶样品中加入醋酐,超声混合,得到溶液一;
8.s300:在溶液一中加入甲苯,混合均匀后,再加入石墨化炭黑和次氯酸,去除色 素,得到溶液二;
9.s400:制作空白样品:分别移取与步骤s200中体积相同的醋酐和与步骤s300中 体积相同的甲苯,将醋酐和甲苯混合均匀,加入结晶紫指示液,得到空白样品;
10.s500:向溶液二中加入结晶紫指示液,再放入所述光热电位分析仪的电极,滴定 液为高氯酸标准滴定溶液,同时使用电位滴定和光度滴定,滴定至终点,记录溶液二 所消耗的高氯酸标准滴定溶液的体积;
11.按照同样方法滴定空白样品,记录空白样品所消耗的高氯酸标准滴定溶液的体积。
12.所述检测方法的计算方法为:
[0013][0014]
或
[0015]
式中:
[0016]
c:高氯酸标准滴定溶液的浓度,mol/l;
[0017]
v:溶液二消耗的高氯酸标准滴定溶液的体积,ml
[0018]
v0:空白样品消耗的高氯酸标准滴定溶液的体积,ml
[0019]
m:称取的样品的质量,g
[0020]
194.2:咖啡因最小反应单元的质量分数,g/mol。
[0021]
可选的,步骤s100中,茶叶研磨后的样品的目数为250
‑
300目。
[0022]
可选的,步骤s200和s300中,茶叶样品、醋酐和甲苯的质量体积比为 1g:(4
‑
6)ml:(6
‑
9)ml。
[0023]
可选的,步骤s200中,超声频率为40
‑
50hz,功率为50
‑
60w,时间为10
‑
20min, 本发明中超声频率、功率和时间不宜过大过长,避免过多色素溶出。
[0024]
可选的,步骤s300中,茶叶样品与石墨化炭黑的质量比为1:(5
‑
8),茶叶样品与 次氯酸的质量体积比为1g:(0.5
‑
1.5)ml。
[0025]
可选的,步骤s500中,所述结晶紫指示液为结晶紫的无水醋酸溶液。
[0026]
可选的,步骤s500中,所述高氯酸标准滴定溶液的浓度为0.1
‑
0.2mol/l。
[0027]
所述光热电位分析仪的电极为ph复合电极和光度电极,利用ph复合电极进行动 态等当点滴定,即用ph复合电极来测量溶液中ph值的变化所带来的电位变化,将电 位变化的突变点为终点的一种滴定方式。ph复合电极和光度电极对同一样品进行检测, 得到两条滴定曲线,便于将两个电极对应曲线所计算出的检测结果进行比较,验证光 度电极检测的准确性。
[0028]
步骤s500中,溶液二滴定时由紫色变为蓝绿色时,即为光度滴定的终点;光度 滴定的光度电极的光线波长为502nm。
[0029]
本发明在研究过程中发现,茶叶中色素的溶出对光度滴定的影响较大,为了提高 滴定准确性,排出茶叶中色素的干扰具有重要作用。茶叶中色素包括水溶性色素和脂 溶性色素,具体到本发明,茶叶样品的溶剂为醋酐和甲苯,即提供了无水环境,所以 溶液二中主要为脂溶性色素,脂溶性色素主要包括叶绿素、类胡萝卜素、叶酸等。
[0030]
可选的,步骤s200包括以下步骤:
[0031]
(1)用乙醚和苯的混合溶液洗涤茶叶样品,叶绿素和类胡萝卜素溶入乙醚和苯的 混合溶液中,然后过滤,得到滤渣,烘干滤渣;
[0032]
(2)用氯仿和醋酐的混合溶液超声洗涤步骤(1)的滤渣,咖啡因溶入氯仿和醋 酐的混合溶液中,然后过滤,得到滤液,即为溶液一。
[0033]
步骤(1)去除色素中的大部分叶绿素和类胡萝卜素,步骤(2)去除色素中的大 部分叶酸。
[0034]
可选的,步骤s300包括以下步骤:
[0035]
(3)向步骤(2)得到的溶液一中加入甲苯,混合均匀;
[0036]
(4)向步骤(3)得到的溶液中加入石墨化炭黑和次氯酸,进一步去除色素;
[0037]
(5)向步骤(4)得到的溶液中加入结晶紫指示液,得到溶液二。
[0038]
可选的,步骤(1)中,茶叶样品与乙醚的质量体积比为1g:(10
‑
13)ml,苯与乙醚 的体积比为1:(5
‑
6)。
[0039]
可选的,步骤(2)中,步骤(1)的滤渣与醋酐的质量体积比为1g:(4
‑
6)ml,氯 仿与醋酐的体积比为1:(2
‑
3)。
[0040]
可选的,步骤(3)中,步骤(1)的滤渣与甲苯的质量体积比为1g:(6
‑
9)ml。
[0041]
可选的,步骤(4)中,步骤(1)的滤渣与石墨化炭黑的质量比为1:(5
‑
8),步骤 (1)的滤渣与次氯酸的质量体积比为1g:(0.5
‑
1.5)ml,由于咖啡因具有弱碱性,本发 明选择了氧化性较强同时酸性很弱的次氯酸,并严格控制次氯酸的用量,达到较好的 脱色效果。
[0042]
茶叶中的色素成本比较复杂,而且脂溶性色素的性质相似,脂溶性色素与咖啡因 的分离比较困难。本发明经过长期实验研究,反复研究对比脂溶性色素和咖啡因的特 性,终于得出上述分离方法,即利用一定比例的乙醚和苯的混合溶液,先分离出叶绿 素和类胡萝卜素,这就解决了茶叶中大部分显色性能较强的脂溶性色素;然后,利用 一定比例的氯仿和醋酐的混合溶液,分离出大部分的叶酸色素,至此,样品溶液中的 绝大部分显色物质得到了有效的去除。最后再通过石墨化炭黑的物理吸附作用和次氯 酸的氧化作用,对残余的色素进行去除,最大程度地去除茶叶中色素显色对光度滴定 的影响。
[0043]
另外,本发明对滴定终点的科学选取,也进行了研究。由于本发明的样品溶液中 含有多种溶剂且含量各异,各种溶剂对咖啡因和高氯酸的反应具有比较复杂的影响, 这些影响的原理现在还没有完全研究清楚,但为了准确测定茶叶中的咖啡因含量,对 计算时,滴定终点的科学选取,并做合理的修正。
[0044]
电位滴定分析时,终点微分值是判定终点的重要依据,通过对电极电位与滴定液 体积之间的关系进行微分得出,现有的滴定终点确定方法有e
‑
v曲线法、一阶微商法 和二阶微商法。具体到本发明的茶叶中咖啡因的检测,e
‑
v曲线具有两个突变点,即 具有两个爬升平台,选择第二个突变点对滴定终点进行分析。
[0045]
一般分析是在e
‑
v曲线的终点突跃线上,选择电位的最大值和最小值之和的1/2 处电位对应的体积为滴定终点的体积。而对于本技术,加入了修正系数,所述修正系 数与苯、乙醚、氯仿、醋酐和甲苯的介电常数和用量体积有关。
[0046]
具体的,滴定终点的体积为e
‑
v曲线的终点突跃线上的最大电位和最小电位之和 的1/2 r处电位对应的体积。
[0047]
所述修正系数为r,计算公式为:
[0048][0049]
式(1)中,a与步骤(1)中的乙醚和苯的介电常数和用量体积有关,a的计算公 式为:
[0050][0051]
式(2)中,ε1和ε2分别为苯和乙醚的介电常数,分别为2.3和4.3;l1为苯与乙 醚的体积比1:(5
‑
6),即l1的取值为0.167
‑
0.2;l2为乙醚与茶叶样品的体积质量比为 (10
‑
13)ml:1g,即l2的取值为10
‑
13。
滴定液为c=0.1mol/l高氯酸标准滴定溶液,同时使用电位滴定和光度滴定,滴定至终 点,记录溶液二所消耗的高氯酸标准滴定溶液的体积v;
[0072]
按照同样方法滴定空白样品,记录空白样品所消耗的高氯酸标准滴定溶液的体积 v0;
[0073]
步骤s500中,溶液二滴定时由紫色变为蓝绿色时,即为光度滴定的终点;光度 滴定使用的光线的波长为502nm。
[0074]
所述检测方法的计算方法为:
[0075][0076]
式中:c:高氯酸标准滴定溶液的浓度,0.1mol/l;v:溶液二消耗的高氯酸标准滴定 溶液的体积,ml;v0:空白样品消耗的高氯酸标准滴定溶液的体积,ml;m:称取的样 品的质量,5.0013g;194.2g/mol为咖啡因最小反应单元的质量分数;v和v0均为各自e
‑
v 曲线的终点突跃线上的最大电位和最小电位之和的1/2处电位对应的体积。
[0077]
本实施例使用ph复合电极和光度电极对同一样品进行检测,得到两条滴定曲线,并 分别按上述计算方法得到计算结果,将两个计算结果进行比较,相对偏差r在5%以内。
[0078][0079]
上式中,r为相对偏差,%;c1为光度电极检测的计算结果,mg/g;c2为ph复合电 极检测的计算结果,mg/g。
[0080]
对比例1
[0081]
本对比例所述的茶叶中咖啡因的检测方法,与实施例1相同,区别在于,步骤s300 中茶叶样品中不加入甲苯,加入结晶紫指示液后,直接滴定。
[0082]
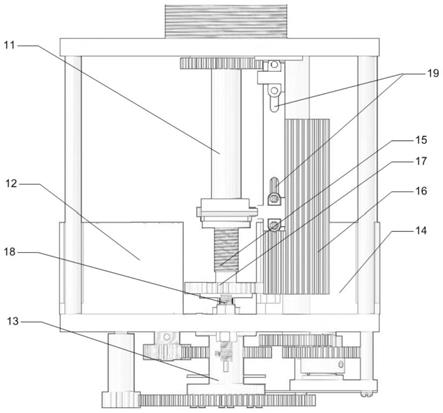
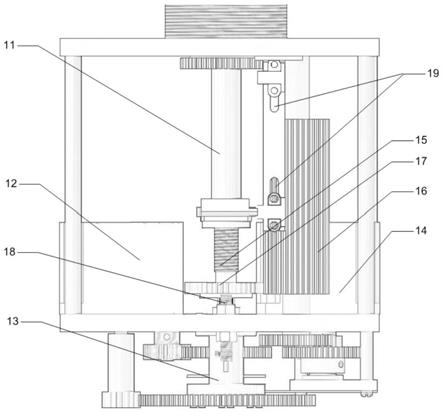
对比例2
[0083]
本对比例所述的茶叶中咖啡因的检测方法,与实施例1相同,区别在于,步骤s300 中不添加石墨化炭黑和次氯酸。
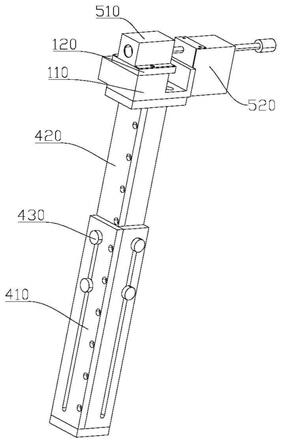
[0084]
实施例2
[0085]
本实施例所述的茶叶中咖啡因的检测方法,与实施例1相同,区别在于,步骤s100 中,茶叶样品研磨至300目;步骤s200和s300中,茶叶样品、醋酐和甲苯的质量体 积比为1g:6ml:9ml;步骤s200中,超声频率为50hz,功率为60w,时间为10min。
[0086]
实施例3
[0087]
本实施例所述的茶叶中咖啡因的检测方法,与实施例2相同,区别在于,步骤s300 中,茶叶样品与石墨化炭黑的质量比为1:8。
[0088]
实施例4
[0089]
本实施例所述的茶叶中咖啡因的检测方法,与实施例2相同,区别在于,步骤s300 中,茶叶样品与石墨化炭黑的质量比为1:9。
[0090]
实施例5
[0091]
本实施例所述的茶叶中咖啡因的检测方法,与实施例3相同,区别在于,步骤(4) 中,茶叶样品与次氯酸的质量体积比为1g:1.5ml。
[0092]
实施例6
[0093]
本实施例所述的茶叶中咖啡因的检测方法,与实施例3相同,区别在于,步骤(4) 中,茶叶样品与次氯酸的质量体积比为1g:1.6ml。
[0094]
实施例7
[0095]
本实施例所述的茶叶中咖啡因的检测方法,与实施例5相同,区别在于,步骤s200 包括以下步骤:
[0096]
(1)用乙醚和苯的混合溶液洗涤茶叶样品,叶绿素和类胡萝卜素溶入乙醚和苯的 混合溶液中,然后过滤,得到滤渣,烘干滤渣;
[0097]
茶叶样品与乙醚的质量体积比为1g:10ml,苯与乙醚的体积比为1:5;
[0098]
(2)用氯仿和醋酐的混合溶液超声洗涤步骤(1)的滤渣,咖啡因溶入氯仿和醋 酐的混合溶液中,然后过滤,得到滤液,即为溶液一;
[0099]
步骤(1)所得的干燥的滤渣与醋酐的质量体积比为1g:4ml,氯仿与醋酐的体积 比为1:2;
[0100]
上述步骤(1)去除色素中的大部分叶绿素和类胡萝卜素,步骤(2)去除色素中 的大部分叶酸。
[0101]
实施例8
[0102]
本实施例所述的茶叶中咖啡因的检测方法,与实施例7相同,区别在于,步骤(1) 中,茶叶样品与乙醚的质量体积比为1g:13ml。
[0103]
实施例9
[0104]
本实施例所述的茶叶中咖啡因的检测方法,与实施例7相同,区别在于,步骤(1) 中,茶叶样品与乙醚的质量体积比为1g:14ml。
[0105]
实施例10
[0106]
本实施例所述的茶叶中咖啡因的检测方法,与实施例8相同,区别在于,步骤(1) 中,苯与乙醚的体积比为1:6。
[0107]
实施例11
[0108]
本实施例所述的茶叶中咖啡因的检测方法,与实施例8相同,区别在于,步骤(1) 中,苯与乙醚的体积比为1:7。
[0109]
实施例12
[0110]
本实施例所述的茶叶中咖啡因的检测方法,与实施例10相同,区别在于,步骤(2) 中,氯仿与醋酐的体积比为1:3。
[0111]
实施例13
[0112]
本实施例所述的茶叶中咖啡因的检测方法,与实施例10相同,区别在于,步骤(2) 中,氯仿与醋酐的体积比为1:1。
[0113]
实施例14
[0114]
本实施例所述的茶叶中咖啡因的检测方法,与实施例10相同,区别在于,步骤 s300包括以下步骤:
[0115]
(3)向步骤(2)得到的溶液一中加入甲苯,混合均匀;步骤(1)所得的干燥的 滤渣与甲苯的质量体积比为1g:6ml;
[0116]
(4)向步骤(3)得到的溶液中加入石墨化炭黑和次氯酸,进一步去除色素;步 骤
(1)所得的干燥的滤渣与石墨化炭黑的质量比为1:5,与次氯酸的质量体积比为 1g:0.5ml;
[0117]
(5)向步骤(4)得到的溶液中加入结晶紫指示液,得到溶液二。
[0118]
实施例15
[0119]
本实施例所述的茶叶中咖啡因的检测方法,与实施例14相同,区别在于,步骤(3) 中,步骤(1)所得的干燥的滤渣与甲苯的质量体积比为1g:9ml。
[0120]
实施例16
[0121]
本实施例所述的茶叶中咖啡因的检测方法,与实施例15相同,区别在于,步骤(4) 中,步骤(1)所得的干燥的滤渣与石墨化炭黑的质量比为1:8。
[0122]
实施例17
[0123]
本实施例所述的茶叶中咖啡因的检测方法,与实施例15相同,区别在于,步骤(4) 中,步骤(1)所得的干燥的滤渣与石墨化炭黑的质量比为1:9。
[0124]
实施例18
[0125]
本实施例所述的茶叶中咖啡因的检测方法,与实施例16相同,区别在于,步骤(4) 中,步骤(1)所得的干燥的滤渣与次氯酸的质量体积比为1g:1.5ml。
[0126]
实施例19
[0127]
本实施例所述的茶叶中咖啡因的检测方法,与实施例16相同,区别在于,步骤(4) 中,步骤(1)所得的干燥的滤渣与次氯酸的质量体积比为1g:1.6ml。
[0128]
实施例20
[0129]
本实施例所述的茶叶中咖啡因的检测方法,与实施例18相同,区别在于,加入了 修正系数,所述修正系数与苯、乙醚、氯仿、醋酐和甲苯的介电常数和用量体积有关。
[0130]
具体的,滴定终点的体积为e
‑
v曲线的终点突跃线上的最大电位和最小电位之和 的1/2 r处电位对应的体积。
[0131]
所述修正系数为r,计算公式为:
[0132][0133]
式(1)中,a与步骤(1)中的乙醚和苯的介电常数和用量体积有关,a的计算公 式为:
[0134][0135]
式(2)中,ε1和ε2分别为苯和乙醚的介电常数,分别为2.3和4.3;l1为苯与乙 醚的体积比1:6,即l1的取值为0.167;l2为乙醚与茶叶样品的体积质量比为13ml:1g, 即l2的取值为13。
[0136]
式(1)中,b与步骤(2)中的氯仿和醋酐的介电常数和用量体积有关,b的计算 公式为:
[0137][0138]
式(3)中,ε3和ε4分别为氯仿和醋酐的介电常数,分别为5.1和20.7;l3为氯仿 与醋酐的体积比为1:2,即l1的取值为0.5;l4为醋酐与步骤(1)的滤渣的体积质量 比为6ml:1g,即l4的取值为6。
[0139]
式(1)中,c与步骤(3)中的甲苯的介电常数和用量体积有关,c的计算公式为:
[0140][0141]
式(4)中,ε5为甲苯的介电常数,为2.4;l5为甲苯与步骤(1)的滤渣的体积 质量比为9ml:1g,即l5的取值为9。
[0142]
上述实施例1
‑
20和对比例1
‑
2均检测福利来茉莉花茶中咖啡因的含量;
[0143]
福利来茉莉花茶,配料为绿茶和茉莉花,类型为烘青茉莉花茶,生产商为成都御 茗春茶茶业有限公司,条形码号为6936160900918。
[0144]
实施例21
[0145]
本实施例所述的茶叶中咖啡因的检测方法,与实施例20相同,区别在于,茶叶样 品为立顿茉莉花茶,取样量为5.0026g。立顿茉莉花茶的生产商为联合利华(中国)有 限公司,产地安徽省黄山市,配料为绿茶和茉莉花,条形码号为6902088802566。
[0146]
实施例22
[0147]
本实施例所述的茶叶中咖啡因的检测方法,与实施例20相同,区别在于,茶叶样 品为忆江南铁观音,取样量为5.0026g;
[0148]
采购于京东的忆江南茗茶官方旗舰店,货号为6923790798701,产地为杭州,采 摘时间为秋季,等级为二级,发酵程度为半发酵,类型为壮结,采摘要求为一芽三四 叶,条形码号为6923790798701。
[0149]
上述三种茶叶先使用液相色谱法检测茶叶中咖啡因的含量,作为标准值,检测方 法按照国标gb 5009.139
‑
2014《食品安全国家标准饮料中咖啡因的测定》,将实施例 1
‑
18和对比例1的检测结果与液相色谱法的检测结果进行对比,评价本发明方法的准 确性。表1中准确性的计算方法为:
[0150][0151]
表1实施例和对比例的检测结果比较
[0152] 准确性(%) 准确性(%)实施例16.0实施例133.9实施例25.6实施例141.3实施例35.4实施例151.2实施例45.8实施例161.0实施例55.2实施例171.4实施例65.4实施例180.9实施例74.0实施例191.2实施例83.7实施例200.6实施例94.5实施例210.5实施例103.3实施例220.6实施例114.0对比例115.8实施例123.5对比例210.4
[0153]
由上表可知,使用本发明所述的茶叶中咖啡因的检测方法,检测结果与使用液相 色谱的检测结果相比偏差较小,大致控制在6%以内。另外,在实施例1
‑
18中,所述 检测方
法使用ph复合电极和光度电极得到的检测结果的相对误差均在5%以内。本发 明的方法适用于多种茶叶的检测,适用性较强,比液相色谱检测方法更为简单、快捷, 具有广泛推广价值。
[0154]
为了便于公众理解本发明所使用的光热电位分析仪,现将其馈液传动单元和滴定 管的结构补充如下:
[0155]
如图1所示,所述滴定管2包括管体21和位于管体内的泵头22,用于抽打滴定 液;所述馈液传动单元1推拉泵头22,从而将滴定液抽取至管体21中或将管体21内 的滴定液注入烧杯中,烧杯中存放有待滴定液体。
[0156]
在泵头22上设置有换向机构,以分别对洗涤液和滴定液抽取。
[0157]
如图2
‑
3所示,所述馈液传动单元1包括伸缩组件11、主电机12、主丝杠13、 细分电机14、细分丝杠15、驱动齿轮16和大丝母17。伸缩组件11的顶端与泵头22 连接,伸缩组件11的底端具有螺纹孔,套设在细分丝杠15上;
[0158]
细分丝杠15的一端与大丝母17顶端固定连接,大丝母的底端具有螺纹孔,使得 大丝母17能够套设在主丝杠13上,在大丝母17的周向侧面设置有与驱动齿轮16相 对应的齿轮,使得驱动齿轮16能够驱动或限制大丝母17的旋转;驱动齿轮16与细分 电机14的输出轴连接;主丝杠13与主电机12的输出轴连接。
[0159]
在伸缩组件11的上部和下部设置有限位开关19,使得伸缩组件11仅能够上下伸 缩,不能旋转。
[0160]
主丝杠13和细分丝杠15的螺纹旋向相同,主丝杠13的导程大于细分丝杠15的 导程。
[0161]
驱动齿轮16的轴线与细分丝杠15的轴线平行,驱动齿轮16的齿宽大于大丝母 17周向齿轮的齿宽,使得大丝母17与驱动齿轮16在轴向上能够相对滑动。
[0162]
主电机12转动,带动主丝杠13旋转,主丝杠13与主电机12的传动比是1:5, 主丝杠的导程是5mm,在该种传动效果下,1/4步细分的主电机即可实现馈液传动单 元的20000/1细分。大丝母17被驱动齿轮16旋转锁定,大丝母17在主丝杠13的驱 动下沿主丝杠13上的螺纹滑动,进而带动细分电机14与伸缩组件11上下移动,实现 泵头22的快速升降。
[0163]
在慢速滴定时,主电机12处于通电锁死状态,此时主丝杠13不转动;细分电机 14转动,带动驱动齿轮16转动,大丝母17在驱动齿轮16的转动下进行周向旋转, 大丝母17沿主丝杠13上的螺纹滑动上升或下降;细分丝杠15随大丝母17旋转,使 得伸缩组件11沿细分丝杠15相对滑动,进行与大丝母17相反方向的相对运动;由于 主丝杠13导程大于与细分电机14的导程,大丝母17的升降与伸缩组件11的升降速 度不同,最终伸缩组件11的实际升降量为大丝母17的升降量与伸缩组件11相对细分 丝杠15升降量之差。在大丝母17下端设置有接近开关18,通过接近开关18复位大 丝母17的初始位置。主电机12工作,使得大丝母17下降,直至大丝母17与接近开 关18接触,此时,大丝母17位置即为初始位置。
[0164]
上述馈液传动单元1的使用如下:
[0165]
s1、确定单次馈液量,获得单次泵头移动量;
[0166]
s2、按泵头移动量划分快速滴定阶段和慢速滴定阶段进行滴定;
[0167]
s3、电极检测,确认下次馈液量,重复上述过程直至滴定结束。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。