一种无标记、均相生物发光的dna损伤检测方法
技术领域
1.本发明属于dna损伤检测技术领域,具体涉及一种无标记、均相生物发光的dna损伤检测方法,基于该检测方法的试剂盒及所述检测方法、试剂盒在疾病诊断、抗肿瘤活性药物筛选领域的应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.保持基因组的完整性和稳定性对人类具有重要意义,细胞dna不断受到各种内源性因素(如水解、活性氧和氮)和外源性因素(如x射线、紫外线和许多化学物质)的影响,每天在单个人体细胞中产生104‑
106个dna损伤事件。基因组不稳定性在癌症、衰老和阿尔茨海默病中起着关键作用。最常见的dna损伤是碱基、核糖和单链断裂损伤,这些损伤是由氧化、烷基化、脱氨基、自发水解等产生的。当这些损伤通过错误的修复或复制转化为突变时,将会是永久性的,并持续在后代细胞中引发一系列影响。突变的一个重要后果是肿瘤抑制基因的丢失和原癌基因的激活,导致细胞不受控制的增殖从而引发癌症。
4.dna损伤有两种类型,包括单碱基的dna损伤和聚集的dna损伤。其中,聚集dna损伤较难检测,因为聚集dna损伤由两个或两个以上的损伤位点组成,这些损伤紧密地分布在dna的一个或两个螺旋内。另外,由于损伤的丰度低、多样性大,位置随机,准确而特异的检测全基因组dna损伤水平仍然是一个巨大的挑战。
5.目前,dna损伤的检测方法有彗星法、酶联免疫吸附法、质谱法、高效液相色谱—质谱法、单分子检测法和荧光法等。彗星试验和酶联免疫吸附试验是目前最常用的方法,但它们费时、涉及多步分离、成本高。单分子检测具有灵敏度高的优点和原位成像dna损伤的能力,但需要昂贵的实验设备。荧光法具有简单、快速、灵敏度高等优点,但涉及复杂的探针设计和昂贵的标记。与荧光法相比,生物发光法具有背景信号低、高通量、无需激发的自发光等显著优点,特别是其信号读出不受自荧光、散射光和光漂白的干扰。在生物体内,细胞具有多种修复机制来抵消dna损伤的有害影响,如碱基切除修复(ber)、核苷酸切除修复(ner)、错配修复(mmr)、同源重组(hr)和非同源末端连接(nhej)等。ber是维持dna稳定性最通用的途径,它可以修复由烷基化、氧化和脱氨基引起的各种dna损伤。ber由dna糖基化酶引发,糖基化酶识别受损和不匹配的碱基,并通过水解碱基和dna主链之间的n
‑
糖苷键生成一个无碱基位点。每种损伤碱基都对应独特的dna糖基化酶,dna糖基化酶能区分特定的核碱基损伤。受dna糖基化酶高特异性的启发,科学家发展了一系列检测dna特异性损伤的方法,包括纳米孔测序法和单分子计数法。基于特异性核苷酸标记和pcr扩增的纳米孔测序方法可以识别多种病变,但需要热循环和复杂的标记步骤;单分子计数方法可以灵敏地检测dna损伤,但需要多步骤核苷酸标记、分离和洗涤步骤。
技术实现要素:
6.基于上述技术背景,维持dna的完整性和稳定性对所有生物体都至关重要,内源性/外源性因素引起的dna损伤可引起多种疾病。全基因组dna损伤水平可能成为临床早期诊断、风险评估和治疗监测的重要生物标志物。然而,由于基因组dna的多样性,很难实现对基因组dna损伤的无标记、均相检测。
7.为了克服上述技术难点,本发明提供了以下技术方案:
8.本发明主要的技术贡献在于提供了一种基于碱基切除修复(ber)途径和末端转移酶(tdt)启动的无模板等温循环扩增的无标记和均相生物发光法检测方法,该检测方法可以检测基因组dna中的单碱基损伤和聚集损伤。
9.本发明提供的方法中,首先封闭待测基因组中所有的3'
‑
oh末端,通过dna糖基化酶识别并且切割待测dna受损部分暴露新的3'
‑
oh末端,在新的3'
‑
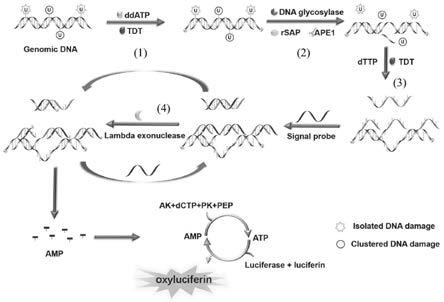
oh末端构建富含腺嘌呤(a)和胸腺嘧啶(t)的杂交链,通过核酸外切酶对所述富含腺嘌呤(a)和胸腺嘧啶(t)的杂交链中的腺嘌呤单链进行剪切产生单碱基形式的腺嘌呤核苷酸从而产生腺嘌呤核糖核苷酸(amp)分子,在萤火虫荧光素酶和荧光素的协助下,通过amp
‑
atp
‑
amp的转化,实现生物发光。
10.上述技术方案中lambda核酸外切酶对富a探针的剪切作用,一方面实现了生物发光,另一方面对dna损伤信号进行了一次放大。经验证,该方法具有很好的选择性,能特异性地检测单碱基的dna损伤和聚集的dna损伤,其聚集dna损伤的检测限为8.26
×
10
‑
10
mol/l。
11.为了进一步提高检测的灵敏度,本发明还设计引入核酸内切酶(ape1)诱导的循环裂解信号再一次的放大。所述核酸内切酶(ape1)诱导的循环裂解的检测方法中,本发明设计了一种具有两个ap位点的ap探针,该探针能被ape1切割并产生更多的3'
‑
oh引物。ap探针的引入可以产生更多的poly
‑
t结构和更多的amp来产生增强的生物发光信号。改进方法的灵敏度比原方法提高了3个数量级,比非天然核苷类似物标记法(1
×
10
‑
10
mol/l)、高效液相色谱
‑
串联质谱法(1
×
10
‑
10
mol/l)提高了4个数量级,比适体荧光法(3
×
10
‑9mol/l)提高5个数量级。该方法可准确检测hela细胞、a549细胞、hek
‑
293和mrc
‑
5细胞dna中的尿嘧啶,检出限为0.011ng。
12.以尿嘧啶dna损伤为模型,检测限为3.26
×
10
‑
14
mol/l,并能区分0.001%的dna损伤。重要的是,它可以进一步应用于细胞样品中dna损伤水平的检测,检测限为0.011ng,通过选择合适的dna糖基化酶,有望成为检测各种dna损伤的通用平台。
13.以上一个或多个技术方案的有益效果是:
14.(1)本发明提供的检测方法显著压缩了现有检测工序,这种生物发光传感器实现了对基因组dna损伤的无标记、均相检测,无需任何分离和洗涤步骤,是dna损伤检测领域的一大创新。
15.(2)本发明方法不仅可以检测基因组dna中的单碱基损伤,而且可以检测基因组dna中的聚集损伤,且这两种损伤检测之间没有干扰。
16.(3)本发明方法不受序列不同的影响,可以检测出基因组中任意序列的损伤。
17.(4)利用tdt催化聚合反应的高效性和生物发光固有的高灵敏度,该方法能灵敏地检测dna损伤,检测灵敏度优于以往报道。以尿嘧啶dna损伤模型为例,所述方法的检测限为3.26
×
10
‑
14
mol/l,对肿瘤细胞dna的检测限为0.011ng。
18.(5)此外,这种生物发光传感器利用dna糖基化酶的高度特异性可以检测特定的损伤,并可以通过适当的选择dna糖基化酶可以扩展到检测多种损伤碱基。它可以成为检测其他dna损伤的通用平台。
19.(6)除富a信号探针、ap探针外,检测过程中不需额外的信号探针标记。
附图说明
20.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
21.图1为无标记、均相生物发光法检测dna损伤的原理;
22.其中,(1)tdt介导的ddatp聚合反应以封闭dna的3'
‑
oh;(2)dna糖基化酶催化切除dna损伤的碱基;(3)tdt介导的无模板等温扩增以形成poly
‑
t结构;(4)lambda外切酶切割产生丰富的amp以产生生物发光信号。
23.图2为改进的生物发光法检测dna损伤的原理图;
24.其中,(1)tdt介导的ddatp聚合反应以封闭dna的3'
‑
oh;(2)dna糖基化酶催化切除dna损伤的碱基;(3)ape1诱导的双链dna循环裂解释放大量3'
‑
oh末端引物;(4)tdt介导的无模板等温扩增以形成poly
‑
t结构;(5)lambda外切酶切割产生丰富的amp以产生生物发光信号。
25.图3为实施例1中尿嘧啶损伤dna检测效果验证图;
26.(a)ddatp封闭反应的凝胶电泳分析,sybr gold作为染色剂;
27.泳道m,dna marker;泳道1,封闭ddatp的u damage dna反应产物分析;泳道2,封闭ddatp的normal dna反应产物分析;泳道3,u damage dna反应产物分析;泳道4,normal dna反应产物分析;泳道5,u damage dna;泳道6,normal dna;
28.(b)dna糖基化酶切除去除dna损伤反应后,u damage dna(箭头)和normal dna的
‑
df/dt值随着温度的变化;
29.(c)tdt介导的无模板扩增产物的凝胶电泳分析;sybr gold作为染色剂;泳道m,dna marker;泳道1,u damage dna的反应产物;泳道2,normal dna的反应产物;
30.(d)实时生物发光监测dna损伤检测;u damage dna(箭头),normal dna。图4为u损伤dna、u
‑
6损伤dna、u
‑
17损伤dna和正常dna产生的生物发光信号;
31.每个dsdna的浓度为100nm;误差棒代表三次实验的标准差。
32.图5为u损伤dna udg ape1、u损伤dna fpg ape1、8
‑
oxog损伤dna udg ape1和8
‑
oxog损伤dna fpg ape1的生物发光信号;
33.每个dsdna的浓度为100nm;udg浓度为1u/μl,fpg浓度为1.6u/μl;误差棒代表三次实验的标准差。
34.图6为不同浓度尿嘧啶损伤模型的生物发光信号测定及浓度关系;
35.其中,(a)不同浓度u
‑
6损伤dna的实时生物发光曲线;(b)不同浓度u
‑
6损伤dna产生的生物发光信号的测定;(c)在1
×
10
‑9~1
×
10
‑7mol/l范围内,生物发光信号与u
‑
6损伤dna浓度的对数呈线性关系;误差棒代表三次实验的标准差。
36.图7为ape1诱导循环裂解优化检测方法中生物发光信号及浓度关系图;
37.(a)改进方法测得的不同浓度u
‑
6损伤dna的实时生物发光曲线;(b)25min时生物
发光强度随u
‑
6损伤dna浓度的变化;(c)在1
×
10
‑
13
~1
×
10
‑8mol/l范围内,生物发光强度与u
‑
6损伤dna浓度的对数呈线性关系;(d)在u
‑
6损伤dna与正常dna的混合物中,实际加入与测量的u
‑
6损伤dna水平的线性相关性;u
‑
6损伤dna和正常dna的总浓度为10nm;误差棒代表三次实验的标准差。
38.图8为ape1诱导循环裂解优化检测方法检测不同肿瘤细胞中dna损伤的信号强度;
39.(a)用改进的方法检测从hela细胞、a549细胞、hek
‑
293细胞和mrc
‑
5细胞提取的基因组dna中的尿嘧啶损伤;每个细胞的基因组dna含量为100ng;(b)生物发光强度与不同质量hela细胞基因组dna的线性关系;误差棒代表三次实验的标准差。
具体实施方式
40.应该指出,以下详细说明都是例示性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
41.需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
42.术语解释
43.poly
‑
t:本技术文件中,表示在受损dna末尾的一段由多个胸腺嘧啶连接而成的核苷酸序列,本技术中文件中所称的poly
‑
t单链,poly
‑
t序列及富含胸腺嘧啶单链等均表示相同含义;
44.富a探针:或称poly
‑
a信号探针,表示一段具有多个腺嘌呤碱基的核苷酸单链;
45.ap探针:本技术文件中,所述ap探针为含有核酸内切酶识别位点的富a探针,所述ap位点为dna双链中的无碱基位点;
46.ap位点:表示dna双链中的无碱基位点,用于核酸内切酶的识别;
47.tdt:末端转移酶,或末端脱氧核苷酸转移酶(terminal deoxyribonucleotidyl transferase,能够不依赖于模板的dna聚合酶,催化脱氧核苷酸结合到dna分子的3
’‑
oh端;
48.ddatp:双脱氧的腺苷三磷酸;
49.ape1酶:核酸内切酶;
50.dttps:胸腺嘧啶的三磷酸脱氧核苷酸;
51.rsap:虾碱性磷酸酶;
52.amp
‑
atp
‑
amp的转化:在萤火虫发光反应中,atp 荧光素 o2=amp 氧化荧光素 ppi 光;在磷酸烯醇式丙酮酸单钠盐水合物(pep)和dctp存在下,通过腺苷酸激酶(ak)和丙酮酸激酶(pk)的酶促反应组合,释放的amps可转化为atps,在萤火虫荧光素酶和荧光素的协助下,atps转化为amps产生了强烈的生物发光信号;
53.amp:磷酸腺苷;
54.ap探针/poly
‑
t双链dna:表示ap探针链与poly
‑
t杂交而成的双链;
55.udg:尿嘧啶dna糖基化酶,用于识别序列中的尿嘧啶碱基修饰并进行剪切;
56.聚集的dna损伤:clustered damage,聚集的dna损伤、聚集性dna损伤、聚集dna损
伤”表示dna损伤位点紧密地分布在dna的一个或两个螺旋内。
57.正如背景技术所介绍的,现有技术中dna损伤检测方法往往需要较多生物标记及洗涤分离的过程,为了解决如上的技术问题,本发明提出了一种基于碱基切除修复(ber)途径和末端转移酶(tdt)启动的无模板等温循环扩增的无标记和均相生物发光法检测基因组中的dna损伤。
58.本发明第一方面,提供一种无标记、均相生物发光的dna损伤检测方法,所述检测方法如下:封闭待测基因组中所有的3'
‑
oh末端,通过dna糖基化酶识别并且切割待测基因组的受损dna部分暴露新的3'
‑
oh末端,在新的3'
‑
oh末端构建富腺嘌呤(a)和胸腺嘧啶(t)的杂交链,加入核酸外切酶对所述杂交链中的富腺嘌呤(a)单链进行剪切产生得到腺嘌呤单碱基从而产生腺嘌呤核糖核苷酸(amp)分子,催化荧光素实现生物发光,通过检测光信号强度实现受损dna的检测。
59.第一方面优选的技术方案中,所述封闭3'
‑
oh的方式包括但不限于向待测样品中加入双脱氧的腺苷三磷酸(ddatp)对3'
‑
oh末端进行封闭。
60.进一步的,所述双脱氧的腺苷三磷酸(ddatp)在末端转移酶(tdt)的催化作用下对3'
‑
oh末端进行标记。
61.上述优选的技术方案的一些实施例中,为了避免过量的ddatp对后续反应产生影响,上述封闭反应还包括向反应后的体系中加入虾碱性磷酸酶(rsap),用于水解过量的ddatp。
62.本领域公知,糖基化酶能够特异性识别dna链中的损伤类型,并对其进行剪切,使原本的dna链留下空位,被剪切的部分则暴露出3'
‑
oh末端。为了确保糖基化酶识别出损伤dna碱基后能够将dna链剪断暴露出3'
‑
oh,本发明优选的方案中,还加入核酸内切酶(ape1)与糖基化酶共同对损伤位点进行识别和剪切。
63.由于大多数情况下,待测基因组中dna损伤的类型是未知的,为了确定dna损伤类型,本领域技术人员可以将待测样品分为多份,分别加入不同类型的糖基化酶及后续试剂进行处理从而确定待测基因组中的dna损伤类型。
64.优选的,所述富含腺嘌呤(a)和胸腺嘧啶(t)的杂交链的构建方式如下:通过末端转移酶催化胸腺嘧啶的三磷酸脱氧核苷酸加入受损dna的3'
‑
oh末端,形成富含胸腺嘧啶(t)的dna单链,并对应的加入富含腺嘌呤(a)的探针,两者杂交形成富含腺嘌呤(a)和胸腺嘧啶(t)的杂交链。
65.进一步的,所述富含腺嘌呤(a)的探针5'端具有磷酸基团修饰。
66.优选的,本发明所采用的核酸外切酶应当能够对上述杂交链中的富a链进行剪切,并得到腺嘌呤单碱基以实现生物发光,因此能够实现上述剪切效果的核酸外切酶即可应用于本发明所述的检测方法,具体的实例中,所述核酸外切酶为lambda核酸外切酶。lambda核酸外切酶将杂交链中的富a单链剪切成单碱基,而剩下的富t碱基单链可以继续与探针结合启动下一个循环。
67.上述优选技术方案的一种具体的实施方式中,所述检测方法如下:通过tdt催化ddatp封闭基因组dna中所有的3'
‑
oh,加入rsap终止反应;dna糖基化酶切割受损的碱基产生新的3'
‑
oh末端;随后,tdt催化dttps重复加入受损dna的3'
‑
oh末端,产生丰富的poly
‑
t结构单链,加入富a的信号探针杂交形成杂交链;所述杂交链在lambda核酸外切酶水解下产
生大量的amp分子,通过萤火虫荧光素酶和荧光素的协助实现amp
‑
atp
‑
amp的转化,产生生物发光。
68.第一方面又一种优选的方案中,为了进一步提高检测的灵敏度,本发明还通过核酸内切酶(ape1)诱导的循环裂解对检测信号进行放大的检测方法,所述ape1诱导的循环裂解检测方法如下:封闭待测基因组中的3'
‑
oh末端,加入糖基化酶剪切受损碱基并产生新的3'
‑
oh,在新的3'
‑
oh末端扩展富含t碱基的单链,并与含有poly
‑
a序列和ap位点的ap探针杂交形成双链,ape1酶识别ap位点并催化所述杂交双链循环裂解,释放出新的3'
‑
oh序列,新释放的引物序列3'
‑
oh末端重复扩增成为富含腺嘌呤及胸腺嘧啶的杂交双链,以实现再一次的信号放大。
69.进一步的,上述方案中,所述ap位点为至少一个无碱基的位点;具体的实例中,所述ap探针含有poly
‑
a序列和两个无碱基位点(ap位点)的ap探针,其3'端被nh2修饰以防止非特异性扩增。
70.具体的实施方式中,所述ape1诱导的循环裂解信号放大的检测方法步骤如下:通过tdt催化将ddatp引入,封闭所有游离3'
‑
oh末端;随后,添加糖基化酶识别并剪切受损碱基以产生新的3'
‑
oh;被切割的受损dna启动tdt催化的扩增,产生poly
‑
t单链;ap探针与poly
‑
t单链杂交以形成ap探针/poly
‑
t双链dna,随后ape1酶启动双链dna的循环裂解,释放出大量3'
‑
oh的引物;tdt催化dttps重复加到新产生的引物的3'
‑
oh末端,形成长链poly
‑
t序列;产生的poly
‑
t与poly
‑
a信号探针杂交,形成poly
‑
t/信号探针双链;双链中的信号探针被lambda核酸外切酶从其5'
‑
磷酸化末端特异性降解以释放大量amp,剩余的poly
‑
t序列可以与新的信号探针杂交形成新的poly
‑
t/信号探针双链,引发信号探针的循环切割和越来越多amp的释放。
71.本发明第二方面,提供一种用于dna损伤检测的试剂盒,所述试剂盒中至少包括三磷酸腺嘌呤双脱氧核苷酸(ddatps)、胸腺嘧啶的三磷酸脱氧核苷酸(dttps)、荧光素、末端转移酶(tdt)、虾碱性磷酸酶、dna糖基化酶、lambda核酸外切酶、荧光素酶、腺苷酸激酶(ak)和丙酮酸激酶(pk)。
72.优选的,所述试剂盒中,还包括核酸内切酶(ape1)及ap探针。
73.本发明第三方面,提供第一方面所述无标记、均相生物发光的dna损伤检测方法或第二方面所述dna损伤检测试剂盒在疾病诊断、抗肿瘤活性成分筛选等领域的应用。
74.上述应用领域中,所述疾病诊断领域的应用包括用于dna损伤相关疾病的诊断,如肿瘤、神经退行性疾病或其他dna损伤相关疾病,所述其他dna损伤相关疾病的具体实例如着色性干皮病、华沙破损综合征或亨廷顿病等。
75.基于现有或将来可能公开的,以dna损伤或修饰作为诊断标准的疾病均可以施用本发明提供的检测方法。
76.另外,本领域公知,肿瘤细胞修复受损dna的能力较强,抑制肿瘤细胞修复dna损伤的能力也是一种抗肿瘤药物开发思路,因此本发明提供的dna损伤检测方法还可能用于评价抗肿瘤活性成分。
77.为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
78.1、生物发光法检测dna损伤的可行性实验。
79.本实施例中,使用dna中的尿嘧啶碱基(u)作为目标物来验证该方法的可行性(图3)。尿嘧啶是一种常见的dna损伤,可引起胞嘧啶脱氨基,并在dutp误入时引起突变性u/g错配或遗传毒性u/a对。首先利用凝胶电泳验证了ddatp对3'
‑
oh的封闭作用(图1)。在ddatp和rsap存在的情况下,在正常dna(图3a,泳道1)或u损伤dna(图3a,泳道2)中仅观察到39bp的条带,未检测到扩增条带,因为dsdna首先经历tdt介导的ddatp标记反应,随后rsap去除游离多余的ddatp。tdt催化dttps重复延伸到dna的3'
‑
oh末端时dsdnas被ddatp阻断。因此,无论是正常dna(图3a,泳道1)还是受损dna(图3a,泳道2)在tdt的催化下都不能形成poly
‑
t结构的扩增条带,条带的位置与未处理的双链dna(图3a,泳道5和泳道6)相同。相反,在没有ddatp和rsap的情况下,正常dna(图3a,泳道3)和u损伤dna(图3a,泳道4)都观察到超过500bp的大条带,表明tdt催化的无模板等温扩增产生了大分子量dna片段。这些结果表明,ddatp可以有效地阻断dsdna的3'
‑
oh末端。
80.为了验证dna糖基化酶是否可以去除受损的dna碱基,本发明测量了正常双链dna和受损双链dna的熔解曲线(图3b)。udg可以通过催化尿嘧啶和dna骨架之间的n
‑
糖苷键断裂,释放受损的碱基并产生ap位点来特异性识别和切除尿嘧啶。ape1在ap位点切割磷酸二酯骨架,产生带有3'
‑
oh和5'
‑
po4的dna单链断裂。经udg和ape1处理后,正常dsdna的熔链温度为78℃(图3b),损伤dna的熔化温度为65℃(图3b)。损伤dsdna的tm值远低于正常dsdna。熔链温度的降低表明,dna糖基化酶在u损伤dna中断裂损伤单链的磷酸二酯键,形成新的3'
‑
oh末端。
81.本实施例进一步使用凝胶电泳来验证dna糖基化酶切割后tdt催化的无模板等温扩增(图3c)。正常dna仅观察到39bp的条带(图3c,泳道1),表明ddatp封闭了双链dna的3'
‑
oh末端,没有出现扩增。相反,在u损伤dna存在的情况下,出现明显的扩增带,表明由dna糖基化酶诱导形成新的3'
‑
oh末端和由tdt催化的poly
‑
t结构(图3c,泳道2)。这一结果清楚地表明,只有损伤的dna才能在tdt的催化下形成poly
‑
t结构。
82.最后,本实施例进行了生物发光测量,以监测lambda核酸外切酶消化产生的裂解产物amp(图3d)。对照组由于缺乏amp产物,未检测到生物发光信号,证明dna损伤(图3d)。相反,在u损伤dna存在的情况下观察到增强的生物发光信号(图3d),表明只有u损伤dna可以诱导lambda核酸外切酶消化介导的amp释放,通过amp到atp的转化产生明显的生物发光信号。
83.2、研究不同数目和位置的dna损伤。
84.本实施例设计了一个含有1个尿嘧啶碱基的损伤dna,即u损伤dna和两个含有2个尿嘧啶碱基的损伤dna,即u
‑
6损伤dna和u
‑
17损伤dna,u
‑
6损伤dna的两个尿嘧啶碱基间距为6bp(即聚集dna损伤),u
‑
17损伤dna的两个尿嘧啶碱基间距为17bp(即单碱基dna损伤)。值得注意的是,聚集的dna损伤通常由两个或两个以上的损伤紧密地分布在dna的一个或两个螺旋内,而在单碱基的dna损伤中,两个损伤的间距相对较远。在没有损伤位点的情况下,没有检测到生物发光信号(图4)。相反,在存在1个损伤碱基的dna(图4)、聚集损伤碱基(图4)和单碱基损伤碱基(图4)的情况下观察到强烈的生物发光信号。无论两个损伤碱基的位置如何,含两个损伤位点的dsdna的生物发光信号都远高于仅含一个损伤位点的dsdna,并且u
‑
6损伤dna的生物发光信号与u
‑
17损伤dna的生物发光信号相同。这些结果表明,该方法能够区分含有不同数目损伤的dsdna,传统方法难以检测的聚集损伤也能准确地检测出。
85.值得注意的是,大多数报道的基于ber的受损碱基检测方法依赖于模板,它们只能检测单碱基的dna损伤而不能检测聚集的dna损伤,因为聚集损伤2个碱基的位置在1个螺旋内,并且断裂的双链dna在切除修复后可能产生不稳定的单链dna,这阻止了dna聚合酶以模板依赖的方式标记聚集损伤位点。tdt可以识别和捕获在dna糖基化酶从聚集损伤dna中去除损伤碱基的过程中产生的3'末端的非常短的寡核苷酸(大约只有4个核苷酸),由于tdt的引入,本实施例的方法可以检测到单碱基损伤和聚集损伤。所获得的单链dna在tdt的催化下以无模板的方式形成poly
‑
t结构,然后与含ra的信号探针配对形成双链。lambda核酸外切酶的加入可以水解双链中的信号探针,释放丰富的amp来启动环amp焦磷酸化
‑
atp去焦磷酸化以产生增强的生物发光信号。
86.表1本实施例中所使用的dna序列
[0087][0088][0089]
注:下划线粗体u表示尿嘧啶脱氧核糖核苷酸,下划线粗体g表示8
‑
氧代鸟嘌呤(8
‑
oxog)脱氧核糖核苷酸。带下划线的粗体字母“x”表示ap位点。信号探针中的ra表示腺嘌呤核苷酸。
[0090]
3、检测选择性。
[0091]8‑
氧代
‑
7,8
‑
二氢鸟嘌呤(8
‑
oxog)是由活性氧(ros)氧化鸟嘌呤(g)杂环产生的最显著的氧化碱基加合物,fpg作为一种双功能dna糖基化酶,可以去除8
‑
氧基,切割dna骨架,形成单核苷酸间隙。本实施例设计了一个在5'端21个碱基上含有一个8
‑
oxog碱基的dna探针(8
‑
oxog损伤dna),作为fpg的底物。用两种dna损伤(u损伤dna和8
‑
oxog损伤dna)和两种dna糖基化酶(udg和fpg)分析该方法的选择性。如图5所示,当udg和ape1用作dna糖基化酶时,u损伤dna(图5,红色柱)产生生物发光信号,而8
‑
oxog损伤dna(图5)没有产生明显的生
物发光信号。当fpg和ape1用作dna糖基化酶时,8
‑
oxog损伤dna(图5)产生高生物发光信号,而u损伤dna(图5)没有产生明显的生物发光信号。结果表明,该方法对特异性dna损伤具有良好的选择性,结合不同的dna糖基化酶可以特异性地检测不同的dna损伤。
[0092]
4、检测灵敏度。
[0093]
为了评估该方法的灵敏度,本实施例在最佳条件下测量了不同浓度的u
‑
6损伤dna产生的生物发光信号。如图6a所示,生物发光信号随着u
‑
6损伤dna浓度的增加和时间延长而增强,并在25分钟内达到最大值。当u
‑
6损伤dna浓度从1.0
×
10
‑9增加到1.0
×
10
‑7mol/l时,生物发光信号也随之不断增强(图6b)。在对数坐标系中,生物发光信号与损伤dna浓度在1.0
×
10
‑9~1.0
×
10
‑7mol/l范围内呈线性相关(图6c),回归方程为b=31976.75log
10
c 294865.51(r2=0.9878),其中c代表损伤dna的浓度(mol/l),b代表生物发光强度。按对照组信号加3倍标准偏差的原则计算,检出限为8.26
×
10
‑
10
mol/l。
[0094]
5、改进方法的灵敏度分析
[0095]
为了验证改进方法的灵敏度,本实施例在最佳条件下测量了不同浓度损伤dna(u
‑
6损伤dna)产生的生物发光信号。当u
‑
6损伤dna浓度从1.0
×
10
‑
13
增加到1.0
×
10
‑8mol/l时,生物发光值随之提高,并在25min内达到平台(图7a、7b)。在对数坐标中,生物发光信号在1.0
×
10
‑
13
至1.0
×
10
‑8mol/l范围内与u
‑
6损伤dna的浓度呈线性相关(图7c)。回归方程为b=19734.53log
10
c 276062.34(r2=0.9900),其中c代表损伤dna的浓度(mol/l),b代表生物发光强度。检出限为3.26
×
10
‑
14
mol/l。改进方法的灵敏度比原方法(图6c)提高了3个数量级;比非天然核苷类似物标记法(1
×
10
‑
10
mol/l)和高效液相色谱
‑
串联质谱法(1
×
10
‑
10
mol/l)提高了4个数量级;比基于适配体的荧光法(3
×
10
‑9mol/l)高5个数量级。为探讨此改进方法检测混合物中尿嘧啶的可行性,我们将u
‑
6损伤dna与正常dna按不同比例混合,测定了人工混合物中尿嘧啶的含量。测量出的u
‑
6损伤dna水平(y)与输入的u
‑
6损伤dna水平(x)呈良好的线性关系(图7d)。回归方程为y=0.9831x 0.0402(r2=0.9993)。值得注意的是,此改进的方法甚至可以区分混合物中低至0.001%的尿嘧啶,优于突增的高度特异性共扩增的低温pcr反应(cold
‑
pcr)方法(0.01%),与单分子计数方法(0.001%)相当。
[0096]
6、细胞基因组dna损伤的检测
[0097]
为了证明改进的方法检测真实生物样本中dna损伤的可行性,本实施例测量了肿瘤细胞和正常细胞基因组中的尿嘧啶碱基水平。肿瘤细胞包括人肺腺癌细胞系(a549细胞)和人宫颈癌细胞系(hela细胞),正常细胞包括人胚肾细胞系(hek
‑
293细胞)和人胚肺成纤维细胞系(mrc
‑
5细胞)。本实施例使用qiaamp dna微量试剂盒(德国qiagen)提取所有细胞基因组dna,使用nanodrop 2000分光光度计(thermo scientific,wilmington,usa)测量基因组dna的浓度(ng/μl)。与包括hek
‑
293细胞(图8a)和mrc
‑
5细胞(图8a)在内的正常细胞相比,在癌细胞hela细胞(图8a)和a549细胞(图8a)中测得更高的基因组尿嘧啶水平,与文献报道的lc
‑
ms/ms方法结果一致。本实施例进一步以hela细胞基因组dna为模型,研究了生物发光信号与dna质量的关系。如图8b所示,生物发光信号随基因组dna含质量从0.05增加到200ng而增强,回归方程为b=24657.92log
10 n 58484.54(r2=0.9951),其中n代表基因组dna含量(ng),b代表生物发光强度。检测限计算为0.011ng。结果表明,改进后的方法可用于尿嘧啶全基因组分析,具有较高的准确性和可靠性。
[0098]
实施例1
[0099]
(1)双链dna底物的制备
[0100]
1微摩尔每升(μm)可以相互杂交的寡核苷酸加入退火缓冲溶液(50mm nacl,10mm tris
‑
hcl,ph 8.0)中,在95℃下孵育5分钟,然后缓慢冷却至室温形成双链dna(dsdna)结构。退火得到的dsdna底物用退火缓冲溶液稀释至不同浓度。获得的所有dsdna底物在4℃中保存,以备后续实验使用。
[0101]
(2)等温信号放大反应
[0102]
等温信号放大反应过程包括四个连续的步骤。第一步,将不同浓度的dsdna、30μm ddatp、16u tdt、0.25mm cocl2和1
×
tdt缓冲液(50mm醋酸钾,20mm tris
‑
醋酸,10mm醋酸镁,ph 7.9)在总体积为10μl的体系中混合,并在37℃中孵育2小时,接着在75℃孵育20分钟以灭活tdt。第二步,将0.3u rsap、1u udg、0.1u ape1和1
×
tdt缓冲液混合并加入到反应体系中,使总体积为15μl。并将混合后的溶液在37℃中孵育60分钟,然后在95℃中反应10分钟。第三步,包含1.5μl 100mm dttp、6u tdt、0.25mm cocl2、1
×
tdt缓冲液以及上一步反应产物的总体积为20μl的反应溶液在37℃中反应60分钟产生ploy t结构,然后在95℃下加热20分钟终止反应。最后一步,将200nm信号探针2、2u lambda核酸外切酶以及1
×
lambda核酸外切酶反应缓冲液(6.7mm glycine
‑
koh,0.25mm mgcl2,5μg/ml bsa,ph 9.4)加入到溶液中,至总体积为50μl。随后将混合物在37℃下孵育30分钟,以产生大量amp。
[0103]
(3)改进方法的步骤如下
[0104]
第一步,将不同浓度的dsdna、30μm ddatp、16u tdt、0.25mm cocl2和1
×
tdt缓冲液在总体积为10μl的体系中混合,并在37℃中孵育2小时,接着在75℃孵育20分钟以灭活tdt。第二步,将0.3u rsap、1u udg、0.1u ape1和1
×
tdt缓冲液混合并加入到反应体系中,使总体积为15μl,并将混合后的溶液在37℃中孵育60min,然后在95℃中反应10分钟。第三步,包含1.5μl 100mm dttp,6u tdt,0.25mm cocl2,1
×
tdt缓冲液以及上一步反应产物的总体积为20μl的反应溶液在37℃反应60分钟产生ploy t结构,然后在95℃下反应20分钟终止反应。接下来,将0.5μl 10μm ap探针、2u的ape1和1
×
tdt缓冲液加入反应体系中,使得总体积为25μl,并在37℃中反应40分钟,产生大量的3'
‑
oh末端。为了产生更多的poly t结构,再次加入6u tdt、1μl 100mm dttp和1
×
tdt缓冲液加至体积为30μl,在37℃中反应60分钟,随后在95℃反应20分钟以终止反应。最后一步,将200nm信号探针2、2u lambda核酸外切酶以及1
×
lambda核酸外切酶反应缓冲液加入到溶液中,至总体积为50μl。随后将混合物在37℃下孵育30分钟,以产生大量amp。
[0105]
(4)生物发光测量与数据分析
[0106]
将上述含有amp的反应溶液转移到atp检测系统中混合后,在室温下用glomax生物/化学发光检测仪实时监测生物发光信号。atp检测系统由4.0μl amp
‑
atp转换缓冲液(1.0μl 1u/μl ak,1.0μl 1u/μl pk,1.0μl 10mm dctp和1.0μl 4.8mm pep)和6.0μl atp检测缓冲液(0.5mm d
‑
荧光素,1.25μg/ml萤火虫荧光素酶,10mg/ml bsa,500mm tricine缓冲液(ph 7.8),100mm mgso4,2mm edta,100mm dtt)组成。仪器的参数设置分别为:积分时间为1s,运行次数为25次。并选取25次检测中的最大值作为生物发光信号进行后续的数据分析。
[0107]
(5)凝胶电泳测量
[0108]
tdt介导的dna扩增产物经sybr gold染色后,在1
×
tbe缓冲液(44.5mm tris
‑
boric acid,1mm edta,ph 8.2)中,用12%的非变性聚丙烯酰胺凝胶,室温110v恒压下电泳45分钟。最后使用bio
‑
rad chemidoc mp成像系统对凝胶可视化成像。
[0109]
(6)荧光测量
[0110]
tdt介导的dna扩增产物用sybr gold染色后,采用日本日立公司的f
‑
7000荧光分光光度计,以氙灯为激发光源,测量反应产物的荧光信号。激发波长为488nm,光谱记录范围为500
‑
700nm。激发和发射狭缝均为5.0nm。采用540nm(sybr gold的最大发射波长)处的荧光强度用于后续数据分析。
[0111]
(7)熔链温度的测定
[0112]
将100nm损伤dsdna和100nm正常dsdna与udg和ape1在37℃孵育1h。反应后,样品用sybr green i染色,用bio
‑
rad实时荧光检测系统对样品在55℃
‑
95℃的温度下每隔30s检测一次荧光值。熔链温度为
‑
df/dt达到最大值时所对应的温度值。(其中f为荧光强度,t为温度)。
[0113]
(8)细胞培养及基因组dna的提取
[0114]
将人宫颈癌细胞系(hela细胞)、人肺腺癌细胞系(a549细胞)、人胚肾细胞系(hek
‑
293细胞)和正常人肺细胞系(mrc
‑
5细胞)在含有10%胎牛血清(fbs)和1%青霉素
‑
链霉素混合液的dulbecco改良的eagle培养基(dmem)中培养。所有细胞均在37℃、含5%co2湿润的培养箱中培养。在指数生长期时,收集细胞,并使用qiaamp dna mini试剂盒依照说明书提取细胞内dna,并用超微量紫外分光光度计测定提取的dna浓度(ng/μl)。
[0115]
(9)生物发光法检测dna损伤的原理(图1)
[0116]
该方案包括四个部分:(1)tdt介导的ddatp标记反应以封闭dna 3'
‑
羟基(3'
‑
oh)末端,(2)dna糖基化酶催化去除dna损伤,(3)tdt介导的无模板扩增形成poly
‑
t结构,(4)amp的产生和生物发光反应。整个反应系统由ddatp,tdt,rsap,dna糖基化酶,信号探针以及lambda核酸外切酶组成。首先在tdt聚合酶的辅助下将ddatp标记在dna上以阻断初始的3'
‑
oh末端,以防止初始的3'
‑
oh末端对后续反应的影响。添加虾碱性磷酸酶(rsap)以去除过量的ddatp,避免过量的ddatp影响后续反应。然后,在dna糖基化酶和ape1的作用下去除基因组中的dna损伤,并产生一个新的3'
‑
oh位点。紧接着tdt以无模板扩增的方式将多个dttp掺入到新生成的3'
‑
oh位点处,形成poly
‑
t序列。本实施例设计了5'端磷酸基团(5'
‑
drp)修饰且中间含有多个腺嘌呤核糖核苷酸的信号探针。该信号探针可以与生成的poly t序列完全杂交形成poly
‑
t/信号探针的双链结构。在lambda核酸外切酶的催化下,信号探针从5'
‑
drp处逐渐降解,释放出大量amp。这是因为lambda核酸外切酶可以从5'
‑
磷酸化(5'
‑
drp)末端特异性消化双链结构中的单链,但对于核苷酸单链甚至无5'
‑
drp末端的序列的催化效率都极低。生成的amp在pep和dctp的存在下,通过腺苷酸激酶(ak)和丙酮酸激酶(pk)的酶促反应转化为atp,随后在萤光素酶和萤光素的帮助下,atp转化成amp并产生强烈的生物发光信号。值得注意的是,amp又可以通过新的循环转化为atp,从而可以产生放大的生物发光信号。若基因组dna中没有损伤,则在dna糖基化酶的催化下不会产生新的3'
‑
oh位点,也就不会产生明显的生物发光信号。
[0117]
(10)改进的dna损伤检测方法(图2)
[0118]
本实施例进一步引入ape1诱导的循环裂解信号放大技术来提高检测灵敏度。本实施例设计了一个含有poly
‑
a序列和两个无碱基位点(ap位点)的ap探针,其3'端被nh2修饰
以防止非特异性扩增(图2)。通过tdt催化将ddatp引入,封闭所有具有游离3'
‑
oh末端的u损伤的dna(图2),然后加入rsap,通过水解剩余的ddatp停止封闭反应。随后,添加udg和ape1以去除尿嘧啶碱基以产生新的3'
‑
oh。被切割的u损伤dna(图2)可以作为启动tdt催化的无模板等温扩增的引物,扩增产生poly
‑
t结构。ap探针(图2)可以与poly
‑
t结构杂交以形成ap探针/poly
‑
t双链dna(图2),随后ape1酶启动双链dna的循环裂解,释放出大量3'
‑
oh的引物。tdt催化dttps重复加到新产生的引物的3'
‑
oh末端,形成长链poly
‑
t序列(图2)。产生的poly
‑
t可与poly
‑
a信号探针杂交,形成poly
‑
t/信号探针双链(图2)。双链中的信号探针可以被lambda核酸外切酶从其5'
‑
磷酸化末端特异性降解以释放大量amp,剩余的poly
‑
t序列可以与新的信号探针杂交形成新的poly
‑
t/信号探针双链,引发信号探针的循环切割和越来越多amp的释放。在磷酸烯醇式丙酮酸单钠盐水合物(pep)和dctp存在下,通过腺苷酸激酶(ak)和丙酮酸激酶(pk)的酶促反应组合,释放的amps可转化为atps,在萤火虫荧光素酶和荧光素的协助下,atps转化为amps产生了强烈的生物发光信号。与以前的方法相比,改进后的方法由于引入了ap探针,可以产生额外的3'
‑
oh,新产生的引物在tdt催化下可以产生更多的聚poly
‑
t序列,形成更多的poly
‑
t/信号探针双链结构并产生更多的amp分子以产生增强的生物发光信号。
[0119]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。