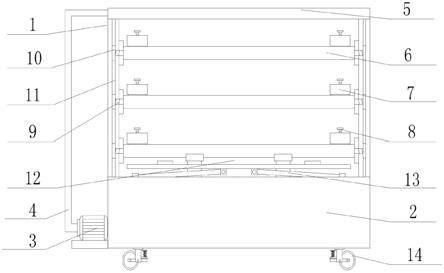
一种茂基铱/铑二聚体的应用
1.本技术为申请号为cn201810305752.7、申请日为2018.04.08、申请人为南京师范大学、发明名称为“一种大黄酸特定基团修饰的有机化合物、其芳基金属配合物、及其制备方法和应用”的分案申请。
技术领域
2.本发明属于医药领域,涉及一种茂基铱/铑二聚体的应用。
技术背景
3.癌症是目前威胁人类生命最严重的疾病之一,根据世界卫生组织公布的数据推测至2020年,全球每年将会新增1500万的癌症患者。目前临床使用的多数铂类及其它药物都具有毒副作用大、选择性差以及耐药性等缺点,因此研究开发新型高效、低毒的抗癌药物已经成为刻不容缓的任务。随着抗癌药物在肿瘤细胞中作用机制的深入研究,近年来已出现不少具有潜在成药的金属抗肿瘤药物,其中,人们发现芳基金属化合物具有较好的抗癌活性,部分芳基金属配合物的抗癌效果与临床使用的顺铂相当甚至超过顺铂,已有个别芳基金属化合物进入临床或临床前试验。但如何更好的解决药物对正常机体的毒副作用成为其是否能成药的关键性因素,因此,设计靶向于肿瘤细胞的药物已成为现代药物的研究热点。在这方面,英国华威大学的peter sadler教授、瑞士洛桑联邦理工学院的paul dyson教授以及奥地利维也纳大学的bernhard keppler教授等人成功地将具有靶向性的分子连接到金属上并获得了具有高效选择性的药物。
4.部分中草药活性成分具有良好的抗癌、抗菌及抗炎活性,如大黄酸等已经在临床使用并取得较好疗效。大黄酸对小鼠黑色素瘤、艾氏腹水癌、肝癌、乳腺癌、p388白血病细胞都有一定的抑制作用;在1.5~25mg/ml深度,大黄酸对葡萄球菌、链球菌、白喉杆菌、枯草杆菌、副伤寒杆菌、痢疾杆菌等均有抑制作用(光谱学与光谱分析,2011,31(9),精细有机化工中间体全书:化学工业出版社,2008)。
5.2007年英国科学家在和ⅱ型糖尿病相关的单核苷酸多态性(snp)研究中发现了和肥胖相关的基因fto(fat mass and obesity associated),fto基因的变异增加了肥胖的概率。由于fto也是一类保守的2og加氧酶,因此通用抑制剂nog也能抑制fto的活性。世界上目前仅有四个研究组发现了6个fto的有机化合物抑制剂。2012年上海药物所的研究人员筛选出大黄酸等3个化合物能抑制fto的活性,其中大黄酸的抑制效果最好,更适合作为候选药物分子。2013年英国科学家用2og加氧酶通用抑制剂即2og类似物进行了筛选,在fto和这些化合物的晶体结构中发现这类抑制剂通过金属离子螯合基团与2og竞争,研究还发现脯氨酰羟化酶抑制剂fg
‑
2216/iox3和fg4592/sellbeckbio均能有效抑制fto,不过这类抑制剂在开发药物时候需避免抑制其他2og加氧酶,提高特异性。2014年美国科学家在研究抗癫痫药物时意外发现所得到的化学物不能有效抑制靶点蛋白phd2,却能有效抑制其2og加氧酶家族的fto,而且ic50值和广谱性2og酶抑制剂nog接近,强于之前报道的大黄酸。2014年底,上海药物所发现甲氯芬那酸可以有效地抑制fto去甲基化单链核酸上的m6a甲基化修
饰,而不影响alkbh5的去甲基化酶活。2014年,新加坡国立大学的研究人员发现了几个新型有效的fto抑制剂,最强的是第12号:4
‑
[n'
‑
(4
‑
benzyl
‑
pyridine
‑3‑
carbonyl)
‑
hydrazino]
‑4‑
oxo
‑
but
‑2‑
enoicacid。目前为止,还未有金属配合物的fto抑制剂的研究报道。基于此,我们将具有抗肿瘤、抗菌等功效的中草药活性成分大黄酸进行特定基团修饰并与金属配位,从而获得了具有较好抑制fto活性、抗癌活性、抗菌活性、且具有其他突出功能的先导化合物。
技术实现要素:
[0006]
本发明目的在于提供一种大黄酸特定基团修饰的有机化合物及其金属配合物、制备方法和应用。该有机化合物由大黄酸的羧酸基团进行化学修饰得到,本身具有良好的抗肿瘤、抗菌功效,以这种有机化合物为配体与金属配位从而得到一系列配合物。该有机化合物及其金属配合物的合成反应单一,简单易得,且产率高。并且通过分析药物的抗癌活性及其抗癌机理,发现不同芳基金属的配合物抗癌效果以及抗癌机理也不同,从而筛选出靶向作用较强的药物,同时具有较好的抑制fto活性。
[0007]
为解决现有技术中的问题,本发明提供的技术方案为:
[0008]
一种大黄酸特定基团修饰的有机化合物,其特征在于具有式(i)及(ii)的结构:
[0009][0010]
其中,r代表ch3,h或
[0011]
一种含有大黄酸特定基团修饰的有机化合物的制备方法,该方法通过在大黄酸分子的羧酸基团进行功能化修饰,所述功能化修饰包括如下步骤:在惰性氛围下,将大黄酸、edci或dcc、hobt(1
‑
羟基苯并三唑)、tea以及桥联配体溶解于有机溶剂中进行反应,将反应生成的粗产物通过柱层析方法分离纯化,即可得到大黄酸特定基团修饰的有机化合物,其中桥联配体为2
‑
(1
‑
咪唑基)乙胺、4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺、4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶或2,2'
‑
联吡啶
‑4‑
甲胺
[0012]
进一步优选,在惰性氛围下,将大黄酸、edci或dcc、hobt(1
‑
羟基苯并三唑)、tea以及桥联配体溶解于有机溶剂中于
‑
20~120℃反应2~72h,将反应生成的粗产物通过柱层析方法分离纯化,即可得到大黄酸特定基团修饰的有机化合物,其中大黄酸与桥联配体的反应摩尔比为10:1~1:10,桥联配体为2
‑
(1
‑
咪唑基)乙胺、4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺、4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶或2,2'
‑
联吡啶
‑4‑
甲胺,有机溶剂为n,n
‑
二甲基甲酰胺或其它
甲酰胺溶剂、乙酰胺溶剂。edci或dcc用于羧基的活化试剂,hobt用于缩合反应的催化剂。
[0013]
上述的大黄酸特定基团修饰的有机化合物在用于制备抗癌药物、抗癌药物组分、抗菌药物、抗菌药物组分的应用。
[0014]
一种基于上述大黄酸特定基团修饰的有机化合物的金属配合物,具有通式(iii)及(iv)的结构:
[0015][0016]
其中,r1代表ch3,h或r2代表x代表cl、br或i,m代表ru,os,ir或rh。
[0017]
一种上述的金属配合物的制备方法,该方法包括如下步骤:在惰性氛围下,将相对应的芳基金属二聚体与上述具有式(ⅰ)或(ii)结构的有机化合物溶解于有机溶剂中进行反应,反应结束后加入乙醚离心得到产物或加入nh4pf6进行冷冻后离心得到产物,所述相对应的芳基金属二聚体为[ru(η6‑
p
‑
cymene)cl2]2、[ru(η6‑
p
‑
cymene)i2]2、[ru(η6‑
p
‑
bip)cl2]2、[os(η6‑
p
‑
cymene)cl2]2、[os(η6‑
p
‑
bip)cl2]2、[ir(η5‑
cp
*
)cl2]2、[ir(η5‑
cp
xph
)cl2]2、[rh(η5‑
cp
*
)cl2]2或[rh(η5‑
cp
xph
)cl2]2。
[0018]
进一步优选,在惰性氛围下,将相对应的芳基金属二聚体与上述具有式(ⅰ)或(ⅱ)结构的大黄酸有机化合物溶解于有机溶剂中于2~90℃回流反应2~72h,反应结束后加入乙醚离心得到产物或加入nh4pf6于
‑
20~4℃冷冻1h后离心得到产物,其中相对应的芳基金属二聚体与具有式(ⅰ)或(ⅱ)的结构的有机化合物的反应摩尔比为10:1~1:20,相对应的芳基金属二聚体为[ru(η6‑
p
‑
cymene)cl2]2、[ru(η6‑
p
‑
cymene)i2]2、[ru(η6‑
p
‑
bip)cl2]2、[os(η6‑
p
‑
cymene)cl2]2、[os(η6‑
p
‑
bip)cl2]2、[ir(η5‑
cp
*
)cl2]2、[ir(η5‑
cp
xph
)cl2]2、[rh(η5‑
cp
*
)cl2]2或[rh(η5‑
cp
xph
)cl2]2,有机溶剂为甲醇、乙腈、二氯甲烷、乙醇、氯仿中的一种或多种混合。
[0019]
上述的金属配合物在用于制备fto抑制剂药物、fto抑制剂药物组分、减肥药物、减肥药物组分、抗癌药物、抗癌药物组分、抗菌药物、抗菌药物组分的应用。
[0020]
上述的金属配合物在dna(核酸)构型转变诱导剂药物、dna构型转变诱导剂药物组分方面的应用。
[0021]
一种相对应的芳基金属二聚体在用于制备fto抑制剂药物、fto抑制剂药物组分、减肥药物、减肥药物组分方面的应用,其中相对应的芳基金属二聚体为[ru(η6‑
p
‑
cymene)cl2]2、[ru(η6‑
p
‑
cymene)i2]2、[ru(η6‑
p
‑
bip)cl2]2、[os(η6‑
p
‑
cymene)cl2]2、[os(η6‑
p
‑
bip)cl2]2、[ir(η5‑
cp
*
)cl2]2、[ir(η5‑
cp
xph
)cl2]2、[rh(η5‑
cp
*
)cl2]2或[rh(η5‑
cp
xph
)cl2]2。
[0022]
上述金属配合物合成方法的反应链为:
[0023][0024]
本发明经桥联配体2
‑
(1
‑
咪唑基)乙胺,4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺,4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶或2,2'
‑
联吡啶
‑4‑
甲胺修饰后的新型含有大黄酸基团的有机化合物可以合成不同具有靶向作用的尾接有机导向分子的芳基金属配合物,且都具有较高的抗癌、抗菌活性。
[0025]
本发明的芳基铑金属抗癌配合物可以较好定位于癌细胞的细胞核中,并且芳基铑配合物与芳基铱/铑二聚体均具有抑制fto活性的作用,对于配合物而言,可能是由于引入大黄酸官能团使其具有抑制fto活性,对于二聚体而言,具有抑制fto活性可能与金属茂基以及金属价态有直接关系,并且配合物可以将dna由b型转变为z型。
[0026]
本发明的芳基铱/铑金属配合物可以较好定位于癌细胞的细胞核中,可以将dna由b型转变为z型,且可以提高癌细胞的活性氧。
[0027]
有益效果:
[0028]
本发明提供了一种由大黄酸羧酸基团经桥联配体2
‑
(1
‑
咪唑基)乙胺,4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺,4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶或2,2'
‑
联吡啶
‑4‑
甲胺修饰后得到的新型含有大黄酸基团有机化合物,并且该化合物可以与芳基金属二聚体合成不同具有靶向作用的抗癌、抗菌及减肥功能的金属配合物,本发明在制备过程中反应单一、简单,这为后续合成分离带来方便,易于提高反应产率。本发明的含有大黄酸基团有机化合物及其金属配合物具有很好的抑制fto活性、抗癌活性、抗菌活性、诱导dna构型转变作用与选择性,具有可能成药的前景,芳基金属二聚体则是很好的fto抑制剂,在减肥方面有突出功能。
附图说明
[0029]
图1是实施例7的芳基铱金属配合物在细胞中的细胞定位、促进细胞中活性氧
(ros)生成的图。
[0030]
图2是实施例8的芳基铑金属配合物在细胞中的细胞定位、实施例8及其二聚体对fto活性的有效抑制图。
[0031]
图3是实施例7、8芳基铱/铑金属配合物诱导dna构型转变图。
[0032]
图4是本发明基于大黄酸特定基团修饰的有机化合物的金属配合物诱导dna构型转变以及该金属配合物、芳基金属二聚体对fto活性抑制的示意图。
具体实施方式
[0033]
以下结合附图和具体实施例对本发明的技术方案做进一步说明,但本发明所要求的保护范围并不局限于此。
[0034]
实施例1
[0035]
在氩气氛围下,将大黄酸(0.14g,0.5mmol),edci(1.05g,5.5mmol),hobt(0.61g,4.5mmol)以及桥联配体4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺(1.0g,5.0mmol)溶解于dmf(n,n
‑
二甲基甲酰胺)中,并滴加100μl的tea(三乙胺),冰浴下搅拌5h,粗产物通过柱层析方法分离纯化,得到经桥联配体修饰后的有机化合物l1,产率为55%。
[0036]1h nmr(400hz,d6‑
dmso):11.93(s,2h),9.66(t,1h),8.63(d,1h),8.52(d,1h),8.38(s,1h),8.23(m,2h),7.85(dd,2h)7.77(d,1h),7.42(t,2h),7.28(d,1h),4.63(d,2h),2.42(s,3h)。
[0037]
上述桥联配体4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺制备方法如下:步骤(1):seo2(二氧化硒),1,4
‑
dioxide(1,4
‑
二氧六环),4,4'
‑
dimethyl
‑
2,2'
‑
bipyridyl(4,4'
‑
二甲基
‑
2,2'
‑
联吡啶)混合于三颈瓶中,回流加热反应0.4
‑
45h,反应结束后,冷却过滤,旋转蒸发溶剂,残留物中加入nahco3溶液,二氯甲烷萃取,旋蒸除去溶剂,残留物中加入一定浓度的na2s2o5,过滤,滤液调ph至4
‑
12,再用二氯甲烷萃取,减压蒸去溶剂,产物为固体粉末;
[0038]
步骤(2):盐酸羟胺,k2co3以及步骤(1)中的产物溶于甲醇与水的混合溶液中,加热条件下反应一段时间,反应结束后冷却至室温,加入适量的水,过滤,得固体粉末;
[0039]
步骤(3):步骤(2)中的产物,醋酸铵以及氨水溶于乙醇与水的混合溶液中,回流加热一段时间,加入锌粉,反应0.2
‑
4h,冷却至室温,过滤,旋蒸除去溶剂,加入强碱溶液,使用二氯甲烷萃取,减压蒸去溶剂,产物为固体粉末状的4
‑
(4'
‑
甲基
‑
2,2'
‑
联吡啶)
‑
甲胺。
[0040]
实施例2
[0041]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[ru(η6‑
p
‑
cymene)cl2]2(0.31g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,结束后加入nh4pf6,
‑
4度冷冻1h,离心得到配合物1,产率为65%。
[0042]1h nmr(400hz,d6‑
dmso):9.72(s,1h),9.44(d,1h),9.36(d,1h),8.60(s,1h),8.51(s,1h),8.17(s,1h),7.90(s,1h)7.81(m,1h),7.74(d,1h),7.66(dd,2h),7.41(d,1h),6.19(d,2h),5.94(d,2h),4.75(d,2h),3.17(s,2h),2.59(s,3h),2.33(m,1h),2.15(s,3h),0.95(d,6h)。esi
‑
ms( ):理论值:[1
‑
pf6‑
]
m/z:736.20,实验值:m/z 736.25。
[0043]
实施例3
[0044]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[ru(η6‑
p
‑
cymene)i2]2(0.489g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到
配合物2,产率为75%。
[0045]1h nmr(400hz,d6‑
dmso):11.95(s,2h),9.68(s,1h),9.40(d,1h),9.32(d,1h),8.61(s,1h),8.54(s,1h),8.25(s,1h)7.88(dd,2h),7.79(d,1h),7.64(dd,2h),7.45(d,1h),6.13(d,2h),6.00(d,2h),4.78(d,2h),2.72(m,1h),2.60(s,3h),2.39(s,3h),0.99(d,6h)。esi
‑
ms( ):理论值:[2
‑
i
‑
]
m/z 828.65,实验值:m/z 828.17。
[0046]
实施例4
[0047]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[ru(η6‑
p
‑
bip)cl2]2(0.326g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入nh4pf6,
‑
4度冷冻1h,离心得到配合物3,产率为62%。
[0048]1h nmr(400hz,d6‑
dmso):9.57(s,2h),9.29(d,1h),9.19(d,1h),8.56(s,2h),8.47(s,2h),7.99(s,1h),7.68(m,5h)7.55(dd,2h),7.41(dd,3h),7.31(d,1h),6.62(s,2h),6.43(dd,2h),6.19(t,1h),4.69(d,2h),2.53(s,3h)。esi
‑
ms( ):理论值:[3
‑
pf6‑
]
m/z 756.17,实验值:m/z756.19。
[0049]
实施例5
[0050]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[os(η6‑
p
‑
cymene)cl2]2(0.395g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入nh4pf6,
‑
4度冷冻1h,离心得到配合物4,产率为65%。
[0051]1h nmr(400hz,d6‑
dmso):11.96(s,2h),9.81(t,1h),9.42(d,1h),9.33(d,1h),8.73(s,1h),8.64(s,1h),8.24(d,1h)7.94(d,1h),7.86(m,1h),7.77(dd,1h),7.65(dd,1h),7.45(dd,1h),6.42(d,2h),6.11(d,2h),4.83(s,2h),2.64(s,3h),2.45(m,1h),2.24(s,3h),0.89(d,6h)。esi
‑
ms( ):理论值:[4
‑
pf6‑
]
m/z 826.36,实验值:m/z 826.33。
[0052]
实施例6
[0053]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[ir(η5‑
cp
*
)cl2]2(0.398g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物5,产率为78%。
[0054]1h nmr(400hz,d6‑
dmso):12.01(s,2h),9.87(t,1h),8.98(d,1h),8.88(d,1h),8.82(s,1h),8.73(s,1h),8.31(d,1h)7.99(d,1h),7.93(m,1h),7.84(m,2h),7.76(d,1h),7.52(m,1h),4.88(d,2h),2.70(s,3h),1.70(s,15h)。esi
‑
ms( ):理论值:[5
‑
cl
‑
]
m/z 828.36,实验值:m/z828.24。
[0055]
实施例7
[0056]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[ir(η5‑
cp
xph
)cl2]2(0.460g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物6,产率为78%。
[0057]1h nmr(400hz,d6‑
dmso):11.95(s,2h),9.76(t,1h),8.71(s,1h),8.65(d,1h),8.52(d,1h),8.24(d,1h),7.92(d,1h)7.87(m,1h),7.79(m,1h),7.73(d,1h),7.65(d,1h),7.59(dd,1h),7.47(m,6h),7.40(m,2h),4.81(d,2h),2.62(s,3h),1.77(d,6h),1.70(d,6h)。esi
‑
ms( ):理论值:[6
‑
cl
‑
]
m/z 738.05,实验值:m/z 738.10。
[0058]
实施例8
[0059]
在氩气氛围下,将有机化合物l1(0.05g,0.1mmol)与[rh(η5‑
cp
*
)cl2]2(0.311g,
(0.489g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物10,产率为75%。
[0075]1h nmr(400hz,d6‑
dmso):11.89(s,2h),9.56(s,1h),9.40(d,1h),9.35(d,1h),8.74(s,1h),8.58(s,1h),8.25(s,1h)7.87(dd,2h),7.76(d,1h),7.62(dd,2h),7.49(d,1h),6.13(d,2h),6.00(d,2h),4.74(d,2h),2.72(m,1h),2.55(s,3h),0.98(d,6h)。esi
‑
ms( ):理论值:[10
‑
i
‑
]
m/z813.78,实验值:m/z 813.53。
[0076]
实施例13
[0077]
在氩气氛围下,将有机化合物l2(0.045g,0.1mmol)与[ru(η6‑
p
‑
bip)cl2]2(0.326g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入nh4pf6,
‑
4度冷冻1h,离心得到配合物11,产率为68%。
[0078]1h nmr(400hz,d6‑
dmso):9.76(s,2h),9.35(d,1h),9.22(d,1h),8.65(s,2h),8.52(s,2h),7.86(s,1h),7.63(m,5h)7.55(dd,2h),7.42(dd,3h),7.36(d,1h),6.59(s,2h),6.40(dd,2h),6.19(t,1h),4.63(d,2h)。esi
‑
ms( ):理论值:[11
‑
pf6‑
]
m/z 741.23,实验值:m/z 741.57。
[0079]
实施例14
[0080]
在氩气氛围下,将有机化合物l2(0.045g,0.1mmol)与[os(η6‑
p
‑
cymene)cl2]2(0.395g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入nh4pf6,
‑
4度冷冻1h,离心得到配合物12,产率为65%。
[0081]1h nmr(400hz,d6‑
dmso):11.89(s,2h),9.81(t,1h),9.48(d,1h),9.35(d,1h),8.73(s,1h),8.68(s,1h),8.33(d,1h)7.98(d,1h),7.83(m,1h),7.72(dd,1h),7.65(dd,1h),7.48(dd,1h),6.48(d,2h),6.09(d,2h),4.85(s,2h),2.45(m,1h),2.24(s,3h),0.89(d,6h)。esi
‑
ms( ):理论值:[12
‑
pf6‑
]
m/z 811.63,实验值:m/z 811.33。
[0082]
实施例15
[0083]
在氩气氛围下,将有机化合物l2(0.045g,0.1mmol)与[ir(η5‑
cp
*
)cl2]2(0.398g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物13,产率为78%。
[0084]1h nmr(400hz,d6‑
dmso):12.11(s,2h),9.83(t,1h),8.95(d,1h),8.85(d,1h),8.82(s,1h),8.69(s,1h),8.40(d,1h)7.99(d,1h),7.89(m,1h),7.82(m,2h),7.76(d,1h),7.50(m,1h),4.82(d,2h),1.73(s,15h)。esi
‑
ms( ):理论值:[13
‑
cl
‑
]
m/z 813.56,实验值:m/z 813.75。
[0085]
实施例16
[0086]
在氩气氛围下,将有机化合物l2(0.045g,0.1mmol)与[ir(η5‑
cp
xph
)cl2]2(0.460g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物14,产率为63%。
[0087]1h nmr(400hz,d6‑
dmso):11.89(s,2h),9.67(t,1h),8.75(s,1h),8.62(d,1h),8.55(d,1h),8.31(d,1h),7.93(d,1h)7.89(m,1h),7.81(m,1h),7.76(d,1h),7.61(d,1h),7.55(dd,1h),7.42(m,6h),7.37(m,2h),4.83(d,2h),1.75(d,6h),1.69(d,6h)。esi
‑
ms( ):理论值:[14
‑
cl
‑
]
m/z 723.11,实验值:m/z 723.23。
[0088]
实施例17
[0089]
在氩气氛围下,将有机化合物l2(0.045g,0.1mmol)与[rh(η5‑
cp
*
)cl2]2(0.311g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物15,产率为68%。
[0090]1h nmr(400hz,d6‑
dmso):11.89(s,2h),9.72(t,1h),8.95(d,1h),8.88(d,1h),8.63(s,1h),8.54(s,1h),8.30(d,1h)7.90(s,1h),7.83(m,1h),7.78(dd,2h),7.71(d,1h),7.51(d,1h),4.72(d,2h),1.66(s,15h)。esi
‑
ms( ):理论值:[15
‑
cl
‑
]
m/z 723.13,实验值:m/z 723.05。
[0091]
实施例18
[0092]
在氩气氛围下,将有机化合物l2(0.045g,0.1mmol)与[rh(η5‑
cp
xph
)cl2]2(0.371g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物16,产率为60%。
[0093]1h nmr(400hz,d6‑
dmso):11.98(s,2h),9.75(t,1h),8.69(s,1h),8.63(d,2h),8.42(d,1h),8.26(d,1h),7.83(m,2h)7.79(d,1h),7.71(d,1h),7.63(d,1h),7.49(m,5h),7.43(d,1h),4.73(m,2h),1.78(d,6h),1.71(d,6h)。
[0094]
实施例19
[0095]
在氩气氛围下,将大黄酸(0.28g,1.0mmol),edci(1.05g,5.5mmol),hobt(0.61g,4.5mmol)以及桥联配体4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶(1.07g,5.0mmol)溶解于dmf中,并滴加100μl的tea(三乙胺),冰浴下搅拌5h,粗产物通过柱层析方法分离纯化,得到经桥联配体修饰后的有机化合物l3,产率为55%。
[0096]1h nmr(400hz,d6‑
dmso):11.98(s,4h),9.61(t,2h),8.63(d,1h),8.52(d,1h),8.38(s,1h),8.26(m,4h),7.81(dd,3h),7.77(d,1h),7.42(t,4h),7.28(d,1h),4.68(d,4h)。
[0097]
上述桥联配体4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶制备方法如下:
[0098]
步骤(1):4,4'
‑
二硝基
‑
2,2'
‑
联吡啶与浓硫酸在冰浴条件下滴加硝酸,混合溶液回流反应一段时间,冷却至室温,再于
‑
40度(液氮)中持续搅拌直至淡黄色沉淀析出,过滤沉淀,水洗后干燥,得固体粉末;
[0099]
步骤(2):步骤(1)中的产物,pd/c(10%)氮气氛围下于乙醇中回流反应至溶液澄清,缓慢滴加水合肼,继续回流反应0.5
‑
15h,反应结束后,趁热过滤,滤饼用热乙醇洗,将滤饼置于冰水中于冰箱过夜,沉淀过滤,冰水洗后干燥,得到产物4,4'
‑
二甲胺
‑
2,2'
‑
联吡啶。
[0100]
实施例20
[0101]
在氩气氛围下,将有机化合物l3(0.075g,0.1mmol)与[ru(η6‑
p
‑
cymene)cl2]2(0.31g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入nh4pf6,
‑
4度冷冻1h,离心得到配合物17,产率为68%。
[0102]1h nmr(400hz,d6‑
dmso):9.81(s,2h),9.43(d,2h),9.37(d,2h),8.68(s,1h),8.57(s,1h),8.20(s,1h),7.95(s,1h),7.81(m,2h),7.74(d,1h),7.65(dd,4h),7.52(d,1h),6.15(d,2h),5.93(d,2h),4.87(d,4h),3.20(s,4h),2.38(m,1h),2.12(s,3h),0.89(d,6h)。esi
‑
ms( ):理论值:[17
‑
pf6‑
]
m/z:1002.68,实验值:m/z 1002.52。
[0103]
实施例21
[0104]
在氩气氛围下,将大黄酸(0.28g,1.0mmol),edci(1.05g,5.5mmol),hobt(0.61g,
4.5mmol)以及桥联配体2
‑
(1
‑
咪唑基)乙胺(0.56g,5.0mmol)溶解于dmf中,并滴加100μl的tea(三乙胺),冰浴下搅拌3h,粗产物通过柱层析方法分离纯化,得到经桥联配体修饰后的有机化合物l4,产率为42%。
[0105]1h nmr(400hz,d6‑
dmso):11.93(s,2h),9.66(t,1h),9.23(d,1h),8.56(d,1h),8.24(s,1h),8.23(m,2h),7.85(dd,1h),7.42(t,2h),4.63(d,2h),4.45(d,2h)。
[0106]
上述桥联配体2
‑
(1
‑
咪唑基)乙胺制备方法如下:
[0107]
步骤(1):将适量的咪唑、丙烯酸乙酯和fecl3·
6h2o溶于水中,加热反应0.1~15h,过滤,滤液用二氯甲烷萃取,减压蒸去溶剂,粗产物通过柱层析法分离纯化,得淡黄色液体;
[0108]
步骤(2):将水合肼与乙醇混合,加热条件下缓慢滴加乙醇与步骤(1)产物的混合溶液,加热回流反应0.2~9h,二氯甲烷萃取,减压蒸去溶剂,粗产物通过柱层析法分离纯化,得黄色油状物;
[0109]
步骤(3):将步骤(2)中的产物、水和浓硫酸加入到三颈瓶中,低温条件下缓慢滴加到含有nano2的溶液中,反应0.1~20h后,加热条件下反应0.3~48h,冷却至室温,加氢氧化钠水溶液调ph至3~13,减压蒸去溶剂,向剩余物中加入甲醇,抽滤,粗产物通过柱层析法分离纯化,得淡黄色油状物2
‑
(1
‑
咪唑基)乙胺。
[0110]
实施例22
[0111]
在氩气氛围下,将有机化合物l4(0.04g,0.1mmol)与[ir(η5‑
cp
*
)cl2]2(0.398g,0.5mmol)溶解于甲醇中,65度条件下回流反应2h,反应结束后加入乙醚,离心得到配合物18,产率为78%。
[0112]1h nmr(400hz,d6‑
dmso):12.01(s,2h),9.87(t,1h),9.05(d,1h),8.98(d,1h),8.73(s,1h),8.38(d,1h),7.93(m,1h),7.84(m,2h),7.52(m,1h),4.88(d,2h),4.58(d,2h),1.70(s,15h)。esi
‑
ms( ):理论值:[18
‑
cl
‑
]
m/z 740.11,实验值:m/z 740.28。
[0113]
实施例23
[0114]
本发明大黄酸特定基团修饰的有机化合物及其金属配合物在制备抗肿瘤药物方面的应用。
[0115]
方法:mtt比色法。本实验测定对人源癌细胞系a2780(卵巢癌),mcf
‑
7(乳腺癌),a549(肺癌)在体外的抗癌活性。a2780,mcf
‑
7和a549细胞在含有10%胎牛血清与1%青霉素
‑
链霉素溶液的dmem培养基中,于37℃,5%co2细胞培养箱中培养。将细胞按照5000细胞/孔的初始密度接种到96孔细胞培养板中,培养24小时后移除培养基,加入不同浓度梯度的药物培养基200μl,继续孵育44小时后,在每个孔中加入20μl的mtt水溶液(5mg/ml),继续孵育4小时,最后吸去培养基,加入150μl的dmso,晃动10分钟后,使用eliasa(tecan infinite m1000 pro)酶标仪读取590nm的吸光度。
[0116]
实施例1、10、19、21制得的有机化合物l1、l2、l3、l4及实施例2
‑
8、12、20、22制得的金属配合物1
‑
7、10、17、18的抗癌活性如表1所示。
[0117]
表1为有机化合物l1、l2、l3、l4,配合物1
‑
7、10、17、18以及顺铂(cddp)的ic
50
(μm)值
[0118][0119][0120]
结果表明,经大黄酸修饰后的新型有机化合物及配合物都具有较好的抗癌活性,部分配合物的活性是顺铂活性的4倍左右。
[0121]
实施例24
[0122]
本发明大黄酸特定基团修饰的有机化合物及其金属配合物在制备抗菌药物方面的应用。
[0123]
方法:微量肉汤稀释法,体外测定具有有机导向分子芳基金属配合物对金黄色葡萄球菌以及白喉杆菌的最低杀菌浓度(mbc)以及最低抑菌浓度(mic)。
[0124]
将对数生长期的菌种接种于普通肉汤培养基中并将其浓度稀释至5
×
105cfu/ml,
将稀释好的等梯度抗菌药物(0.125
‑
256μg/ml)加入到96孔平板中,每孔100μl,然后将提前稀释好的菌液加入到96孔平板中,每孔的最终体积为200μl,如此孔中抗菌药物的最终浓度为0.125
‑
256μg/ml,将96孔平板置于垫有湿脱脂棉的托盘内,不震摇的情况下37℃培养18h。最低抑菌浓度在瑞士tecantm 10m多功能酶标仪上读取600nm处的吸光度。取mic终点以上未长菌的培养液5μl移种于另外的琼脂平板上,将琼脂平板置于37℃的恒温箱中培养18h,则培养后平板上无菌生长的药物最低浓度即是最低杀菌浓度(mbc)。
[0125]
实施例1、10、19、21制得的有机化合物l1、l2、l3、l4及实施例8、12及20、22制得的金属配合物7、10、17、18的抗菌性如表2所示。
[0126]
表2为有机化合物l1、l2、l3、l4,配合物7、10、17、18的最低抑菌浓度(mic)和最低杀菌浓度(mbc)值
[0127][0128]
结果表明,经大黄酸修饰后的新型有机化合物及配合物都具有较好的抗菌活性,这与引入具有抗菌作用的大黄酸基团有关。
[0129]
实施例25
[0130]
本发明配合物6在细胞中的定位、促进活性氧(ros)生成的应用。
[0131]
方法(定位):将10μm的配合物6加入到事先培养好的a549细胞中,于37℃,5%co2细胞培养箱中孵化6h,将含有药物的培养基吸掉,使用胰酶消化贴壁细胞后,离心收集细胞,使用试剂盒分别提取细胞质、线粒体及细胞核,通过电感耦合等离子质谱icp
‑
ms测试不同细胞器中摄取的ir的含量。
[0132]
方法(ros):将10μm的配合物6加入到事先培养好的a549细胞中,于37℃,5%co2细胞培养箱中孵化2h,然后向内加入10μm dcfhda继续避光孵化30min。孵化结束后使用离心的方法获取细胞,并使用无血清的培养基洗涤细胞三次,除去未进入细胞内的dcfhd。最后上流式细胞仪(flowjo 7.6software(tree star,or,usa))检测细胞内活性氧。激发光波长设为488nm,发射光波长设为530
±
30nm。
[0133]
实施例7的芳基铱配合物6的细胞定位、促进活性氧(ros)生成如图1所示。
[0134]
结果表明,配合物6可以在短时间内大量通过细胞膜被a549细胞摄取,主要定位于细胞核中,且有20%左右分布于线粒体中。配合物6可以明显有效地诱发细胞内活性氧含量升高,这说明配合物可以通过诱发细胞活性氧生成导致癌细胞发生凋亡。
[0135]
实施例26
[0136]
本发明配合物7在细胞中的定位,配合物7与二聚体[rh(η5‑
cp
*
)cl2]2抑制fto活性的应用。
[0137]
方法(定位):将10μm的配合物7加入到事先培养好的a549细胞中,于37℃,5%co2细胞培养箱中孵化6h,然后向内加入10μm核染料hoechst33343继续避光孵化30min,孵化结束后使用无血清的培养基洗涤细胞三次,随后立刻使用共聚焦显微镜观察,激发光波长设为353nm,发射光波长设为465
±
20nm。
[0138]
方法(抑制fto活性):将0.1nmol含有m6a的ssdna,0.1nmol的fto,300μm的2og,280μm的(nh4)2fe(so4)2,2mm的l
‑
ascorbic acid以及50μm的配合物7或二聚体[rh(η5‑
cp
*
)cl2]2溶于100μl的tris
‑
hcl(50mm,ph=7.5)缓冲溶液,室温孵育2h后,于90度条件下煮5min后退火,加入20%的非还原page,进行电泳跑样后使用核酸染料gelred染色,比较条带强度,强度越强,抑制效果越明显。
[0139]
实施例8的含有尾接有机导向分子的芳基铑配合物7的细胞定位以及配合物7与二聚体[rh(η5‑
cp*)cl2]2抑制fto活性如图2所示。
[0140]
结果表明,通过共聚焦显微镜显示,配合物7可以穿过核膜较好的定位于细胞核中,并且配合物7与二聚体[rh(η5‑
cp*)cl2]2都具有抑制fto活性,配合物7与阳性对照的甲氯芬那酸(ma)的抑制效果相当,二聚体[rh(η5‑
cp*)cl2]2则具有比甲氯芬那酸更强的抑制作用。
[0141]
实施例27
[0142]
本发明配合物6、7与dna作用的应用:
[0143]
方法:cd/ld色谱法。通过cd/ld光谱在pbs缓冲溶液(5mm,ph=7.2)中研究配合物6,7与ct
‑
dna的相互作用。配制一系列不同摩尔比的(r
i
=[配合物]/[ct
‑
dna],r
i
值为0~0.2)样品,其中,ct
‑
dna碱基对浓度设定为200μm,于37℃恒温箱中孵化24小时。
[0144]
实施例7,8含有尾接导向分子的芳基金属配合物6,7与dna的作用如图3所示。
[0145]
结果表明:cd色谱中,随着[配合物]/[ct
‑
dna]摩尔比的增加,即配合物6,7比例的增加,245nm处的圆二信号由负逐渐变为正,而280nm处的正信号也逐渐增强,并且300nm左右出现一个新的负峰,这说明配合物6,7可以诱导dna的构型由b型转为z型。ld色谱中,随着[配合物]/[ct
‑
dna]摩尔比的增加,450nm左右出现新的负信号,该波长为大黄酸的吸收波长,这说明配合物通过配体大黄酸与dna发生插入作用。
[0146]
上述实施例26和27中所述的金属配合物诱导dna构型转变过程以及该金属配合物、芳基金属二聚体对fto活性抑制的过程如图4所示。
[0147]
以上所述只为说明本发明的技术构思及特点,并非对本发明作任何形式上的限制,在不脱离上述专利范围与精神下可进行任何修饰或变化,凡依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均属于本发明技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。