1.本发明涉及基因检测领域,具体涉及一种基于甲基化测序数据进行变异检测的方法及装置。
背景技术:
2.人类基因组中存在各种各样的突变,包括但核苷酸变异(snv)、插入缺失变异(indel)等。其中相当一部分跟肿瘤的形成和发展息息相关。通过基因组测序,从测序的序列数据中,快速准确地鉴定出这些变异,对肿瘤的研究以及治疗有非常大的帮助。
3.最近,甲基化测序技术在肿瘤基因组中的应用越来越多。对比于普通的测序技术,甲基化测序技术除了能提供丰富的甲基化修饰信息,同时还能提供基因组变异信息。
4.但是甲基化测序会导致dna上面的碱基信息改变。市面上主流的使用重亚硫酸盐处理的甲基化测序会导致非甲基化的c碱基变换成t。而最近出现的新甲基化测序方法,tet酶和吡啶硼烷结合处理的方法(taps)会导致甲基化的c碱基变换成t。这种碱基变换的干扰会影响序列比对以及突变检测。对于普通基因组测序的变异检测软件并不能很好地在甲基化测序数据上面兼容使用。对于单碱基变异(snp),现时已经有多种方法和软件针对甲基化数据进行处理,例如bis
‑
snp、bs
‑
snper、gembs等。但对于插入缺失突变(indel),并没有成熟的方法。其中主要有三方面的问题:第一、甲基化测序的干扰会影响序列比对,特别是容易出现与插入缺失变异密切相关的gap比对错误,导致插入缺失变异错位;第二、甲基化测序会导致插入序列的改变;第三、不同于snp,插入缺失突变无法通过修改基因型概率模型,兼容碱基转化的情况,对其进行处理。
技术实现要素:
5.根据第一方面,在一实施例中,提供一种基于甲基化测序数据进行变异检测的方法,包括:
6.候选变异提取步骤,包括提取待测样本测序数据中的候选变异;
7.比对文件提取步骤,包括锁定候选变异所在的基因组区域,提取基因组区域位置的比对文件;
8.重比对步骤,包括将所述比对文件进行重比对,生成重比对之后的比对文件,即重比对文件;
9.鉴定及修正步骤,包括对重比对文件进行转化位点的鉴定,修正转化位点的碱基信息,并重新生成修正碱基之后的比对文件,即修正比对文件;
10.变异检测步骤,包括根据修正比对文件,进行变异检测,获得检测结果。
11.根据第二方面,在一实施例中,提供一种基于甲基化测序数据进行变异检测的系统,包括:
12.候选变异提取装置,用于提取待测样本测序数据中的候选变异;
13.比对文件提取装置,用于锁定候选变异所在的基因组区域,提取基因组区域位置
的比对文件;
14.重比对装置,用于将所述比对文件进行重比对,生成重比对之后的比对文件,即重比对文件;
15.鉴定及修正装置,用于对重比对文件进行转化位点的鉴定,修正转化位点的碱基信息,并重新生成修正碱基之后的比对文件,即修正比对文件;
16.变异检测装置,用于根据修正比对文件,进行变异检测,获得检测结果。
17.根据第三方面,在一实施例中,提供一种基于甲基化测序数据进行变异检测的装置,包括:
18.存储器,用于存储程序;
19.处理器,用于通过执行所述存储器存储的程序以实现如第一方面所述的方法。
20.根据第四方面,在一实施例中,提供一种计算机可读存储介质,所述介质上存储有程序,所述程序能够被处理器执行以实现如第一方面所述的方法。
21.依据上述实施例的一种基于甲基化测序数据进行变异检测的方法及装置,通过重比对、转化位点的鉴定及修复,实现基于甲基化测序数据进行变异检测。
附图说明
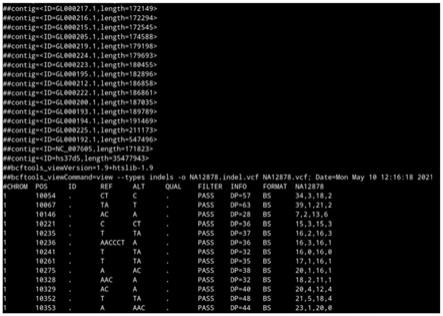
22.图1为实施例1中的部分indel候选集合截图;
23.图2为实施例1中的一个indel位点的比对图;
24.图3为实施例1中的重比对图;
25.图4为实施例1中位点修复比对图。
具体实施方式
26.下面通过具体实施方式结合附图对本发明作进一步详细说明。其中不同实施方式中类似元件采用了相关联的类似的元件标号。在以下的实施方式中,很多细节描述是为了使得本技术能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本技术相关的一些操作并没有在说明书中显示或者描述,这是为了避免本技术的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
27.另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
28.本文中为部件所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。而本技术所说“连接”、“联接,”如无特别说明,均包括直接和间接连接(联接)。
29.定义
30.本文中,“突变”(mutation)是指生物体、病毒或染色体外dna基因组核苷酸序列的改变。突变包括小规模突变和大规模突变,小规模突变影响基因中的一个或几个核苷酸,其
中,只影响到一个核苷酸的突变称为点突变。小规模突变包括:1)插入:将一个或多个额外的核苷酸添加到dna中;2)缺失:从dna中去除一个或多个核苷酸;3)替换:通常是由化学物质或dna复制失常引起的替换突变,将基因中的单个核苷酸替换为另一个核苷酸。大规模突变涉及到染色体结构的突变,包括:1)扩增(或基因复制):导致染色体所有区域拷贝数增加,从而增加了染色体中基因的剂量;2)缺失:大片段染色体缺失,导致该区域内基因的丢失。按照遗传性质改变分类,突变分为生殖细胞突变、体细胞突变。本文中,“突变”与“变异”可互换使用。
31.本文中,“胚系变异”是指在动物体(尤其指人)的胚胎发育期已经携带的变异(几乎全部遗传自父母)。如果胚系变异存在于体细胞内,则不具有遗传性,如果胚系变异存在于生殖细胞内,则具有遗传性。胚系突变亦称生殖细胞突变,是来源于精子或卵子等生殖细胞的突变,因此,通常生物体中所有细胞都带有胚系突变。
32.本文中,“体细胞突变”亦称获得性突变,是在生物体生长、发育过程中或者环境因素影响下后天获得的突变,是指除性细胞外的体细胞发生的突变,通常,生物体中只有部分细胞带有体细胞突变。本文中,“体细胞突变”与“体细胞变异”可互换使用。
33.本文中,“dna甲基化”是dna化学修饰的一种,可以在不改变dna序列的前提下,改变遗传物质。早在1925年,dna甲基化修饰已经被发现。大量研究表明,dna甲基化在基因调控中具有表现遗传作用。dna甲基化中,研究最多的是5
‑
甲基胞嘧啶(5mc),该修饰通常被认为是基因表达的一种稳定的抑制性调控因子。
34.本文中,tet辅助吡啶硼烷的甲基化测序法(tet
‑
assisted pyridine borane sequencing),简称taps,无需亚硫酸氢盐,利用tet(ten
‑
eleven translocation,简称tet)酶将5mc(5
‑
甲基胞嘧啶)和5hmc(5
‑
羟甲基胞嘧啶)氧化为5cac(5
‑
羧甲基胞嘧啶),随后使用有机硼烷(包括但不限于吡啶硼烷、2
‑
甲基吡啶硼烷等等)将5cac还原为二氢尿嘧啶(dihydrou racil,dhu),而后的pcr再将dhu转化为胸腺嘧啶(t),可对目标序列直接进行dna甲基化测序,是一种破坏性更小、效率更高的单碱基分辨率dna甲基化测序方法。
35.本文中,基于重亚硫酸盐转化的甲基化测序法是一种基于二代测序技术的dna甲基化检测方法,通过重亚硫酸氢盐将未甲基化的胞嘧啶(c)转化为尿嘧啶(u),然后在pcr过程中,采用u耐受的聚合酶,将u识别为胸腺嘧啶(t),实现c到t的转化,分析时将测序数据分别比对到c到t和g到a转化的参考基因组,识别样本dna甲基化水平。在正常的人类dna中,约有3%至6%的c被甲基化,因此经过重亚硫酸氢盐转化的测序数据超过90%的c转化为t。
36.本文中,术语“参考序列”指的是可将候选序列与其比较的已知序列,例如来自公共或内部数据库的序列。参考序列可以是参考基因组序列。
37.术语“基因组位置”或“基因组区域”可互换使用,用于指基因组(例如,动物或植物基因组,具体例如人类、猴子、大鼠、鱼或昆虫或植物的基因组)的区域。
38.根据第一方面,在一实施例中,提供一种基于甲基化测序数据进行变异检测的方法,包括:
39.候选变异提取步骤,包括提取待测样本测序数据中的候选变异;
40.比对文件提取步骤,包括锁定候选变异所在的基因组区域,提取基因组区域位置的比对文件;
41.重比对步骤,包括将比对文件进行重比对,生成重比对之后的比对文件,即重比对
文件;
42.鉴定及修正步骤,包括对重比对文件进行转化位点的鉴定,修正转化位点的碱基信息,并重新生成修正碱基之后的比对文件,即修正比对文件;
43.变异检测步骤,包括根据修正比对文件,进行变异检测,获得检测结果。
44.在一实施例中,重比对步骤中,重比对的方法包括但不限于多重比对、基于一致性序列的比对中的至少一种。
45.在一实施例中,多重比对具体包括:对比对文件进行无参考基因组辅助的多重比对,将多重比对结果与参考基因组进行对齐处理,得到新的比对结果。
46.在一实施例中,基于一致性序列的比对具体包括:
47.将测序序列进行组装,获得组装图,将组装图中的路径比对到参考基因组,获得路径
‑
参考基因组位置对应关系,将测序序列比对到组装图中的所有路径,获得测序序列
‑
路径位置对应关系,选取符合筛选规则的测序序列
‑
路径
‑
参考基因组位置作为新的比对结果。
48.上述两种方法都能重新调节测序序列与参考基因组的比对结果,包括测序序列比对位置和gap位置、个数和长度。该步的目的在于消除因为甲基化测序导致的错误比对,从而产生的假阳性插入缺失突变。
49.在一实施例中,将测序序列进行组装的方法包括但不限于德布鲁恩图组装法、先重叠后扩展组装法等组装方法中的至少一种。
50.德布鲁恩图组装法、先重叠后扩展组装法均可参照现有的文献,例如,德布鲁恩图组装法可参考文献《soapdenovo2:an empirically improved memory
‑
efficient short
‑
read de novo assembler》(ruibang luo,binghang liu,yinlong xie等,gigascience,volume 1,issue 1,december 2012,2047
‑
217x
‑1‑
18,https://doi.org/10.1186/2047
‑
217x
‑1‑
18,公开日:2012年12月27日)。
51.先重叠后扩展组装法可参考文献《a consistency
‑
based consensus algorithm for de novo and reference
‑
guided sequence assembly of short reads》(tobias rausch,sergey koren,等,b ioinformatics,volume 25,issue 9,1may 2009,pages 1118
–
1124,https://doi.org/10.1093/bio informatics/btp131,公开日:2009年3月5日)。
52.上述文献仅仅是示例性列举,也可参考其他现有文献中的德布鲁恩图组装法或先重叠后扩展组装法。或者使用开源软件,将比对文件输入其中,得到重比对之后的比对文件。
53.在一实施例中,筛选规则包括但不限于如下规则中的至少一种:
54.1)综合比对分值最高;
55.2)综合最优比对概率最大。
56.在一实施例中,可以以综合最优比对概率最大作为筛选规则,即保留综合最优比对概率最大的测序序列
‑
路径
‑
参考基因组位置作为新的比对结果,通常情况下,比对分值和最优比对概率具有相关性。例如,a个序列可能比对到b个组装片段,两两组合,每种组合概率为pab(有a
×
b个概率),同理,b个组装片段可能比对到c个基因组位置,每种组合概率为pbc(有b
×
c个概率),最后计算总概率为pab
×
pbc,挑选最高概率的结果为最终比对结
果。
57.在一实施例中,鉴定及修正步骤中,转化位点的鉴定方法包括:读取重比对文件,根据每一个基因组位置的比对情况,判断是否呈现出甲基化测序特有的转化模式,鉴定出测序数据中的转化位点位置。
58.在一实施例中,根据每一个基因组位置的比对情况,判断是否呈现出甲基化测序特有的转化模式,通过过滤,鉴定出测序数据中的转化位点位置。
59.在一实施例中,可以根据支持甲基化测序特有的转化模式的测序序列的支持数进行过滤,鉴定出测序数据中的转化位点位置。
60.在一实施例中,根据支持甲基化测序特有的转化模式的测序序列的支持数与阈值的大小关系进行过滤,鉴定出测序数据中的转化位点位置。
61.在一实施例中,如果支持甲基化测序特有的转化模式的测序序列的支持数>阈值,则判断为测序数据中的转化位点位置。
62.在一实施例中,阈值包括但不限于3,此处的具体阈值3为经验值,阈值越高越可靠,越低越灵敏,阈值可以根据实际需要进行确定。
63.在一实施例中,甲基化测序特有的转化模式包括但不限于tet辅助吡啶硼烷的甲基化测序法中特有的碱基转化模式、基于重亚硫酸盐转化的甲基化测序法中特有的碱基转化模式中的至少一种。
64.在一实施例中,甲基化测序特有的转化模式包括如下模式中的至少一种:
65.模式1):
66.比对到参考基因组正向的测序序列中的甲基化的胞嘧啶(c)变成胸腺嘧啶(t);
67.比对到参考基因组反向的测序序列中的鸟嘌呤(g)变成腺嘌呤(a),鸟嘌呤(g)与甲基化的胞嘧啶(c)配对;此模式为tet辅助吡啶硼烷的甲基化测序法中特有的碱基转化模式;
68.模式2):
69.比对到参考基因组正向的测序序列中未甲基化的胞嘧啶(c)变成胸腺嘧啶(t);
70.比对到参考基因组反向的测序序列中的鸟嘌呤(g)变成腺嘌呤(a),鸟嘌呤(g)与未甲基化的胞嘧啶(c)配对。此模式为基于重亚硫酸盐转化的甲基化测序法中特有的碱基转化模式。
71.在一实施例中,鉴定及修正步骤中,修正转化位点的碱基信息的方法包括:将测序数据中转化后的碱基修正为转化前的碱基。例如,对于模式1),将比对到参考基因组正向的测序序列中的胸腺嘧啶(t)修正为转化前的胞嘧啶(c),将比对到参考基因组反向的测序序列中的腺嘌呤(a)修正为鸟嘌呤(g)。再例如,对于模式2),将比对到参考基因组正向的测序序列中的胸腺嘧啶(t)修正为胞嘧啶(c),将比对到参考基因组反向的测序序列中的腺嘌呤(a)修正为鸟嘌呤(g)。
72.在一实施例中,候选变异包含插入变异、缺失变异中的至少一种。插入变异是指至少一个核苷酸插入到dna中,缺失变异是指dna中缺失至少一个核苷酸。
73.在一实施例中,本发明也适用于snp(单核苷酸多态性)检测。
74.在一实施例中,候选变异为胚系变异、体细胞变异中的至少一种。
75.在一实施例中,候选变异为胚系变异。
76.在一实施例中,候选变异提取步骤中,待测样本测序数据为甲基化测序数据。甲基化测序数据是指通过甲基化测序法获得的测序数据。
77.在一实施例中,甲基化测序包括但不限于氧化亚硫酸氢盐测序(oxbs
‑
seq)、tet辅助吡啶硼烷测序(taps)等等。
78.在一些实施例中,甲基化测序在不超过约40
×
的深度进行。在一些实施例中,甲基化测序在不大于约30
×
的深度进行。在一些实施例中,甲基化测序在不大于约25
×
的深度进行。在一些实施例中,甲基化测序在不超过约20
×
的深度进行。在一些实施例中,甲基化测序在不大于约12
×
的深度进行。在一些实施例中,甲基化测序在不大于约10
×
的深度进行。在一些实施例中,甲基化测序在不超过约8
×
的深度进行。在一些实施例中,甲基化测序在不超过约6
×
的深度进行。在一些实施例中,甲基化测序在不大于约5
×
,不大于约4
×
,不大于约3
×
,不大于约2
×
或不大于约1
×
的深度进行。
79.在一实施例中,待测样本包括但不限于组织样本、体液样本中的至少一种。
80.在一实施例中,组织样本包括但不限于肿瘤组织样本。
81.在一实施例中,体液样本包括但不限于血液样本、血浆样本中的至少一种。
82.在一实施例中,待测样本为dna。
83.在一实施例中,候选变异提取步骤中,待测样本测序数据为靶向甲基化测序数据、全基因组甲基化测序数据中的至少一种。靶向甲基化测序数据可以是全外显子组范围的甲基化测序数据或区域捕获范围的甲基化测序数据。测序方法可以是第一代测序法(荧光标记的sang er法)、第二代测序法(循环阵列合成测序法)、第三代测序法(不需要化学处理的第三代测序方法除外)等等。
84.在一实施例中,候选变异提取步骤中,包括通过分析比对文件中的比对信息,提取出候选变异。
85.在一实施例中,比对信息包括但不限于cigar字符串、md字符串中的至少一种。
86.cigar字符串记录了测序序列与参考基因组序列之间的差异,例如单碱基变换、碱基插入缺失等。
87.在一实施例中,候选变异提取步骤中,还包括在提取出候选变异之后,通过对包含变异的测序序列支持数、测序序列频率初步过滤,得到候选变异集合。
88.在一实施例中,初步过滤时,保留满足如下条件的数据:
89.待测样本中包含变异的测序序列支持数(即支持变异的测序序列频数)≥3;
90.待测样本中包含变异的测序序列频率≥0.15。测序序列频率=变异位点的测序序列支持数/变异位点的测序深度。此处的包含变异的测序序列支持数阈值、测序序列频率阈值仅仅是示例性列举,可以根据需要设置其他阈值。
91.在一实施例中,通过分析比对文件中的比对信息,提取出候选变异时,比对文件为待测样本测序数据比对到参考基因组所得的比对文件。比对文件包括但不限于bam文件、cra m文件、sam文件。
92.在一实施例中,候选变异所在的基因组区域是指包含候选变异集合所在位置以及包含变异上下游部分碱基的区域。生成只包含变异集合所在范围的比对文件,有利于加快后续分析速度。
93.在一实施例中,候选变异所在的基因组区域是指包含候选变异集合所在位置以及
包含变异上下游200bp碱基的区域。即包含变异位点上游200bp、下游200bp碱基的区域。变异上下游碱基的数量不限于200bp,可以是其他任意数量的碱基。
94.根据第二方面,在一实施例中,提供一种基于甲基化测序数据进行变异检测的系统,包括:
95.候选变异提取装置,用于提取待测样本测序数据中的候选变异;
96.比对文件提取装置,用于锁定候选变异所在的基因组区域,提取基因组区域位置的比对文件;
97.重比对装置,用于将比对文件进行重比对,生成重比对之后的比对文件,即重比对文件;
98.鉴定及修正装置,用于对重比对文件进行转化位点的鉴定,修正转化位点的碱基信息,并重新生成修正碱基之后的比对文件,即修正比对文件;
99.变异检测装置,用于根据修正比对文件,进行变异检测,获得检测结果。
100.根据第三方面,在一实施例中,提供一种基于甲基化测序数据进行变异检测的装置,包括:
101.存储器,用于存储程序;
102.处理器,用于通过执行存储器存储的程序以实现如第一方面的方法。
103.根据第四方面,在一实施例中,提供一种计算机可读存储介质,介质上存储有程序,程序能够被处理器执行以实现如第一方面的方法。
104.在一实施例中,提供一种基于甲基化测序数据进行胚系插入缺失变异检测的方法,包括:快速提取样本中的候选插入缺失变异;锁定候选插入缺失变异所在的基因组区域,提取区域位置的比对文件;对提取出来的比对文件进行重比对,生成重比对之后的比对文件;对重新生成的比对文件进行转化位点的鉴定,修正转化位点的碱基信息,并重新生成修正碱基之后的比对文件;对于修正过后的比对文件,使用基因组插入缺失检测软件进行变异检测。该方法可以应用于靶向甲基化测序和全基因组甲基化测序数据。
105.在一实施例中,提供一种基于甲基化测序数据进行胚系插入缺失变异检测的方法,具体可以包括如下步骤:
106.(1)快速提取样本中的候选插入缺失变异。通过分析比对文件(例如,可以是bam文件、cram文件等等)中的cigar字符串提取出候选变异。cigar字符串记录了测序序列与参考基因组序列之间的差异,例如单碱基变换、碱基插入缺失等。通过对测序序列支持数、测序序列频率初步过滤得到候选的插入缺失变异集合。
107.(2)锁定候选插入缺失变异所在的基因组区域,提取区域位置的比对文件。为加快后续分析速度,提取候选变异集合所在位置及旁边一定范围测序序列,例如
±
200bp。生成只包含变异集合所在范围的比对文件。
108.(3)对提取出来的比对文件进行重比对,生成重比对之后的比对文件。重比对可通过两种方法中的其中一种获得。第一种是对测序序列进行无参考基因组辅助的多重比对(multipl e sequence alignment)。将多重比对结果与参考基因组进行对齐处理,即得新的比对结果。第二种为基于一致性序列的比对(consensus alginment)。具体方法如下:先将测序序列进行组装,包括德布鲁恩图(de brujin graph)或先重叠后扩展等组装方法(overlap
‑
layout
‑
cons ensus)。将组装图中的所有路径比对到参考基因组,获得路径
‑
参
考基因组位置对应关系。将测序序列比对到组装图中的所有路径,获得测序序列
‑
路径位置对应关系。综合考虑三者两两间对应关系,例如综合比对分值最高或综合最优比对概率最大,选取出最优的测序序列
‑
路径
‑
参考基因组位置作为最终新的比对结果。上面两种方法都能重新调节测序序列与参考基因组的比对结果,包括测序序列比对位置和gap位置,个数和长度。该步的目的在于消除因为甲基化测序导致的错误比对,从而产生的假阳性插入缺失突变。
109.(4)对重新生成的比对文件进行转化位点的鉴定,修正转化位点的碱基信息,并重新生成修正碱基之后的比对文件。甲基化测序会产生特有的甲基化碱基转化模式,例如taps测序,会将比对到参考基因组正向的pair 1(f1)或比对到参考基因组反向的pair2(r2)上面的甲基化碱基c变成t,会将比对到参考基因组正向的pair 2(f2)或比对到参考基因组反向的pair 1(r1)上面的测序碱基g(与甲基化碱基c配对)变成a。而重亚硫酸盐测序转化模式一致,但会对非甲基化碱基c进行转化。读取重比对文件,对每一个基因组位置的比对情况进行判断是否呈现出这种特有的转化模式。通过过滤,例如支持该种甲基化特有模式的测序序列支持数大于3。最终鉴定出测序数据中的转化位点位置。对转化位点进行碱基修复处理,将受碱基化转化t碱基或者a碱基转换成原来的c碱基或者g碱基。生成碱基修复过后的比对文件。该步的目的在于消除因为甲基化测序产生的错误的插入序列。
110.(5)对于修正过后的比对文件,使用基因组插入缺失检测软件进行变异检测。该步骤可以使用通用的用于一般基因组测序数据的变异检测软件,包括但不限于gatk、samtools、freebayes等等。
111.实施例1
112.本实施例检测的样本为coriell人类基因组dna标准品na12878(样本信息参见网址:http://www.f
‑
biology.com/pd.jsp?id=10384)。
113.本实施例对样本进行高深度全基因组甲基化测序。
114.对样本进行taps测序,文库构建方法参照申请号为201911159400.6的中国专利《全基因组甲基化非重亚硫酸氢盐测序文库及构建》的实施例4进行,dna片段化时,是将coriel l人类基因组dna标准品na12878与阳性内参(甲基化的puc19)混合打断。上机测序所用的测序仪为mgiseq
‑
t7(华大智造),测序方式:pe100,测序深度47
×
。
115.下机数据经过预处理和参考基因组(hs37d5)比对,具体是使用bwa软件进行比对,得到压缩比对文件cram/bam(本实施例为bam文件)。cram/bam文件为本方法的输入文件。
116.下机数据的预处理包括:低质量序列(reads)过滤(过滤去除低质量碱基占比过大、n碱基占比过大的序列),接头序列污染reads过滤。
117.检测方法如下:
118.(1)读取cram/bam文件,使用变异检测程序,通过cigar信息快速提取indel候选集合。过滤indel,条件为去掉read支持数小于3或者reads支持频率小于0.15的indel变异。如图1所示。
119.变异检测程序参照申请号为202011158198.8的中国专利《一种检测体细胞突变的方法及装置》说明书第81、82段进行。
120.(2)将所有indel位置拓展前后100bp(具体是在基因组序列上,以indel位置为起点,上下游各自延伸100bp),获得候选处理区间。
121.(3)使用samtools软件中的view模块按步骤(2)中的区间对比对文件bam进行测序序列的提取。图2为其中一个indel位点的比对情况。
122.该点的突变信息如下表,存在两个不同碱基型的插入突变。
123.表1
124.#chromposidrefalt13533921.aaccggct13533921.aactggct
125.(4)使用bis
‑
snp软件(此为开源软件,参见网址https://sourceforge.net/projects/bissnp/files/bissnp
‑
0.82.2/)中的重比对模块bisulfiteindelrealigner,输入步骤(3)中的bam文件进行重比对,得到重比对之后的bam文件。该步骤具体是基于一致性序列的比对,使用olc(overlap
‑
layout
‑
consensus)组装方法。olc组装方法包括:首先构建重叠图(重叠图是指前后有若干个相同碱基的序列),然后将重叠图收束成contig,然后每一个contig选择最有可能的核苷酸序列。
126.图3为表1中示例位点的重比对情况。
127.(5)读取步骤(4)中的文件,使用碱基修复软件,对甲基化位点进行鉴定并修复。图4为表1中示例位点修复比对的情况。
128.(6)使用胚系突变检测程序platypus(此为开源软件,参见网址:https://www.well.ox.ac.uk/research/research
‑
groups/lunter
‑
group/lunter
‑
group/platypus
‑
a
‑
haplotype
‑
based
‑
variant
‑
caller
‑
for
‑
next
‑
generation
‑
sequence
‑
data)对步骤(5)中的bam文件进行变异检测,其中产生的in del集合为最终结果。结果中因为甲基化测序产生的错误插入突变actggct被去除,只有真实存在的accggct被保留。
129.表2
130.#chromposidrefalt13533921.aaccggct
131.为了对比本实施例的高深度全基因组甲基化测序结果与现有的普通dna测序结果,对样本进行了高深度全基因组普通dna测序。
132.对于高深度全基因组普通dna测序(测序深度为100
×
),文库构建方法为ta克隆连接接头建库,经过末端修复、加“a”、加接头和引入index,再经过磷酸化、pcr后,构建得到测序仪可识别的dna wgs文库,测序方法为二代测序,变异检测软件为platypus。
133.对于高深度全基因组甲基化测序,下机数据经过预处理和参考基因组比对(使用bwa软件)得到压缩比对文件cram/bam(本实施例具体为bam文件)。cram/bam文件为本实施例方法的输入文件。使用本实施例的甲基化测序检测方法对比对文件进行分析,得到indel变异集合。通过比较全基因组/外显子组区域内,甲基化测序得到的变异、普通dna测序得到的变异、高可信度标准品答案集3者之间的一致性,评估方法性能。高可信度标准品答案来自于因美纳白金基因组数据(参见网址:https://www.illumina.com/platinumgenomes.html)以及瓶中基因组数据(参见网址:https://www.nist.gov/programs
‑
projects/genome
‑
bottle)。
134.表3标准品na12878检测结果比较(全基因组indel一致性)
[0135][0136]
“‑”
表示没有数据。
[0137]
一致性的计算方法如下:以甲基化全基因组测序一致性的计算方法为例,甲基化全基因组测序一致性=共有位点数目/甲基化全基因组位点数
×
100%=(549480/608462)*100%=90.30%。
[0138]
答案集一致性是指答案检出数占答案总数的百分比。
[0139]
从表3可以看出,以共有位点数目占基于测序数据检测得到的变异数目的百分比计,普通全基因组测序一致性为84.55%,甲基化全基因组测序一致性达到90.30%。
[0140]
普通全基因组测序的答案集一致性为94.20%,甲基化全基因组测序的答案集一致性为92.02%。
[0141]
两种测序数据检测结果与答案集一致性较高,证明两种方法的灵敏性较高,均达到90%以上。甲基化全基因组测序的答案集一致性与普通全基因组测序的答案集一致性接近。全基因组范围内与全基因组测序一致性较高,表明准确度较高,达84%以上。上述结果表明,本实施例基于甲基化全基因组测序数据的变异检测方法具有较高的准确性。
[0142]
表4标准品na12878检测结果比较(全外显子组indel一致性)
[0143][0144]
“‑”
表示没有数据。
[0145]
从表4可以看出,以共有位点数目占基于测序数据检测得到的变异数目的百分比计,普通全外显子组测序一致性为94.02%,甲基化全外显子组测序一致性为96.75%。
[0146]
普通全外显子组测序的答案集一致性为97.61%,甲基化全外显子组测序的答案集一致性为95.86%。
[0147]
相对于全基因组测序,全外显子组测序的灵敏度更高,准确度更高,而且突变可解释性更强,因此,全外显子组测序为医学检测(例如遗传疾病的检测)中常用的测序手段。本实施例基于全外显子组测序的一致性都达到94%以上,证明本实施例基于甲基化全外显子组测序的变异检测方法具有较高的准确性。
[0148]
实施例2
[0149]
选择3例肿瘤患者血浆cfdna样本进行高深度全基因组捕获甲基化测序,文库构建方法、测序所用仪器、测序方法同实施例1。
[0150]
表3中,190038718bpd样本对应的受试者患肝癌,使用streck采血管,常温运输。
[0151]
200037881bp1d样本对应的受试者患肝癌,使用streck采血管,常温运输。
[0152]
208002626bp1d样本对应的受试者患左乳腺癌,骨、肝转移;既往检测:免疫组化/左乳,her2(阳性)。使用streck采血管,常温运输。
[0153]
下机数据经过预处理和参考基因组比对(使用bwa软件)得到压缩比对文件cram/bam(本实施例具体为bam文件)。cram/bam文件为本实施例方法的输入文件。使用与实施例1相同的甲基化测序检测方法对比对文件进行分析,得到indel变异集合。通过比较捕获测序区域内,甲基化测序得到的变异和普通dna测序得到的变异之间一致性,评估方法性能。
[0154]
普通dna测序方法同实施例1,文库构建时,具体使用了hieff ngs ultima dna libr ary prep kit for mgi文库构建试剂盒。
[0155]
表5
[0156][0157][0158]
从表5可以看出,虽然肿瘤cfdna样本可能会对胚系突变检测带来影响,但甲基化测序一致性和全基因组重测序(whole genome sequencing,wgs)一致性都达到80%以上,灵敏度和准确度均较高。部分样本接近90%,接近普通全基因组重测序一致性水平。上述结果表明,本实施例基于甲基化测序数据的变异检测具有较高的准确性。
[0159]
本领域技术人员可以理解,上述实施方式中各种方法的全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现上述功能。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现上述全部或部分功能。另外,当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器中的程序时,即可实现上述实施方式中全部或部分功能。
[0160]
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。