1.本发明涉及一种自修复铂金属凝胶材料及其制备方法与应用,属于自修复金属凝胶材料领域。
背景技术:
2.作为现代高科技战争重要手段的激光技术,已经被广泛应用于军事领域。随着激光技术的发展,相应的激光防护材料以及器件的研究已经引起全世界的高度重视。其中,光限幅材料作为激光防护的重要材料之一,其研究有着极其重要的意义。
3.理想的光限幅材料可将强激光束的传输强度调制到对所需保护光学传感器安全的最大值,但在低强度环境光下则需要其展现出较高透过率,应具有线性透射率高、限幅阈值及限幅幅值低、损伤防护光谱范围窄、动态范围宽和热稳定性高等特点(《nonlinear optical materials for the smart filtering of optical radiation》,danilo d.等,chem.rev.,2016,116,13043
‑
13233.)。例如,d8电子结构的二价铂配合物,由于重原子效应影响,易产生较强的旋轨耦合作用,更容易发生单线态与三线态之间的系间窜越(《supramolecular self
‑
assembly of amphiphilic anionic platinum(ii)complexes:a correlation between spectroscopic and morphological properties》,po c.等人,j.am.chem.soc.,2011,133:12136
‑
12143.),有利于提升三重激发态吸收,延长三重激发态寿命。因此,铂配合物是一类优良的磷光材料,并在光限幅领域得到广泛应用。但是,在实际应用中还存在诸多不足,特别是应用于人眼和设备的激光防护方面,基于溶液态的材料往往难以胜任,而玻璃掺杂的光限幅材料往往会因为老化、损坏、碎裂等原因而无法长期使用(《efficient nonlinear absorbing platinum(ii)acetylide chromophores in solid pmma matrices》,robert w.等,adv.funct.mater.,2008,18:1939
‑
1948.)。因此,如何设计形状规整、结构均一、加工性好、对外力破坏有响应修复的功能器件,成为影响光限幅材料发展的重要因素之一。
4.相比而言,具有一定形状,介于固态与液态的软物质,兼具光限幅性能和自修复能力的凝胶材料,更能满足制备新型光限幅器件的需要。自修复材料具有自我修复的能力,在一定的条件下(如光照,加热,调节ph等)情况下实现其修复的过程。作为一种新型的智能材料,自修复材料是目前科学研究中的热点。除了添加修复剂方式的外援型自修复材料外,本征型自修复利用可逆键的高度可逆性,可以重复多次发生自修复过程,也更加有效高效的用于材料的器件化。目前已经有少量的文献报道,利用本征自修复凝胶作为功能分子的器件载体,提升整体材料的实用价值。例如,wang等人(《self
‑
healable,stretchable,transparent triboelectric nanogenerators as soft power sources》,sun j.等,acs nano,2018,12(6):6147
‑
6155.)制备了同时具有自愈性、可拉伸性和透明性聚(二甲基硅氧烷)(pdms)凝胶,用于摩擦电纳米发电机软电源的封装材料;shikler课题组(《fluorescent self
‑
healing carbon dot/polymer gels》,bhattacharya s.等,acs nano,2019,13(2):1433
‑
1442.)用支化聚乙烯亚胺对不同醛类前体制备的碳点进行反应,制备了多色荧光自
愈凝胶;唐本忠院士团队(《fluorescence turn
‑
on visualization of microscopic processes for self
‑
healing gels by aiegens and anticounterfeiting application》,sun j.等,chem.mater.,2019,31(15):5683
‑
5690.)通过共价引入aie活性tpe
‑
4cho作为探针合成自愈合pdms凝胶,并通过对多色荧光自愈凝胶进行拼接,成功地制备出具有隐身效应的荧光编码。同时,这些凝胶器件都具有拉伸性、耐磨性、可复用和良好的抗损伤性能等特点。
5.而到目前为止,激光防护领域还鲜有利用凝胶器件来延长材料使用寿命的报道。长期以来,光限幅材料大多是基于溶液态的研究与讨论,并不符合实际应用的标准,但基于前人系统工作的分析总结,可以预期一些材料分子具有优越的功能性质。自1967年leite首次观察到光限幅现象之后,铂配合物的光限幅研究就已经开始,铂的重原子效应易产生较强的旋轨耦合,有效提升单线态与三线态之间的系间窜越、三重激发态吸收,延长三重激发态寿命,是一类优良的光限幅材料。因此,将铂配合物与具有自修复性能的凝胶材料进行结合,有望开发出一类新型激光防护器件。
技术实现要素:
6.为解决现有技术中的缺陷,本发明公开一种自修复铂金属凝胶及其制备方法与应用,通过对铂配合物和凝胶因子的筛选与结构改造,制备基于自修复凝胶的光限幅材料。
7.本发明是采用下述技术方案实现的:
8.一种自修复铂金属凝胶材料,具有式(ⅰ)所述的结构:
[0009][0010]
其中,r1为醛基或者异氰酸酯基;r2为含碳原子数为c1
‑
c12的烷基。
[0011]
本发明第二个目的是提供一种自修复铂金属凝胶材料的制备方法,包括以下步骤:
[0012]
在保护气氛中,化合物ⅰ与化合物ⅱ溶解在有机溶剂中,并添加催化剂,进行sonogashira偶联反应,制得化合物ⅲ;
[0013]
在保护气氛中,化合物ⅲ与含铂的有机金属试剂,在有机溶剂中发生配位反应,生成化合物ⅳ;
[0014]
化合物ⅳ与化合物ⅰ在催化剂催化下,在有机溶剂中发生sonogashira偶联反应,制得铂金属凝胶材料;
[0015]
所述化合物ⅰ的结构式为
[0016][0017]
所述化合物ⅱ的结构式为
[0018][0019]
所述化合物ⅲ的结构式为
[0020][0021]
所述化合物ⅳ的结构式为
[0022][0023]
进一步的,所述化合物ⅰ与化合物ⅱ进行sonogashira偶联反应的有机溶剂为干燥的thf和三乙胺的混合溶液,所述保护气氛为氮气,所述催化剂为cui、pdcl2(pph3)2。
[0024]
进一步的,所述化合物ⅱ的质量为1g,作为当量标准,所述化合物ⅰ为2
‑
3eq,所述cui为0.1
‑
0.2eq,所述pdcl2(pph3)2为0.1
‑
0.2eq,所述反应温度为20
‑
100℃,所述反应时间为10
‑
30h。
[0025]
进一步的,所述化合物ⅲ与含铂的有机金属试剂进行配位反应中,有机溶剂为乙腈和二氯甲烷,保护气氛为氮气,含铂的有机金属试剂为pt(dmso)2cl2。
[0026]
进一步的,所述化合物ⅲ物质的量为0.022mmol,为当量基准,所述乙腈为200
‑
300eq,二氯甲烷为200
‑
300eq,pt(dmso)2cl2为0.5
‑
2.4eq;所述反应温度为20
‑
100℃,所述反应时间为10
‑
30h。
[0027]
进一步的,所述化合物ⅳ与化合物ⅰ进行的sonogashira偶联反应中,保护气氛为氮气,催化剂为cui,有机溶剂为n,n
‑
二异丙基乙胺和二氯甲烷的混合溶剂。
[0028]
进一步的,所述化合物ⅳ物质的量为0.0084mmol,作为当量基准,所述cui为0.1
‑
0.2eq,所述n,n
‑
二异丙基乙胺为30~50eq,所述二氯甲烷为50
‑
100eq;所述反应温度为20
‑
100℃,所述反应时间为10
‑
30h。
[0029]
本发明第三个目的是提供一种自修复铂金属凝胶材料在光限幅防护领域的应用。
[0030]
进一步的,所述光限幅防护领域为激光的限幅防护领域。
[0031]
与现有技术相比,本发明所达到的有益效果是:
[0032]
本发明提供的铂金属凝胶材料将光限幅材料与自修复凝胶以可逆共价键的形式结合起来,能够在溶液状态和凝胶状态下,对光限幅性能进行研究同时,具有良好的自修复性能、光学透明性、力学性能和光限幅性能,激光防护性能优异,而且由于激光在凝胶内的非线性散射,进一步提升了其激光防护性能。
附图说明
[0033]
图1为测试例1中pt
‑
4imine在甲苯中的紫外
‑
可见吸收光谱;
[0034]
图2为测试例1中pt
‑
4imine在不同溶剂中的紫外
‑
可见归一化吸收光谱;
[0035]
图3为测试例2中pt
‑
4imine在空气饱和和脱气的甲苯溶液中的发射光谱,ex=454nm;
[0036]
图4为测试例2中pt
‑
4imine在不同溶剂中的归一化发射,ex=454nm;
[0037]
图5为测试例3中pt
‑
4imine溶液和凝胶态的紫外
‑
可见吸收光谱;
[0038]
图6为测试例3中pt
‑
4imine在溶液和凝胶态的发射吸收光谱;
[0039]
图7为测试例3中pt
‑
4imine凝胶态的透射光谱;
[0040]
图8为测试例3中邻二甲苯中pt
‑
4imine的光学极限曲线;
[0041]
图9为测试例3凝胶中pt
‑
4imine@pdms的光限幅曲线;
[0042]
图10为测试例3中532nm激光脉冲下的光限幅曲线合并图;
[0043]
图11为不同透过率铂凝胶的光限幅性质曲线图;
[0044]
图12为测试例4中凝胶制备过程;
[0045]
图13为测试例4中凝胶制备初始在不同浓度的pt
‑
4cho样品图;
[0046]
图14为测试例4中凝胶制备初始在不同浓度的pt
‑
4cho样品倒立后的样品图;
[0047]
图15为测试例4中凝胶制备结束在不同浓度的pt
‑
4cho样品图;
[0048]
图16为测试例4中凝胶制备结束在不同浓度的pt
‑
4cho样品倒立后的样品图;
[0049]
图17为测试例4中pt
‑
4cho在甲苯:dmf=3:1中的pdms凝胶自修复实验;
[0050]
图18为测试例5中凝胶在二甲苯、邻二甲苯、甲苯、1,2
‑
二氯苯、1,4
‑
二氧六环、乙酸乙酯6种溶剂中的拉伸应力应变曲线;
[0051]
图19为测试例5中不同的线性聚合物和交联聚合物的比例制备的凝胶的拉伸性能曲线图;
[0052]
图20为测试例5中凝胶的起始状态和拉伸后的自修复能力曲线;
[0053]
图21为测试例5中凝胶多次拉伸后自修复能力曲线;
[0054]
图22为测试例6中使用激光共聚焦扫描显微镜(clsm)观察在pt
‑
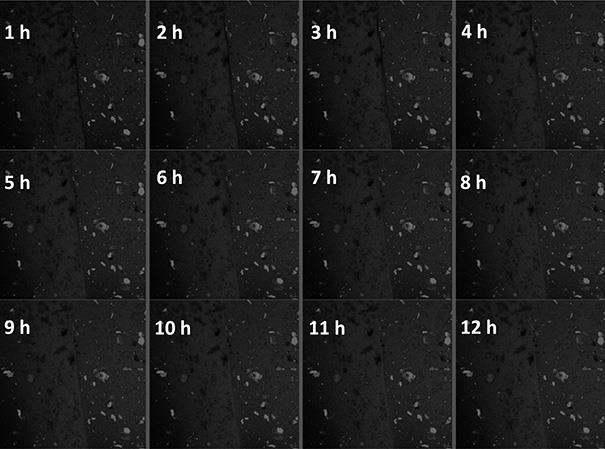
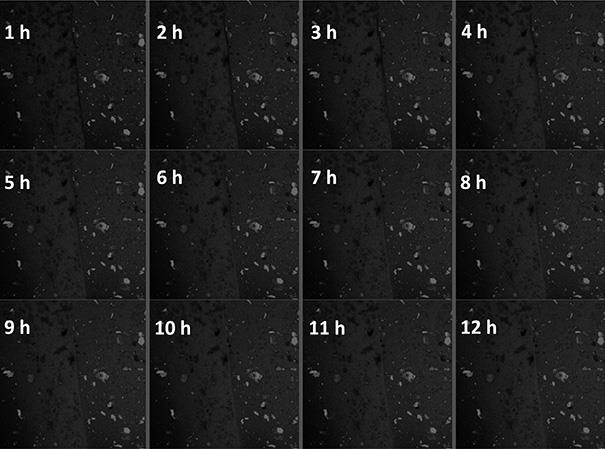
4cho@pdms凝胶中形成的裂纹处的自愈合过程图。
具体实施方式
[0055]
本发明提供了一种自修复铂金属凝胶材料,具有式ⅰ所示的结构:
[0056][0057]
其中,r1为醛基或者异氰酸酯基;r2为含碳原子数为c1
‑
c12的烷基。
[0058]
本发明还提供了对于式ⅰ化合物的制备方法,具体的包括以下步骤:
[0059]
在保护气氛中,化合物ⅰ与化合物ⅱ溶解在有机溶剂中,并添加催化剂和有机碱,进行sonogashira偶联反应,制得化合物ⅲ;
[0060]
在保护气氛中,化合物ⅲ与含铂的有机金属试剂,在有机溶剂中发生配位反应,生成化合物ⅳ;
[0061]
化合物ⅳ与化合物ⅰ在催化剂催化下,在有机溶剂中发生sonogashira偶联反应,制得铂金属凝胶材料;
[0062]
在以上制备步骤中,所述化合物ⅰ的结构式为
[0063][0064]
所述化合物ⅱ的结构式为
[0065][0066]
所述化合物ⅲ的结构式为
[0067][0068]
所述化合物ⅳ的结构式为
[0069][0070]
而在本发明中,化合物ⅰ的由2,7
‑
二溴
‑
9h
‑
芴依次经过取代反应、sonogashira偶联反应和脱保护反应制得,具体制备步骤如下:
[0071]
将2,7
‑
二溴
‑
9h
‑
芴与溴代烷、无机碱试剂在有机溶剂中混合发生取代反应得到化合物1;
[0072]
将化合物1、有机溶剂、有机碱以及dmf混合发生取代反应,制得化合物2;
[0073]
在保护气氛中,将化合物2、三甲基乙炔基硅、催化剂、有机碱和有机溶剂混合,进行sonogashira偶联反应制得化合物3;
[0074]
将化合物3、无机碱和有机溶剂混合,进行脱保护反应,得到化合物ⅰ。
[0075]
化合物ⅰ的反应路线如下所示:
[0076][0077]
对于由2,7
‑
二溴
‑
9h
‑
芴制备化合物1的反应:无机碱试剂优选为氢氧化钾和碘化钾,有机溶剂优选为dmso,其中2,7
‑
二溴
‑
9h
‑
芴、溴代烷、氢氧化钾、碘化钾以及dmso的摩尔比为1:(1
‑
3):(1
‑
10):(0.1
‑
1):80
‑
110。具体方法为将2,7
‑
二溴
‑
9h
‑
芴、溴代烷、氢氧化钾、碘化钾溶解在dmso中,体系在0~30℃不断搅拌8
‑
12小时后,点板跟踪反应,反应结束后,抽滤得液体,用300~500当量(以下用eq表示)的乙酸乙酯稀释混合物,分离水相并用100~300eq乙酸乙酯再次萃取水相。将合并的有机相用无水硫酸钠干燥并浓缩。残余固体通过硅胶柱提纯,得到淡绿色油状的化合物1。
[0078]
对于由化合物1制备化合物2的反应:有机溶剂优选thf,本发明对于thf的干燥方式没有特殊的限定,采用本领域公知的干燥方式即可,比如对thf进行多次重蒸;有机碱优选正丁基锂,正丁基锂溶解在己烷中,浓度为1.6mol/1mol己烷。其中化合物1、thf、dmf的摩尔比为1:(80
‑
100):(0.8
‑
3)。具体方法为将化合物1溶解在重蒸后的thf中,在
‑
20~
‑
80℃不断搅拌1~3小时后,加入正丁基锂,正丁基锂在7~30分钟内逐滴加入,滴加速度按照加入的正丁基锂的量和滴加时间确定。滴加完后加入dmf,在冷却浴中搅拌1~3小时,在浴外搅拌1~3小时。然后将反应物冷却至5~20℃,并用水淬灭反应。用300~500eq乙酸乙酯稀释混合物,分离水相并用100~300eq乙酸乙酯再次萃取。将合并的有机相用无水硫酸钠干燥并浓缩。残余固体通过硅胶柱提纯,得到淡绿色油状化合物2。
[0079]
对于化合物2合成化合物3的反应:保护气氛优选氮气,催化剂为cui和pdcl2(pph3)2,有机碱为三乙胺,有机溶剂为thf,本发明对于thf的干燥方式没有特殊的限定,采用本领域公知的干燥方式即可,比如对thf进行多次重蒸。其中化合物2、三甲基乙炔基硅、cui、pdcl2(pph3)2、三乙胺和thf的摩尔比为1:(3
‑
10):(0
‑
1):(0
‑
1):(30
‑
100):(30
‑
100)。具体反应方法为将化合物2、三甲基乙炔基硅、cui(30mg,0~1eq)、pdcl2(pph3)2和三乙胺在干燥的thf中的溶液及氮气的保护下下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,得到淡黄色固体化合物3。
[0080]
对于化合物3合成化合物ⅰ的反应:无机碱优选为碳酸钾,有机溶剂优选为甲醇。其中化合物3、碳酸钾和甲醇的摩尔比为1:(0.8
‑
1.5):(80
‑
120)。具体方法为化合物3在碳酸钾存在下于甲醇中搅拌悬浮,除去三甲基硅基,得白色固体化合物ⅰ。
[0081]
完成原料化合物ⅰ的制备后,对于化合物ⅰ和化合物ⅱ制备化合物ⅲ的反应:保护
气氛优选氮气,催化剂为cui和pdcl2(pph3)2,有机碱为三乙胺,有机溶剂为thf,本发明对于thf的干燥方式没有特殊的限定,采用本领域公知的干燥方式即可,比如对thf进行多次重蒸。其中化合物ⅰ、化合物ⅱ、cui、pdcl2(pph3)2、三乙胺和thf的摩尔比为(2
‑
3):1:(0.1
‑
0.2):(0.1
‑
0.2):(30
‑
100):(30
‑
100)。具体方法为化合物ⅰ、化合物ⅱ、cui、pdcl2(pph3)2和三乙胺溶解在30~100eq干燥的thf中的溶液,于氮气保护下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,得到黄色固体化合物ⅲ。
[0082]
对于化合物ⅲ制备化合物ⅳ的反应:有机溶剂优选为乙腈和二氯甲烷(以下简称dcm),保护气氛为氮气,含铂的有机金属试剂为二(二甲基亚砜)二氯化铂,其中化合物ⅲ、二(二甲基亚砜)二氯化铂、乙腈、dcm的摩尔比为1:(0.5
‑
2.4):(200
‑
300):(200
‑
300)。具体方法为化合物ⅲ、二(二甲基亚砜)二氯化铂、乙腈和二氯甲烷在氮气保护下,在20~100℃加热10~30小时,混合物通过硅胶柱,得到橘黄色固体化合物ⅳ。
[0083]
对于化合物ⅳ制备得到最终产物铂金属凝胶材料的反应:催化剂为cui,有机碱为n,n
‑
二异丙基乙胺,有机溶剂为dcm,其中化合物ⅳ、化合物ⅰ、cui、n,n
‑
二异丙基乙胺、dcm的摩尔比为1:(2
‑
3):(0.1
‑
0.2):(30
‑
50):(50
‑
100)。具体的反应方法为将化合物ⅳ、化合物ⅰ、cui、n,n
‑
二异丙基乙胺在干燥的二氯甲烷中和氮气的保护下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,得到红色固体铂金属凝胶材料。
[0084]
以下实施例仅用于更加清楚、完整地说明本发明的技术方案,只是本发明一部分实施例,而不能以此来限定本发明的保护范围。基于本发明中的实施例,本领域技术人员在没有做出实质的创造性劳动的前提下所获得的所有其他实施方式,均属于本发明所保护的范围
[0085]
实施例1
[0086]
(1)化合物1的合成
[0087][0088]
2,7
‑
二溴
‑
9h
‑
芴(15.2g,45.4mmol,1eq),溴代正己烷(45.4
‑
136.2mmol,1
‑
3eq),氢氧化钾(45.4
‑
454mmol,1
‑
10eq),碘化钾(4.5
‑
45.4mmol,0.1
‑
1eq)溶解在dmso(80~110eq)中,体系在0~30℃不断搅拌8
‑
12小时后,点板跟踪反应,反应结束后,抽滤得液体,用300~500eq乙酸乙酯稀释混合物,分离水相并用100~300eq乙酸乙酯再次萃取水相。将合并的有机相用无水硫酸钠干燥并浓缩。残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为100:1,得到淡绿色油状化合物1,产率89%。1h nmr(400mhz,cdcl3):δ=7.52(d,j=7.7hz,2h),7.46(d,j=6.8hz,4h),1.95
–
1.87(m,4h),1.17
–
0.97(m,12h),0.78(t,j=7.1hz,6h),0.65
–
0.52(m,4h)。
[0089]
(2)化合物2的合成
[0090][0091]
化合物1(2.45g,5mmol,1eq)溶解在重蒸后的thf(80~110eq)中,体系,在
‑
20~
‑
80℃不断搅拌1~3小时后,加入正丁基锂(3.44ml,1.6m在己烷中的溶液,5.6mmol,0.8~3eq)在7~30分钟内逐滴加入。加入dmf(0.5~3eq),在冷却浴中搅拌1~3小时,在浴外搅拌1~3小时。然后将反应物冷却至5~20℃。并用水淬灭反应。用300~500eq乙酸乙酯稀释混合物,分离水相并用100~300eq乙酸乙酯再次萃取。将合并的有机相用无水硫酸钠干燥并浓缩。残余固体通过硅胶柱提纯,洗脱剂石油醚和乙酸乙酯的体积比优选为25:1,得到淡绿色油状化合物2,产率52%。1h nmr(400mhz,cdcl3):δ=10.08(s,1h),7.88(s,3h),7.64(d,j=7.8hz,1h),7.53(s,2h),2.04(m,4h),1.22(m,12h),0.82(t,j=7.2hz,6h),0.58(m,4h)。
[0092]
(3)化合物3的合成
[0093][0094]
将化合物2(1.5g,3.4mmol,1eq),三甲基乙炔基硅(1.5g,15.3mmol,3~10eq),cui(30mg,0~1eq),将pdcl2(pph3)2(100mg,0~1eq)和三乙胺(10ml,30~100eq)在30~100eq干燥的thf中的溶液在氮气下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,洗脱剂石油醚和乙酸乙酯的体积比优选为30:1,得到淡黄色固体化合物3,产率63%。1h nmr(400mhz,cdcl3)δ10.08(d,j=2.3hz,1h),7.87
–
7.80(m,3h),7.77(dd,j=7.7,0.6hz,1h),7.51(dd,j=7.7,1.5hz,2h),2.09
–
1.94(m,4h),1.07(dd,j=14.8,7.4hz,4h),0.66(td,j=7.3,3.9hz,12h),0.60
–
0.47(m,6h),0.29(s,9h).
[0095]
(4)化合物ⅰ的合成
[0096][0097]
化合物3(1g,2.48mmol,1eq)在300~700mg(0.8~1.5eq)碳酸钾存在下,30~90ml(80~120eq)甲醇中搅拌悬浮,除去三甲基硅基,得白色固体化合物ⅰ,产率95%。1h nmr(400mhz,cdcl3)δ10.07(d,j=2.3hz,1h),7.89
–
7.81(m,3h),7.74(dd,j=7.7,0.6hz,1h),7.53(dd,j=7.7,1.5hz,2h),3.20(s,1h),2.09
–
1.94(m,4h),1.07(dd,j=14.8,7.4hz,4h),0.66(td,j=7.3,3.9hz,12h),0.60
–
0.47(m,6h).
[0098]
(5)化合物ⅲ的合成
[0099][0100]
化合物ⅱ(1g,0.32mmol,1eq),化合物ⅰ(2.47g~3.71g,2
‑
3eq),cui(6~9mg,0.1
‑
0.2eq),将pdcl2(pph3)2(23~34mg,0.1~0.2eq)和三乙胺(30~100eq)在30~100eq干燥的thf中的溶液在氮气下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,洗脱剂为石油醚和乙酸乙酯的体积比优选为12:1,得到黄色固体化合物ⅲ,产率52%。1h nmr(400mhz,cdcl3):δ=10.11(s,2h),8.74(d,j=5.0hz,2h),8.64(s,2h),7.96
–
7.86(m,6h),7.83(d,j=8.4hz,2h),7.68
–
7.58(m,4h),7.55
–
7.44(m,2h),2.18
–
1.99(m,8h),1.20
–
1.00(m,24h),0.79(t,j=7.1hz,12h),0.70
–
0.51(m,8h)。
[0101]
(6)化合物ⅳ的合成
[0102][0103]
化合物ⅲ(0.2g,0.022mmol,1eq),二(二甲基亚砜)二氯化铂(0.5
‑
2.4eq),乙腈(200
‑
300eq)和二氯甲烷(200
‑
300eq)在氮气保护下在20~100℃加热10~30小时,混合物通过硅胶柱,洗脱剂为石油醚和乙酸乙酯的体积比优选为10:1,得到橘黄色固体化合物ⅳ,产率43%。1h nmr(400mhz,cdcl3):δ=10.12(s,2h),8.87(d,j=5.1hz,2h),8.70(s,2h),8.01
–
7.96(m,6h),7.87(d,j=8.2hz,2h),7.70
–
7.60(m,4h),7.57
–
7.44(m,2h),2.19
–
1.99(m,8h),1.21
–
1.03(m,24h),0.80(t,j=7.1hz,12h),0.71
–
0.53(m,8h)。
[0104]
(7)pt
‑
4cho的合成
[0105][0106]
化合物ⅳ(0.1g,0.0084mmol,1eq),化合物ⅰ(2
‑
3eq),cui(6~9mg,0.1
‑
0.2eq),n,n
‑
二异丙基乙胺(30~50eq)在50~100eq干燥的二氯甲烷中的溶液在氮气下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,洗脱剂为石油醚和二氯甲烷的体积比优选为2:1,得到红色固体,产率32%。1h nmr(400mhz,cdcl3):δ=10.12(s,2h),10.07(s,2h),9.95(s,2h),8.25(s,2h),7.96
–
7.92(m,6h),7.89
–
7.84(m,6h),7.79(m,24h),7.74
–
7.57(m,10h),2.17
–
2.00(m,16h),1.21
–
0.99(m,48h),0.79(t,j=7.1hz,24h),0.73
–
0.50(m,16h)。
[0107]
实施例2
[0108]
根据实施例1合成的目标产物pt
‑
4cho,为了更为直观方便的测试目标产物pt
‑
4cho的光物理性质以及自修复性能,本发明在此基础上合成模型分子pt
‑
4imine,具体合成步骤如下:
[0109]
[0110]
四醛基铂配合物pt
‑
4cho(0.1g,0.053mmol,1eq),乙胺(1
‑
10eq),无水硫酸钠(1
‑
5eq),在200
‑
300eq的二氯甲烷中,氮气下在20~50℃反应10~30小时。得到红色固体,产率90%。1h nmr(400mhz,cdcl3):δ=10.10(s,2h),10.05(s,2h),9.93(s,2h),8.23(s,2h),7.93
–
7.90(m,6h),7.87
–
7.82(m,6h),7.81
–
7.73(m,4h),7.72
–
7.57(m,10h),2.16
–
1.97(m,16h),1.38
–
1.20(m,8h),1.20
–
0.95(m,48h),0.80
‑
0.72(m,36h),0.70
–
0.47(m,16h)。
[0111]
实施例3
[0112]
与实施例1不同的是,本实施例中将化合物1的卤素溴用异氰酸酯基取代,继续进行实施例1中的反应,合成端位带有异氰酸酯基的pt
‑
4nco的目标产物。
[0113]
具体合成步骤如下:
[0114]
(1)7
‑
溴
‑
9,9
‑
二己基芴
‑2‑
异氰酸酯的合成
[0115][0116]7‑
溴
‑
9h
‑
芴
‑2‑
异氰酸酯(12.9g,45.4mmol,1eq),溴代正己烷(45.4
‑
136.2mmol,1
‑
3eq),氢氧化钾(45.4
‑
454mmol,1
‑
10eq),碘化钾(4.5
‑
45.4mmol,0.1
‑
1eq)溶解在dmso(80~110eq)中,体系在0~30℃不断搅拌8
‑
12小时后,点板跟踪反应,反应结束后,抽滤得液体,用300~500eq乙酸乙酯稀释混合物,分离水相并用100~300eq乙酸乙酯再次萃取水相。将合并的有机相用无水硫酸钠干燥并浓缩。残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为85:1,得到白色固体,产率74%。
[0117]
(2)9,9
‑
二己基
‑7‑
((三甲基硅)乙炔基)芴
‑2‑
异氰酸酯的合成
[0118][0119]
将7
‑
溴
‑
9,9
‑
二己基芴
‑2‑
异氰酸酯(1.6g,3.4mmol,1eq),三甲基乙炔基硅(1.6g,15.3mmol,3~10eq),cui(30mg,0~1eq),将pdcl2(pph3)2(100mg,0~1eq)和三乙胺(10ml,30~100eq)在30~100eq干燥的thf中的溶液在氮气下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,洗脱剂为石油醚和乙酸乙酯的体积比优选为100:1,得到淡黄色固体,产率52%。
[0120]
(3)9,9
‑
二己基
‑7‑
乙炔基芴
‑2‑
异氰酸酯的合成
[0121][0122]
9,9
‑
二己基
‑7‑
((三甲基硅)乙炔基)芴
‑2‑
异氰酸酯(1.1g,2.48mmol,1eq)在300~700mg(0.8~1.5eq)碳酸钾存在下,30~90ml(80~120eq)甲醇中搅拌悬浮,除去三甲基硅基,得白色固体,产率88%。
[0123]
(4)
[0124][0125]
4,4
’‑
二溴
‑
2,2
’‑
联吡啶(1g,0.32mmol,1eq),9,9
‑
二己基
‑7‑
乙炔基芴
‑2‑
异氰酸酯(2.5g~3.71g,2
‑
3eq),cui(6~9mg,0.1
‑
0.2eq),将pdcl2(pph3)2(23~34mg,0.1~0.2eq)和三乙胺(30~100eq)在30~100eq干燥的thf中的溶液在氮气下在20~100℃加热10~30小时。冷却后,混合物通过硅胶柱,洗脱剂为石油醚和乙酸乙酯的体积比优选为20:1,得到黄色固体,产率45%。
[0126]
实施例4
[0127]
与实施例1不同的是,步骤1的反应中原料产物溴代烷还可以替换成碘甲烷(化学式ch3i),其余步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为50:1,得到白色固体2,7
‑
二溴
‑
9,9
‑
二甲基芴,产率78%。
[0128]
步骤2的反应与实施例1步骤2的反应步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为10:1,得到白色固体7
‑
溴
‑
9,9
‑
二甲基芴
‑2‑
甲醛,产率61%。
[0129]
实施例5
[0130]
与实施例1不同的是,步骤1的反应中原料产物溴代烷还可以替换成溴代丁烷(化学式c4h9br),其余步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为70:1,得到白色固体2,7
‑
二溴
‑
9,9
‑
二丁基芴,产率78%。
[0131]
步骤2的反应与实施例1步骤2的反应步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为20:1,得到白色固体7
‑
溴
‑
9,9
‑
二丁基芴
‑2‑
甲醛,产率59%。
[0132]
实施例6
[0133]
与实施例1不同的是,步骤1的反应中原料产物溴代烷还可以替换成溴代辛烷(化学式c8h
17
br),其余步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为100:1,得到无色油状物2,7
‑
二溴
‑
9,9
‑
二辛基芴,产率85%。
[0134]
步骤2的反应与实施例1步骤2的反应步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为60:1,得到无色油状物7
‑
溴
‑
9,9
‑
二辛基芴
‑2‑
甲醛,产率58%。
[0135]
实施例7
[0136]
与实施例1不同的是,步骤1的反应中原料产物溴代烷还可以替换成溴代十二烷(化学式c
12
h
25
br),其余步骤相同,残余固体通过硅胶柱提纯,洗脱剂为纯石油醚,得到无色油状物2,7
‑
二溴
‑
9,9
‑
二(十二烷)基芴,产率85%。
[0137]
步骤2的反应与实施例1步骤2的反应步骤相同,残余固体通过硅胶柱提纯,洗脱剂为石油醚和乙酸乙酯的体积比优选为200:1,得到无色油状物7
‑
溴
‑
9,9
‑
二(十二烷)基芴
‑2‑
甲醛,产率59%。
[0138]
测试例1
[0139]
紫外
‑
可见光谱吸收光谱是通过使用一种tu
‑
1900uv
‑
vis分光光度计(agilent)获得的。光致发光光谱是在一台日立f
‑
4600荧光分光光度计上进行的。不同浓度的pt
‑
4imine甲苯溶液(1
×
10
‑4‑5×
10
‑6mol/l)紫外
‑
可见吸收光谱如图1
‑
图4所示,具体数据列于表1。如图5
‑
图11所示,在所研究的浓度范围内,pt
‑
4imine的吸收遵循beer定律,说明在该浓度范围内没有发生基态聚集。通过查阅相关文献,将300
‑
400nm的吸收峰归结为π
‑
π*跃迁和n
‑
π*跃迁。如图2所示,pt
‑
4imine的溶剂化效应可以看出,400
‑
500nm处的吸收峰展现出一定的蓝移现象,这可以归结为单重态金属到配体的电荷转移(1mlct)和单重态配体到配体的电荷转移(1llct)。接着又对500nm以后的尾部吸收进行归属,我们发现600
‑
650nm依旧有着弱的吸收,将其归属为三重态金属到配体的电荷转移(3mlct)和三重态配体到配体的电荷转移(3llct),这代表在这一区域依然可以发生激发态吸收。
[0140]
表1.pt
‑
4imine在不同溶液中的光学性质。
[0141][0142]
a
在不同的脱气溶剂中测定量子产率,硫酸奎宁为参比样品(φ
f
=0.042in h2o,e
x
=436nm)
[0143]
测试例2
[0144]
为了研究pt
‑
4imine的发射来源,对比了空气饱和和氮气吹扫两个发射光谱,发现氮气吹扫后发射明显增强,如图3所示,结合其大的斯托克斯位移115nm,判断其来源于磷光发射。随后研究了pt
‑
4imine的发射态,对其溶致变色效应展开了系统的实验。如图4所示,首先在5种不同溶剂中配制1
×
10
‑5mol
·
l
‑1浓度的溶液,分别对其扫吹氮气前后的发射光谱做了比较,发现随着溶剂极性的增加,发射光谱并没有展现出明显移动,说明了其主要的发射态主要来源于π
‑
π*态,可能混合着一些金属到配体的电荷转移和配体到配体的电荷转移。
[0145]
测试例3
[0146]
为了进一步研究铂配合物在凝胶状态下的光物理性能,依旧采用模型分子pt
‑
4imine做为对比进行探讨。如图5所示,对比了两相中铂配合物的紫外
‑
可见光吸收光谱,他们在400nm之后展现出相同的吸收带,说明发挥作用的分子结构也一致,模型分子的设计和
对比是合理的;而400nm之前吸收谱带的差异是由于高浓度均三苯甲醛的吸收,以及300nm以上的吸收可能来源于聚二甲基硅氧烷的n
‑
π*跃迁;之后又比较了两相中铂配合物的归一化发射光谱,其峰型和最大发射波长均完全一致,说明来源于同一发射态,进一步表明凝胶中的铂配合物结构与模型分子高度一致,如图6所示。在此之后,研究了凝胶状态下的透射光谱,如图7所示。透射光谱展现从紫外光区到可见光区的透过率情况,625nm之前透过率逐步上升,是因为铂配合物在该光区中的吸收逐渐减弱;而625nm之后展现出接近100%的透过率,说明在该区域的光学透明性极好。最关键的,研究对比了两相下的光限幅性能。为在同一级别上对比,在制备凝胶时计算好pt
‑
4cho的浓度,以保证形成凝胶后的透过率与模型分子pt
‑
4imine的透过率一致为80%。对比发现两相中,铂配合物都展现出在低能量的入射光条件下有较高的透光率,随着入射光能量的增加,透过率下降,起到光伏限制的作用。相比之下,在凝胶态中的铂配合物展现出更优越的光限幅性能。这主要是因为,凝胶中错综复杂的三维网络结构在强激光干预下会产生非线性散射效应,从而进一步抑制光线的透过,使得透过率下降,如图8
‑
图11所示。
[0147]
测试例4
[0148]
如图12所示,为了进一步研究其在凝胶状态下的光物理性质,引入氨基封端的聚二甲基硅氧烷作为柔性聚合物链,均苯三甲醛和pt
‑
4cho与末端氨基形成可以亚胺键实现交联,形成动态三维交联网络,制备光功能自修复凝胶。将0.1g双(胺)端聚二甲基硅氧烷(nh2‑
pdms
‑
nh2)与0.4ml的20mg pt
‑
4cho复合物在二甲酰胺(简称为dmf)中混合,合成自修复的pt
‑
4cho@pdms凝胶。混合物在容器中室温干燥12小时,在70摄氏度下加热5h,制备pdms凝胶。
[0149]
为制备透明均一的光功能化自修复凝胶,改进实验方案。如图13
‑
图14所示,将0.8g双(胺)端聚二甲基硅氧烷(nh2‑
pdms
‑
nh2)与0.4ml的0.23m均苯三甲醛和不同浓度的pt
‑
4cho配合物溶解在甲苯:dmf=3:1,合成自修复的pt
‑
4cho@pdms凝胶。混合物在容器中室温干燥12小时,在70摄氏度下加热5h,得到pdms凝胶。数码照片观察到,试管倒置不滑落,说明凝胶已经形成。从试管中剥离后能直立不瘫软,说明该凝胶材料具有一定的自支撑能力,使得材料器件化成为可能。而对凝胶模块进行切割,再闭合伤口,短短1小时后可以达到一定的自修复效果,如图15
‑
图17所示,说明在凝胶断裂平面均三苯甲醛或pt
‑
4cho与末端氨基形成的亚胺破坏断裂,而随着伤口的闭合,断裂平面紧密接触,可逆亚胺键再次形成,交联网络结构恢复,达到自修复的效果。
[0150]
测试例5
[0151]
机械拉伸试验采用cmt4202/zwick/z020系统进行。测试样本的测量尺寸是大约10毫米
×
1毫米
×
5毫米。拉伸试验中,速度为2mm/min。为进一步优化凝胶性能,我们首先筛选了6种溶剂,溶剂选择依据是兼具铂配合物、均苯三甲醛、以及氨基封端的pdms优异溶解性。如图18可知,在二甲苯、邻二甲苯、甲苯、1,2
‑
二氯苯、1,4
‑
二氧六环、乙酸乙酯中的拉伸应力应变曲线可知,其在邻二甲苯中的凝胶力学性能最为优越,因此,之后的实验将以此为溶剂条件。而在凝胶体系的制备过程中,会有意识调整线性聚合物和交联聚合物的比例以谋求最韧结构,因此在本工作中通过1,4
‑
苯二醛和1,3,5
‑
均苯三甲醛作为连接分子,改变其交联度,已调节拉伸性能最优。如图可以看出,随1,4
‑
苯二醛增加,线性聚合物比例提升,凝胶本身的流动性有所提高,因此展现出更好的拉伸性能,在三醛比二醛6:4时达到最佳,在
之后,二醛比例进一步提升则会使凝胶体系的流动性不足以支持它的回弹性能,导致不可逆形变。自修复后的拉伸性能是衡量凝胶体系自修复性能的重要参数之一,从破坏前和修复后的拉伸性能对比可以看出,拉伸的自修复效率高达95%,显示其优越的自修复性能。而最后,为了验证其拉伸回弹效果,进行了应力应变循环实验,可以看出在4次循环后,应变依旧能达到原先的99%以上,但是应力衰减严重,这可能是内部的高分子链发生伸展,短时间内无法恢复至弯曲盘踞状态所致。以上实验展现了材料良好的拉伸性能和机械性能,有望在软材料器件上得以应用,结果如图18
‑
图21所示。
[0152]
测试例6
[0153]
采用共聚焦激光扫描显微镜(lext ols4100)测量自愈动态。pt
‑
4cho@pdms膜和空白膜分别削减商业刀片,把它们放在一起,紧随其后的是激光点关注空白膜的边界在哪里旁边的pdms膜,然后测量了pl光谱在愈合过程中每1小时在室温下。如图22所示,裂缝处缓慢修复,在12小时后修复完成,并恢复一定的光学性能和力学性能,这也表明动态可逆亚胺键能够在被破坏时,主动扩散、缠绕、可逆反应的自我修复,实现整体材料结构的自愈合。本课题拟构筑新型金属有机凝胶材料,通过将材料加工成型,得到具有自修复特性的功能涂层或镜片等光学器件,拓展光限幅材料在激光防护、激光探测保护等领域的实际应用。
[0154]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。