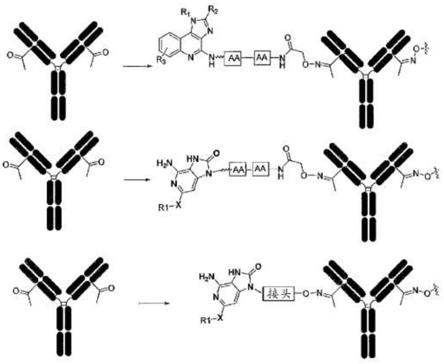
包含抗体
‑
tlr激动剂缀合物的组合物、方法和用途
1.相关申请的引用
2.本技术要求美国临时申请第62/804,742号的权益,其各自的标题为“compositions containing,methods and uses of antibody
‑
tlr agonist conjugates”,提交于2019年2月12日,其内容通过引用完整并入本文。
3.序列表
4.本技术含有已经以ascii格式经由efs
‑
web提交并且通过引用完整并入本文的序列表。创建于2020年2月7日的ascii副本被命名为ambx_0230_pct_sl.txt,大小为30,527个字节。
发明领域
5.本发明公开涉及tlr激动剂化合物和tlr激动剂缀合物(tc)及其用途。本发明进一步涉及含有作为治疗剂或预防剂的(tc)的药物组合物。
6.发明背景
7.使用非天然编码的氨基酸,通过位点特异性缀合,可以将诸如抗体及其片段的靶向分子或多肽与tlr激动剂化合物缀合在一起,从而产生新型的tlr激动剂缀合物(tc)。可以以这样的方式构建新型的tc,即,在全身治疗期间,循环tc可以将tlr激动剂靶向至肿瘤部位,并局部刺激有益的免疫应答,从而将全身性细胞因子释放综合征降至最低限度。
技术实现要素:
8.本发明涉及使用一个或多个非天然编码氨基酸与tlr的激动剂化合物缀合的靶向多肽,所述tlr包括但不限于tlr7和/或tlr8。这样的缀合物在本文中称为tlr激动剂缀合物(tc)。本发明的tc包括靶向生物分子或多肽和tlr激动剂化合物,它们通过位点特异性缀合使用非天然编码氨基酸缀合在一起,产生新型的生物tlr激动剂缀合物(btc)。靶向生物分子或多肽可以是肿瘤靶向生物分子或多肽。
9.在另外的实施方案中,本发明还涉及进一步与水溶性聚合物缀合形成稳定的二聚体或多聚体的tc。
10.本发明提供了抑制或降低肿瘤或癌症生长的方法,其包括使肿瘤与有效量的本发明tc接触,以刺激患者肿瘤附近的免疫系统。本发明提供了抑制或降低肿瘤或癌症生长的方法,其包括使肿瘤与有效量的本发明的聚乙二醇化tc或tc的稳定二聚体或多聚体接触。在一个实施方案中,tc是非聚乙二醇化或单聚乙二醇化的。在一个实施方案中,tc是二聚乙二醇化的。在一个实施方案中,tc具有与之附接的多于一个和/或不同的tlr激动剂分子。在一个实施方案中,tc具有与之附接的多于一个和/或相同的tlr激动剂分子。本发明的另一个实施方案提供了使用本发明的tc来调节对肿瘤细胞的免疫应答的方法。在某些实施方案中,tc与至少一种化疗剂和/或至少一种免疫治疗剂共同施用。化学治疗剂可选自由以下组成的组:替莫唑胺、吉西他滨、多柔比星、环磷酰胺、紫杉醇、顺铂、氟嘧啶、紫杉烷、蒽环类、拉帕替尼、卡培他滨、来曲唑、帕妥珠单抗、多西他赛、ifn
‑
α。在本发明的另一个实施方案
中,tc与至少一种化疗剂和/或至少一种免疫治疗剂共同施用。
11.在一些实施方案中,tc包含靶向多肽,所述靶向多肽包括但不限于包含一个或多个非天然编码氨基酸的抗原结合多肽(abp)。在一些实施方案中,abp包含完整的抗体重链。在一些实施方案中,abp包含完整的抗体轻链。在一些实施方案中,abp包含抗体轻链的可变区。在一些实施方案中,abp包含抗体重链的可变区。在一些实施方案中,abp包含抗体轻链的至少一个cdr。在一些实施方案中,abp包含抗体重链的至少一个cdr。在一些实施方案中,abp包含轻链的至少一个cdr和重链的至少一个cdr。在一些实施方案中,abp包含fab。在一些实施方案中,abp包含两个或更多个fab。在一些实施方案中,abp包含(fab’)2。在一些实施方案中,abp包含两个或更多个(fab’)2。在一些实施方案中,abp包含scfv。在一些实施方案中,abp包含两个或更多个scfv。在一些实施方案中,abp包含微型抗体。在一些实施方案中,abp包含两个或更多个微型抗体。在一些实施方案中,abp包含双抗体。在一些实施方案中,abp包含两个或更多个双抗体。在一些实施方案中,abp包含轻链的可变区和重链的可变区。在一些实施方案中,abp包含完整的轻链和完整的重链。在一些实施方案中,abp包含一个或多个fc结构域或其部分。在一些实施方案中,abp包含任何上述实施方案的组合。在一些实施方案中,abp包含任何上述实施方案的同二聚体、异二聚体、同源多聚体或异源多聚体。在一些实施方案中,abp包含与结合配偶体结合的多肽,其中结合配偶体包括抗原、多肽、核酸分子、聚合物或其他分子或物质。在一些实施方案中,abp与非抗体支架分子或物质缔合。在一些实施方案中,抗原是肿瘤抗原。
12.toll样受体(tlr)检测广泛的保守的病原体相关分子模式(pamp)。它们在感知入侵病原体和随后启动先天免疫应答方面发挥重要作用。在人中有10个已知的tlr家族成员,它们是i型跨膜蛋白,特征为细胞外富含亮氨酸结构域和含有保守的toll/白细胞介素(il)
‑
l受体(tir)结构域的胞质尾。在这个家族中,tlr3、tlr7、tlr8和tlr9位于内体中。通过与特异性小分子配体(即,tlr7激动剂或tlr8激动剂)或其天然配体(即,单链rna,ssrna)结合,tlr7和tlr8可被激活。在激动剂与tlr7或tlr8结合后,处于其二聚化形式的受体被认为经历了结构变化,导致随后在其胞质结构域募集衔接蛋白,包括髓样分化初级应答基因88(myd88)。在通过myd88途径启动受体信号传导级联后,诸如干扰素调节因子7(irf
‑
7)和核因子κb(nf
‑
κβ)的细胞质转录因子被激活。这些转录因子然后转位到细胞核,并启动各种基因例如干扰素
‑
α和其他抗病毒细胞因子基因的转录。tlr7主要在浆细胞样细胞和b细胞上表达。免疫细胞应答性的改变可能导致癌症患者先天免疫应答的降低。因此,激动剂诱导的与靶向部分(比如抗体或其片段)缀合的tlr7和/或tlr8的激活可代表一种新的癌症治疗方法。利用包含tlr7或tlr8激动剂的tc进行治疗代表一种有希望的解决方案,从而提供更好的效果和更好的耐受性。用于本发明中制备tc的适合的tlr7和/或tlr8激动剂可以下美国专利中找到,将其各自通过引用并入本文:美国专利第6,825,350号;美国专利第6,656,389号;美国专利第6,656,398号;美国专利第6,683,088号;美国专利第6,756,382号;美国专利第6,825,350号;美国专利第6,667,312号;美国专利第6,677,347号;美国专利第7,598,382号;美国专利第8,673,932号。
13.在一些实施方案中,tc包含的靶向多肽进一步包含氨基酸取代、添加或缺失,与没有取代、添加或缺失的相应野生型tc的相容性相比,所述氨基酸取代、添加或缺失增加了tc多肽与药物防腐剂(例如,间甲酚、苯酚、苯甲醇)的相容性。这种增加的相容性将使得能够
制备在储存期间保持蛋白质的理化性质和生物活性的保存的药物制剂。
14.在一些实施方案中,用一个或多个非天然氨基酸产生一个或多个经工程改造的键。可以以多种方式产生分子内键,这些方式包括但不限于在适合条件下蛋白质中两个氨基酸之间的反应(一个或两个氨基酸可以是非天然氨基酸);在适合条件下两个氨基酸(每个氨基酸都可以是天然编码的或非天然编码的)与接头、聚合物或其他分子等的反应。
15.在一些实施方案中,tc多肽中的一个或多个氨基酸取代可以是用一个或多个天然存在或非天然存在的氨基酸。在一些实施方案中,tc中的氨基酸取代可以是用天然存在或非天然存在的氨基酸,前提是至少一个取代是用非天然编码氨基酸。在一些实施方案中,tc多肽中的一个或多个氨基酸取代可以是用一个或多个天然存在的氨基酸,并且另外至少一个取代是用非天然编码氨基酸。在一些实施方案中,tc多肽可以是抗体或抗体片段。在一些实施方案中,tc多肽可以是肿瘤靶向多肽。
16.在一些实施方案中,非天然编码氨基酸包含羰基、乙酰基、氨基氧基、肼基、酰肼基、氨基脲基、叠氮基或炔基。
17.在一些实施方案中,非天然编码氨基酸包含羰基。在一些实施方案中,非天然编码氨基酸具有以下结构:
[0018][0019]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基或取代的芳基;r2是h、烷基、芳基、取代的烷基和取代的芳基;r3是h、氨基酸、多肽或氨基末端修饰基团,并且r4是h、氨基酸、多肽或羧基末端修饰基团。
[0020]
在一些实施方案中,非天然编码氨基酸包含氨基氧基。在一些实施方案中,非天然编码氨基酸包含酰肼基。在一些实施方案中,非天然编码氨基酸包含肼基。在一些实施方案中,非天然编码氨基酸残基包含氨基脲基。
[0021]
在一些实施方案中,非天然编码氨基酸残基包含叠氮基。在一些实施方案中,非天然编码氨基酸具有以下结构:
[0022][0023]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基、取代的芳基或不存在;x是o、n、s或不存在;m为0
‑
10;r2是h、氨基酸、多肽或氨基末端修饰基团,并且r3是h、氨基酸、多肽或羧基末端修饰基团。
[0024]
在一些实施方案中,非天然编码氨基酸包含炔基。在一些实施方案中,非天然编码氨基酸具有以下结构:
[0025][0026]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基或取代的芳基;x是o、n、s或不存在;m为0
‑
10;r2是h、氨基酸、多肽或氨基末端修饰基团,并且r3是h、氨基酸、多肽或羧基末端修饰基
团。
[0027]
在一些实施方案中,多肽是包含与水溶性聚合物连接的非天然编码氨基酸的tc。在一些实施方案中,水溶性聚合物包含聚(乙二醇)部分。在一些实施方案中,tc包含非天然编码氨基酸和一种或多种翻译后修饰、接头、聚合物或生物活性分子。
[0028]
本发明还提供了包含编码tc的靶向多肽的多核苷酸的分离的核酸,并且本发明提供了包含在严格条件下与所述多核苷酸杂交的多核苷酸的分离的核酸。本发明还提供了包含编码靶向多肽的多核苷酸的分离的核酸,其中所述多核苷酸包含至少一个选择密码子(selector codon)。对于本领域普通技术人员显而易见的是,许多不同的多核苷酸可以编码本发明的任何多肽。
[0029]
在一些实施方案中,选择密码子选自由以下组成的组:琥珀密码子、赭石密码子、蛋白石密码子、独特密码子、稀有密码子、五碱基密码子和四碱基密码子。
[0030]
本发明还提供了制备与水溶性聚合物连接或与一种或多种tc多肽连接以形成同二聚体或同多聚体的tc多肽的方法。在一些实施方案中,所述方法包括使包含非天然编码氨基酸的分离的tc多肽与水溶性聚合物或包含与非天然编码氨基酸反应的部分的接头接触。在一些实施方案中,掺入tc多肽中的非天然编码氨基酸对水溶性聚合物或接头具有反应性,而所述水溶性聚合物或接头对20种常见氨基酸中的任何一种都不具有反应性。在一些实施方案中,掺入tc多肽中的非天然编码氨基酸对接头、聚合物或生物活性分子具有反应性,而所述接头、聚合物或生物活性分子对20种常见氨基酸中的任何一种都不具有反应性。
[0031]
在一些实施方案中,通过使包含含羰基氨基酸的tc多肽与聚(乙二醇)分子或包含氨基氧基、肼、酰肼或氨基脲基的接头反应,制备与水溶性聚合物或接头连接的tc多肽。在一些实施方案中,氨基氧基、肼、酰肼或氨基脲基通过酰胺键与聚(乙二醇)分子或接头连接。在一些实施方案中,氨基氧基、肼、酰肼或氨基脲基通过氨基甲酸酯键与聚(乙二醇)分子或接头连接。
[0032]
在一些实施方案中,通过使聚(乙二醇)分子或包含羰基的接头与包含非天然编码氨基酸的多肽反应,制备与水溶性聚合物连接的tc多肽,所述非天然编码氨基酸包含氨基氧基、肼基、酰肼基或氨基脲基。
[0033]
在一些实施方案中,通过使包含含炔氨基酸的tc与包含叠氮化物部分的聚(乙二醇)分子反应,制备与水溶性聚合物或接头连接的tc多肽。在一些实施方案中,叠氮化物或炔基通过酰胺键与聚(乙二醇)分子或接头连接。
[0034]
在一些实施方案中,通过使包含含叠氮化物的氨基酸的tc多肽与包含炔部分的聚(乙二醇)分子反应,制备与水溶性聚合物或接头连接的tc多肽。在一些实施方案中,叠氮化物或炔基通过酰胺键与聚(乙二醇)分子或接头连接。
[0035]
在一些实施方案中,聚(乙二醇)分子或接头具有介于约0.1kda和约100kda之间的分子量。在一些实施方案中,聚(乙二醇)分子或接头具有介于0.1kda和50kda之间的分子量。在一些实施方案中,聚(乙二醇)分子或接头是支链聚合物或接头。在一些实施方案中,聚(乙二醇)支链聚合物或接头的每个分支具有介于1kda和100kda之间或介于1kda和50kda之间的分子量。
[0036]
在一些实施方案中,与tc多肽连接的水溶性聚合物包含聚亚烷基二醇部分。在一
些实施方案中,掺入tc中的非天然编码氨基酸残基包含羰基、氨基氧基、酰肼基、肼、氨基脲基、叠氮基或炔基。在一些实施方案中,掺入tc多肽中的非天然编码氨基酸残基包含羰基部分,并且水溶性聚合物包含氨基氧基、酰肼、肼或氨基脲部分。在一些实施方案中,掺入tc多肽中的非天然编码氨基酸残基包含炔部分,并且水溶性聚合物包含叠氮化物部分。在一些实施方案中,掺入tc多肽中的非天然编码氨基酸残基包含叠氮化物部分,并且水溶性聚合物包含炔部分。本发明还提供了包含含有非天然编码氨基酸的tc多肽和药学上可接受的载体的组合物。在一些实施方案中,非天然编码氨基酸与水溶性聚合物连接。
[0037]
本发明还提供了包含编码包含选择密码子的tc的靶向多肽的多核苷酸的细胞。在一些实施方案中,所述细胞包含正交rna合成酶和/或正交trna,用于将非天然编码氨基酸取代到tc的靶向多肽中。
[0038]
本发明还提供了制备包含非天然编码氨基酸的tc的靶向多肽的方法。在一些实施方案中,所述方法包括在允许表达tc的靶向多肽或其变体的条件下培养包含编码tc的靶向多肽的一种或多种多核苷酸、正交rna合成酶和/或正交trna的细胞;以及从细胞和/或培养基纯化所述tc多肽。
[0039]
本发明还提供了延长tc的治疗半衰期、血清半衰期或循环时间的方法。本发明还提供了调节tc的免疫原性的方法。在一些实施方案中,所述方法包括用非天然编码氨基酸取代天然存在的tc的靶向多肽中的任何一个或多个氨基酸,以及/或者,将所述靶向多肽与接头、聚合物、水溶性聚合物或生物活性分子连接。
[0040]
本发明还提供了用有效量的本发明tc分子治疗需要这种治疗的患者的方法。在一些实施方案中,所述方法包括向患者施用治疗有效量的包含含有非天然编码氨基酸的tc和药学上可接受的载体的药物组合物。在一些实施方案中,非天然编码氨基酸与水溶性聚合物连接。在一些实施方案中,tc是糖基化的。在一些实施方案中,tc是未糖基化的。
[0041]
本发明还提供了这样的tc,其包含在单个氨基酸处通过共价键与tc连接的水溶性聚合物或接头。在一些实施方案中,水溶性聚合物包含聚(乙二醇)部分。在一些实施方案中,与水溶性聚合物或接头共价连接的氨基酸是存在于tc的靶向多肽中的非天然编码氨基酸。
[0042]
本发明提供了包含至少一个接头、聚合物或生物活性分子的tc多肽,其中所述接头、聚合物或生物活性分子通过经核糖体掺入tc的靶向多肽中的非天然编码氨基酸的官能团与所述多肽附接。在tc缀合物中,peg或其他水溶性聚合物、另一种tc、多肽或生物活性分子可以经由接头与tc直接缀合。在一个实施方案中,接头足够长以容许柔性以及允许二聚体形成。在一个实施方案中,接头在长度上为至少3个氨基酸或18个原子,从而允许形成二聚体。在一些实施方案中,多肽与接头连接以允许多聚体形成。在一些实施方案中,接头是双官能接头。在一些实施方案中,本发明的组合物和/或tc可以包含多个接头。在其他实施方案中,每个接头可包括附接的一个或多个化合物。接头还可包含亚烷基、亚烯基、亚炔基、聚醚、聚酯、聚酰胺基团以及还有聚氨基酸、多肽、可裂解肽或氨基苄基氨基甲酸酯。在一些实施方案中,接头可以是相同或不同的接头。适合的接头包括,例如,可裂解和不可裂解的接头。适合的可裂解接头包括,例如,可被细胞内蛋白酶如溶酶体蛋白酶或内体蛋白酶裂解的肽接头。可裂解接头可包括缬氨酸
‑
瓜氨酸接头或缬氨酸
‑
丙氨酸肽。在一些实施方案中,接头可以是二肽接头,比如缬氨酸
‑
瓜氨酸或苯丙氨酸
‑
赖氨酸接头。含缬氨酸
‑
瓜氨酸或缬
氨酸
‑
丙氨酸的接头可含有马来酰亚胺或琥珀酰亚胺基团。含缬氨酸
‑
瓜氨酸或缬氨酸
‑
丙氨酸的接头可含有对氨基苄醇(paba)基团或对氨基苄基氨基甲酸酯(pabc)。其他适合的接头包括在低于5.5的ph下可水解的接头,比如腙接头。另外的适合的可裂解接头包括二硫化物接头。在一些实施方案中,可裂解接头可包括在肿瘤微环境比如肿瘤浸润性t细胞处被裂解的接头。在一些实施方案中,不可裂解的接头包括,但不限于,马来酰亚胺基己酰基接头。马来酰亚胺基己酰基接头可包含n
‑
马来酰亚胺甲基环己烷
‑1‑
甲酸酯、琥珀酰亚胺基团、五氟苯基和/或一种或多种peg分子,但不限于此。在一些实施方案中,本发明组合物、化合物或其盐中的任一者可通过接头与多肽连接。在一些实施方案中,在本文表3、4、5、6和7中公开的化合物或其盐中的任一者可通过接头与多肽连接。在一些实施方案中,所述多肽是靶向多肽或生物靶向多肽或肿瘤靶向多肽。在一些实施方案中,靶向多肽是抗体或抗体片段。
[0043]
在一些实施方案中,tc多肽是单聚乙二醇化的。本发明还提供了一种包含与一个或多个非天然编码氨基酸附接的接头、聚合物或生物活性分子的tc,其中所述非天然编码氨基酸在预选位点经核糖体掺入到多肽中。
[0044]
在一些实施方案中,本发明提供了包含具有掺入的一个或多个非天然编码氨基酸的一种或多种靶向多肽的组合物,其中所述多肽中的至少一种通过共价键合到所述多肽的非天然氨基酸的接头与tlr激动剂分子连接。
[0045]
在另一个实施方案中,本发明提供了一种组合物,其中所述一种或多种靶向多肽是相同或不同的靶向多肽。在另一个实施方案中,本发明提供了一种组合物,其中所述一种或多种靶向多肽与细胞表面靶标、肿瘤细胞靶标或癌细胞靶标结合。在另一个实施方案中,所述一种或多种靶向多肽是单特异性、双特异性或多特异性靶向多肽。
[0046]
在其他实施方案中,所述单特异性、双特异性或多特异性靶向多肽包含药物缀合物或检查点抑制剂。考虑将任何适合的免疫检查点抑制剂与本发明的组合物或tc一起使用。在一些实施方案中,免疫检查点抑制剂降低一种或多种免疫检查点蛋白的表达或活性。在另一个实施方案中,免疫检查点抑制剂降低一种或多种免疫检查点蛋白和其配体之间的相互作用。还可以将降低免疫检查点分子的表达和/或活性的抑制性核酸用于本发明中。在一些实施方案中,免疫检查点抑制剂是ctla4、tigit、糖皮质激素诱导的tnfr相关蛋白(gitr)、诱导型t细胞共刺激物(icos)、cd96、脊髓灰质炎病毒受体相关2(pvrl2)、pd
‑
1、pd
‑
ll、pd
‑
l2、lag
‑
3、b7
‑
h4、杀伤细胞免疫球蛋白受体(kir)、ox40、ox40
‑
l、吲哚胺
‑
2,3
‑
双加氧酶
‑
1(ido
‑
1)、吲哚胺
‑
2,3
‑
双加氧酶
‑
2(ido
‑
2)、ceacam1、cd272、tevi3、腺苷a2a受体和vista蛋白。在一些实施方案中,免疫检查点抑制剂是ctla4、pd
‑
1或pd
‑
l1的抑制剂。
[0047]
在另一个实施方案,靶向多肽包含抗体或抗体片段。在其他实施方案中,靶向多肽是与细胞抗原结合的抗体或抗体片段。在另一个实施方案中,靶向多肽是与靶标结合的抗体或抗体片段,所述靶标选自由以下组成的组:her2、her3、pd
‑
1、pdl
‑
1、egfr、trop2、psma、vegfr、ctla
‑
4、epcam、muc1、muc16、c
‑
met、gpc3、enpp3、tim
‑
1、folr1、steap1、间皮素、5t4、cea、ca9、钙粘蛋白6、ror1、slc34a2、slc39a6、slc44a4、ly6e、dll3、epha2、gpnmb、slitrk6、cd3、cd19、cd22、cd24、cd25、cd30、cd33、cd38、cd44、cd47、cd52、cd56、cd70、cd96、cd97、cd99、cd117、cd123、cd179、cd223和cd276。在一些实施方案中,靶向多肽包含与her2结合的抗体或抗体片段。在另一个实施方案中,靶向多肽是曲妥珠单抗。
[0048]
在另一个实施方案中,抗体或抗体片段包括igg、fab、(fab’)2、fv或单链fv
(scfv)。在一些实施方案中,抗体或抗体片段包含一个或多个fab、(fab’)2、fv或单链fv(scfv)突变。在一些实施方案中,抗体或抗体片包含一个或多个fc突变。在其他实施方案中,抗体或抗体片段包含一个至六个fc突变。在一些实施方案中,抗体或抗体片段包含两个或更多个fc突变。在其他实施方案中,抗体或抗体片段包含三个或更多个fc突变。在一些实施方案中,抗体或抗体片段包含四个或更多个fc突变。在其他实施方案中,抗体或抗体片段包含五个或更多个fc突变。在其他实施方案中,抗体或抗体片段包含六个fc突变。
[0049]
在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸。在另一个实施方案中,抗体或抗体片段包含掺入重链和轻链中的一个或多个非天然编码氨基酸。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸,并且进一步包含一个或多个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链和轻链每一者中的非天然编码氨基酸,所述抗体或抗体片段进一步包含一个或多个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸,并且进一步包含至少两个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链和轻链每一者中的非天然编码氨基酸,所述抗体或抗体片段进一步包含至少两个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸,并且进一步包含至少三个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链和轻链每一者中的非天然编码氨基酸,所述抗体或抗体片段进一步包含至少三个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸,并且进一步包含至少四个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链和轻链每一者中的非天然编码氨基酸,所述抗体或抗体片段进一步包含至少四个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸,并且进一步包含至少五个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链和轻链每一者中的非天然编码氨基酸,所述抗体或抗体片段进一步包含至少五个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸,并且进一步包含至少六个fc突变。在另一个实施方案中,抗体或抗体片段包含一个或多个掺入重链和轻链每一者中的非天然编码氨基酸,所述抗体或抗体片段进一步包含至少六个fc突变。
[0050]
在另一个实施方案中,靶向多肽包含一个或多个选自以下氨基酸的组的非天然编码氨基酸:对乙酰基苯丙氨酸、对硝基苯丙氨酸、对磺基酪氨酸、对羧基苯丙氨酸、邻硝基苯丙氨酸、间硝基苯丙氨酸、对硼羰基苯丙氨酸、邻硼羰基苯丙氨酸、间硼羰基苯丙氨酸、对氨基苯丙氨酸、邻氨基苯丙氨酸、间氨基苯丙氨酸、邻酰基苯丙氨酸、间酰基苯丙氨酸、对ome苯丙氨酸、邻ome苯丙氨酸、间ome苯丙氨酸、对磺基苯丙氨酸、邻磺基苯丙氨酸、间磺基苯丙氨酸、5
‑
硝基his、3
‑
硝基tyr、2
‑
硝基tyr、硝基取代的leu、硝基取代的his、硝基取代的de、硝基取代的trp、2
‑
硝基trp、4
‑
硝基trp、5
‑
硝基trp、6
‑
硝基trp、7
‑
硝基trp、3
‑
氨基酪氨酸、2
‑
氨基酪氨酸、o
‑
磺基酪氨酸、2
‑
磺氧基苯丙氨酸、3
‑
磺氧基苯丙氨酸、邻羧基苯丙氨酸、间羧基苯丙氨酸、对乙酰基
‑
l
‑
苯丙氨酸、对炔丙基
‑
苯丙氨酸、o
‑
甲基
‑
l
‑
酪氨酸、l
‑3‑
(2
‑
萘基)丙氨酸、3
‑
甲基
‑
苯丙氨酸、o
‑4‑
烯丙基
‑
l
‑
酪氨酸、4
‑
丙基
‑
l
‑
酪氨酸、三
‑
o
‑
乙酰基
‑
glcnacβ
‑
丝氨酸、左旋多巴(l
‑
dopa)、氟化苯丙氨酸、异丙基
‑
l
‑
苯丙氨酸、对叠氮基
‑
l
‑
苯丙氨酸、对酰基
‑
l
‑
苯丙氨酸、对苯甲酰基
‑
l
‑
苯丙氨酸、l
‑
磷酸丝氨酸、膦酰基丝氨酸、膦酰基酪氨酸、对碘
‑
苯丙氨酸、对溴苯丙氨酸、对氨基
‑
l
‑
苯丙氨酸、对炔丙基氧基
‑
l
‑
苯丙氨酸、4
‑
叠氮基
‑
l
‑
苯丙氨酸、对叠氮基乙氧基苯丙氨酸和对叠氮基甲基
‑
苯丙氨酸。在另一个实施方案中,非天然氨基酸选自由以下组成的组:对乙酰基
‑
苯丙氨酸、4
‑
叠氮基
‑
l
‑
苯丙氨酸、对叠氮基乙氧基苯丙氨酸或对叠氮基甲基
‑
苯丙氨酸。在其他实施方案中,非天然编码氨基酸被位点特异性地掺入一种或多种靶向多肽中。
[0051]
在另一个实施方案中,tlr激动剂是tlr7激动剂、tlr8激动剂或tlr7/tlr8双重激动剂。在其他实施方案中,tlr激动剂是包含根据图1的结构1、2、3、4或5中任一者的分子结构的tlr激动剂。在另一个实施方案中,tlr激动剂是选自根据本发明表3、4、5、6、7的结构的组的tlr激动剂中的任一者。
[0052]
在其他实施方案中,将靶向多肽与一个或多个接头、聚合物或生物活性分子缀合。在一些实施方案中,将靶向多肽直接或间接地与一个或多个接头、聚合物或生物活性分子缀合。在一些实施方案中,一个或多个接头是可裂解或不可裂解的接头。
[0053]
在一些实施方案中,一个或多个接头为0.1kda至50kda。在其他实施方案中,一个或多个接头为0.1kda至10kda。在其他实施方案中,一个或多个接头或聚合物是线性的、支链的、多聚体的或树枝状聚合物的(dendrimeric)。在另一个实施方案中,一个或多个接头或聚合物是双官能或多官能接头或双官能或多官能聚合物。
[0054]
在其他实施方案中,一个或多个聚合物是水溶性聚合物。在其他实施方案中,水溶性聚合物是聚乙二醇(peg)。在一些实施方案中,peg具有介于0.1kda和100kda之间的分子量。在其他实施方案中,peg具有介于0.1kda和50kda之间的分子量。在其他实施方案中,peg具有介于0.1kda和40kda之间的分子量。在其他实施方案中,peg具有介于0.1kda和30kda之间的分子量。在其他实施方案中,peg具有介于0.1kda和20kda之间的分子量。在其他实施方案中,peg具有介于0.1kda和10kda之间的分子量。在一些实施方案中,聚(乙二醇)分子具有介于约0.1kda和约100kda之间的分子量。在一些实施方案中,聚(乙二醇)分子具有介于0.1kda和50kda之间的分子量。在一些实施方案中,聚(乙二醇)具有介于1kda和25kda之间、或介于2kda和22kda之间、或介于5kda和20kda之间的分子量。例如,聚(乙二醇)聚合物的分子量可以是约5kda、或约10kda、或约20kda或约30kda。例如,聚(乙二醇)聚合物的分子量可以是5kda或10kda或20kda或30kda。在一些实施方案中,聚(乙二醇)分子是支链peg。在一些实施方案中,聚(乙二醇)分子是支链的5kpeg。在一些实施方案中,聚(乙二醇)分子是支链的10k peg。在一些实施方案中,聚(乙二醇)分子是支链的20k peg。在一些实施方案中,聚(乙二醇)分子是线性peg。在一些实施方案中,聚(乙二醇)分子是线性的5k peg。在一些实施方案中,聚(乙二醇)分子是线性的10k peg。在一些实施方案中,聚(乙二醇)分子是线性的20k peg。在一些实施方案中,聚(乙二醇)分子是线性的30k peg。在一些实施方案中,聚(乙二醇)聚合物的分子量是平均分子量。在某些实施方案中,平均分子量是数均分子量(mn)。可以使用gpc或sec、sds/page分析、rp
‑
hplc、质谱或毛细管电泳来测定或测量平均分子量。
[0055]
在另一个实施方案中,至少一个接头、聚合物或生物活性分子与至少一个非天然编码氨基酸连接。在一些实施方案中,接头是peg。在其他实施方案中,接头是具有介于
0.1kda和50kda之间的分子量的peg。在其他实施方案中,接头是具有介于0.1kda和40kda之间的分子量的peg。在其他实施方案中,接头是具有介于0.1kda和30kda之间的分子量的peg。在其他实施方案中,接头是具有介于0.1kda和20kda之间的分子量的peg。在其他实施方案中,接头是具有介于0.1kda和10kda之间的分子量的peg。在其他实施方案中,接头是具有介于0.1kda和5kda之间的分子量的peg。
[0056]
在另一个实施方案中,靶向多肽包含一个或多个增加组合物的稳定性或溶解度的氨基酸取代、添加或缺失。在另一个实施方案中,靶向多肽包含一个或多个增强/降低adcp或adcc活性的氨基酸取代、添加或缺失。在另一个实施方案中,靶向多肽包含一个或多个增强组合物的药代动力学的氨基酸取代、添加或缺失。在其他实施方案中,组合物包含一个或多个增加靶向多肽在重组宿主细胞中的表达或体外合成的氨基酸取代、添加或缺失。
[0057]
在另一个实施方案中,非天然编码氨基酸对接头、聚合物或生物活性分子具有反应性,而所述接头、聚合物或生物活性分子对多肽中20种常见氨基酸中的任何一种都不具有反应性。在另一个实施方案中,非天然编码氨基酸包含羰基、氨基氧基、肼基、酰肼基、氨基脲基、叠氮基或炔基。在其他实施方案中,非天然编码氨基酸包含羰基。
[0058]
在另一个实施方案中,靶向多肽与细胞毒性剂或免疫刺激剂连接。在另一个实施方案中,本发明的tc或btc与细胞毒性剂或免疫刺激剂连接。在另一个实施方案中,靶向多肽包含细胞毒剂或免疫刺激剂。在另一个实施方案中,本发明的tc或btc包含细胞毒性剂或免疫刺激剂。
[0059]
在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与tlr激动剂缀合的抗her2抗体或抗体片段,所述tlr激动剂包含根据图1的任何结构的结构,其中tlr激动剂通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合。在另一个实施方案中,tlr激动剂是tlr7激动剂、tlr8激动剂或tlr7/tlr8双重激动剂。在另一个实施方案中,tlr激动剂包含根据图1的结构1的结构。在另一个实施方案中,包含根据结构1的结构的tlr激动剂选自下组:axc
‑
621、axc
‑
622、axc
‑
625、axc
‑
626、axc
‑
627、axc
‑
638、axc
‑
639、axc
‑
640、axc
‑
642、axc
‑
662、axc
‑
665、axc
‑
666、axc
‑
667、axc
‑
668、axc
‑
669、axc
‑
670、axc
‑
671、axc
‑
672、axc
‑
675、axc
‑
678、axc
‑
679、axc
‑
681、axc
‑
687、axc
‑
688、axc
‑
689、axc
‑
690、axc
‑
691、axc
‑
696、axc
‑
697、axc
‑
698、axc
‑
699、axc
‑
700、axc
‑
701、axc
‑
702、axc
‑
709、axc
‑
710、axc
‑
711、axc
‑
712、axc
‑
713、axc
‑
714、axc
‑
715、axc
‑
716、axc
‑
717、axc
‑
718、axc
‑
719、axc
‑
722、axc
‑
723、axc
‑
724、axc
‑
725、axc
‑
726、axc
‑
727、axc
‑
729、axc
‑
731、axc
‑
732、axc
‑
733、axc
‑
734、axc
‑
735、axc
‑
736、axc
‑
737、axc
‑
738、axc
‑
739、axc
‑
740、axc
‑
741、axc
‑
743、axc
‑
742、axc
‑
747、axc
‑
748、axc
‑
749、axc
‑
750、axc
‑
751、axc
‑
752、axc
‑
754、axc
‑
755、axc
‑
756、axc
‑
757、axc
‑
758、axc
‑
759、axc
‑
760、axc
‑
761、axc
‑
762、axc
‑
764、axc
‑
771、axc
‑
772、axc
‑
773、axc
‑
777、axc
‑
778、axc
‑
779、axc
‑
789、axc
‑
793、axc
‑
799、axc
‑
800、axc
‑
801、axc
‑
802、axc
‑
803、axc
‑
804、axc
‑
805、axc
‑
806、axc
‑
807、axc
‑
808、axc
‑
809、axc
‑
810、axc
‑
831和axc
‑
910化合物。在另一个实施方案中,本发明提供了下列任一tlr激动剂:进一步包含接头的axc
‑
621、axc
‑
622、axc
‑
625、axc
‑
626、axc
‑
627、axc
‑
638、axc
‑
639、axc
‑
640、axc
‑
642、axc
‑
662、axc
‑
665、axc
‑
666、axc
‑
667、axc
‑
668、axc
‑
669、axc
‑
670、axc
‑
671、axc
‑
672、axc
‑
675、axc
‑
678、axc
‑
679、axc
‑
681、axc
‑
687、axc
‑
688、axc
‑
689、axc
‑
690、axc
‑
691、axc
‑
696、axc
‑
697、axc
‑
698、axc
‑
699、axc
‑
700、axc
‑
701、axc
‑
702、axc
‑
709、axc
‑
710、axc
‑
711、axc
‑
712、axc
‑
713、axc
‑
714、axc
‑
715、axc
‑
716、axc
‑
717、axc
‑
718、axc
‑
719、axc
‑
722、axc
‑
723、axc
‑
724、axc
‑
725、axc
‑
726、axc
‑
727、axc
‑
729、axc
‑
731、axc
‑
732、axc
‑
733、axc
‑
734、axc
‑
735、axc
‑
736、axc
‑
737、axc
‑
738、axc
‑
739、axc
‑
740、axc
‑
741、axc
‑
743、axc
‑
742、axc
‑
747、axc
‑
748、axc
‑
749、axc
‑
750、axc
‑
751、axc
‑
752、axc
‑
754、axc
‑
755、axc
‑
756、axc
‑
757、axc
‑
758、axc
‑
759、axc
‑
760、axc
‑
761、axc
‑
762、axc
‑
764、axc
‑
771、axc
‑
772、axc
‑
773、axc
‑
777、axc
‑
778、axc
‑
779、axc
‑
789、axc
‑
793、axc
‑
799、axc
‑
800、axc
‑
801、axc
‑
802、axc
‑
803、axc
‑
804、axc
‑
805、axc
‑
806、axc
‑
807、axc
‑
808、axc
‑
809、axc
‑
810、axc
‑
831或axc
‑
910化合物。在另一个实施方案中,tlr激动剂包含进一步包含接头的根据结构1的结构。
[0060]
在其他实施方案中,包含根据结构1的结构的tlr激动剂选自下组:axc
‑
625、axc
‑
626、axc
‑
638、axc
‑
639、axc
‑
640、axc
‑
642、axc
‑
662、axc
‑
667、axc
‑
668、axc
‑
669、axc
‑
670、axc
‑
671、axc
‑
672、axc
‑
675、axc
‑
681、axc
‑
687、axc
‑
688、axc
‑
689、axc
‑
690、axc
‑
691、axc
‑
697、axc
‑
699、axc
‑
700、axc
‑
701、axc
‑
702、axc
‑
709、axc
‑
710、axc
‑
711、axc
‑
713、axc
‑
714、axc
‑
717、axc
‑
719、axc
‑
722、axc
‑
723、axc
‑
724、axc
‑
725、axc
‑
726、axc
‑
727、axc
‑
731、axc
‑
732、axc
‑
733、axc
‑
734、axc
‑
735、axc
‑
736、axc
‑
737、axc
‑
738、axc
‑
739、axc
‑
740、axc
‑
741、axc
‑
743、axc
‑
742、axc
‑
747、axc
‑
748、axc
‑
750、axc
‑
751、axc
‑
752、axc
‑
754、axc
‑
755、axc
‑
756、axc
‑
757、axc
‑
758、axc
‑
759、axc
‑
760、axc
‑
761、axc
‑
762、axc
‑
764、axc
‑
771、axc
‑
772、axc
‑
773、axc
‑
777、axc
‑
778、axc
‑
779、axc
‑
789、axc
‑
793、axc
‑
800、axc
‑
801、axc
‑
802、axc
‑
803、axc
‑
804、axc
‑
805、axc
‑
806、axc
‑
807、axc
‑
808、axc
‑
809、axc
‑
810、axc
‑
831和axc
‑
910化合物。在其他实施方案中,包含根据结构1的结构的tlr激动剂选自下组:axc
‑
801、axc
‑
802、axc
‑
831和axc
‑
910化合物。在其他实施方案中,包含根据结构1的结构的tlr激动剂选自下组:axc
‑
801、axc
‑
802、axc
‑
831和axc
‑
910化合物,所述化合物进一步包含接头。
[0061]
在另一个实施方案中,抗her2抗体或抗体片段包含一个或多个掺入重链、轻链或重链和轻链两者中的非天然编码氨基酸。在另一个实施方案中,所述一个或多个非天然编码氨基酸选自以下氨基酸的组:对乙酰基苯丙氨酸、对硝基苯丙氨酸、对磺基酪氨酸、对羧基苯丙氨酸、邻硝基苯丙氨酸、间硝基苯丙氨酸、对硼羰基苯丙氨酸、邻硼羰基苯丙氨酸、间硼羰基苯丙氨酸、对氨基苯丙氨酸、邻氨基苯丙氨酸、间氨基苯丙氨酸、邻酰基苯丙氨酸、间酰基苯丙氨酸、对ome苯丙氨酸、邻ome苯丙氨酸、间ome苯丙氨酸、对磺基苯丙氨酸、邻磺基苯丙氨酸、间磺基苯丙氨酸、5
‑
硝基his、3
‑
硝基tyr、2
‑
硝基tyr、硝基取代的leu、硝基取代的his、硝基取代的de、硝基取代的trp、2
‑
硝基trp、4
‑
硝基trp、5
‑
硝基trp、6
‑
硝基trp、7
‑
硝基trp、3
‑
氨基酪氨酸、2
‑
氨基酪氨酸、o
‑
磺基酪氨酸、2
‑
磺氧基苯丙氨酸、3
‑
磺氧基苯丙氨酸、邻羧基苯丙氨酸、间羧基苯丙氨酸、对乙酰基
‑
l
‑
苯丙氨酸、对炔丙基
‑
苯丙氨酸、o
‑
甲基
‑
l
‑
酪氨酸、l
‑3‑
(2
‑
萘基)丙氨酸、3
‑
甲基
‑
苯丙氨酸、o
‑4‑
烯丙基
‑
l
‑
酪氨酸、4
‑
丙基
‑
l
‑
酪氨酸、三
‑
o
‑
乙酰基
‑
glcnacβ
‑
丝氨酸、左旋多巴、氟化苯丙氨酸、异丙基
‑
l
‑
苯丙氨酸、对叠氮基
‑
l
‑
苯丙氨酸、对酰基
‑
l
‑
苯丙氨酸、对苯甲酰基
‑
l
‑
苯丙氨酸、l
‑
磷酸丝氨酸、膦酰基丝氨酸、膦酰基酪氨酸、对碘
‑
苯丙氨酸、对溴苯丙氨酸、对氨基
‑
l
‑
苯丙氨酸、对炔丙基氧基
‑
l
‑
苯丙氨酸、4
‑
叠氮基
‑
l
‑
苯丙氨酸、对叠氮基乙氧基苯丙氨酸和对叠氮基甲基
‑
苯丙氨酸。在其他实施方案中,非天然氨基酸是对乙酰基
‑
苯丙氨酸、4
‑
叠氮基
‑
l
‑
苯丙氨酸、对叠氮基甲基
‑
苯丙氨酸或对叠氮基乙氧基苯丙氨酸。
[0062]
在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的一个或多个突变。在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的两个或更多个突变。在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的三个或更多个突变。在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的四个或更多个突变。在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的五个或更多个突变。在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的六个或更多个突变。在另一个实施方案中,抗her2抗体或抗体片段进一步包含在fc区中的六个突变。
[0063]
在另一个实施方案中,一个或多个接头是可裂解或不可裂解的接头。在其他实施方案中,一个或多个接头是双官能或多官能接头。
[0064]
在另一个实施方案中,tlr激动剂包含根据图1的结构2的结构。在另一个实施方案中,tlr激动剂包含选自axc
‑
745、axc
‑
746和axc
‑
753化合物的组的根据结构2的结构。在另一个实施方案中,tlr激动剂包含根据以下任一者的结构:axc
‑
745、axc
‑
746和axc
‑
753化合物,所述化合物进一步包含接头。在另一个实施方案中,tlr激动剂包含进一步包含接头的根据结构2的结构。
[0065]
在另一个实施方案中,tlr激动剂包含根据图1的结构3的结构。在另一个实施方案中,包含根据结构3的结构的tlr激动剂是axc
‑
837或axc
‑
847化合物。在另一个实施方案中,tlr激动剂包含根据axc
‑
837或axc
‑
847化合物的结构,所述化合物进一步包含接头。在另一个实施方案中,tlr激动剂包含根据axc
‑
847化合物的结构,所述化合物进一步包含接头。在另一个实施方案中,tlr激动剂包含进一步包含接头的根据结构3的结构。
[0066]
在另一个实施方案中,tlr激动剂包含根据图1的结构4的结构。在另一个实施方案中,包含根据结构4的结构的tlr激动剂选自下组:axc
‑
844、axc
‑
842、axc
‑
843、axc
‑
845、axc
‑
846、axc
‑
836或axc
‑
841化合物。在另一个实施方案中,tlr激动剂包含根据以下任一者的结构4的结构:axc
‑
844、axc
‑
842、axc
‑
843、axc
‑
845、axc
‑
846、axc
‑
836或axc
‑
841化合物,所述化合物进一步包含接头。在另一个实施方案中,tlr激动剂包含进一步包含接头的根据结构4的结构。
[0067]
在另一个实施方案中,tlr激动剂包含根据图1的结构5的结构。在另一个实施方案中,包含根据结构5的结构的tlr激动剂选自下组:axc
‑
862、axc
‑
863、axc
‑
867、axc
‑
868、axc
‑
869、axc
‑
872、axc
‑
873、axc
‑
876、axc
‑
877、axc
‑
878、axc
‑
879、axc
‑
880、axc
‑
881、axc
‑
882、axc
‑
883、axc
‑
884、axc
‑
885、axc
‑
886、axc
‑
887、axc
‑
888、axc
‑
889、axc
‑
890、axc
‑
891、axc
‑
892、axc
‑
893、axc
‑
895、axc
‑
896、axc
‑
897、axc
‑
898、axc
‑
901、axc
‑
903、axc
‑
904、axc
‑
905、axc
‑
906、axc
‑
907、axc
‑
908、axc
‑
909、axc
‑
911、axc
‑
912、axc
‑
913、axc
‑
914、axc
‑
915或axc
‑
916化合物。在其他实施方案中,包含根据结构5的结构的tlr激动剂选自下组:axc
‑
862、axc
‑
863、axc
‑
867、axc
‑
868、axc
‑
869、axc
‑
873、axc
‑
876、axc
‑
879、axc
‑
880、axc
‑
882、axc
‑
889、axc
‑
893、axc
‑
896、axc
‑
897、axc
‑
901、axc
‑
907、axc
‑
909、axc
‑
913和axc
‑
914化合物。在另一个实施方案中,tlr激动剂包含根据以下任一者的结构:进一步包含接头的axc
‑
862、axc
‑
863、axc
‑
867、axc
‑
868、axc
‑
869、axc
‑
872、axc
‑
873、axc
‑
876、axc
‑
877、axc
‑
878、axc
‑
879、axc
‑
880、axc
‑
881、axc
‑
882、axc
‑
883、axc
‑
884、axc
‑
885、axc
‑
886、axc
‑
887、axc
‑
888、axc
‑
889、axc
‑
890、axc
‑
891、axc
‑
892、axc
‑
893、axc
‑
895、axc
‑
896、axc
‑
897、axc
‑
898、axc
‑
901、axc
‑
903、axc
‑
904、axc
‑
905、axc
‑
906、axc
‑
907、axc
‑
908、axc
‑
909、axc
‑
911、axc
‑
912、axc
‑
913、axc
‑
914、axc
‑
915或axc
‑
916化合物。在另一个实施方案中,tlr激动剂包含进一步包含接头的根据结构5的结构。
[0068]
在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:1
‑
13中至少一者的氨基酸序列。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:1
‑
13中至少两者的氨基酸序列。在另一个实施方案中,抗her2抗体或抗体片段包含a)seq id no:1或2;和b)seq id no:3、4、5、6、7、8、9、10、11、12和13中的任一者。在另一个实施方案中,抗her2抗体或抗体片段包含a)seq id no:1或2的重链;和b)seq id no:3、4、5、6、7、8、9、10、11、12和13中的任一者的轻链。在另一个实施方案中,抗her2抗体或抗体片段包含a)seq id no:1;和b)seq id no:3、4、5、6、7、8、9、10、11、12和13中的任一者。在另一个实施方案中,抗her2抗体或抗体片段包含a)seq id no:2;和b)seq id no:3、4、5、6、7、8、9、10、11、12和13中的任一者。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:3。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:4。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:5。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:6。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:7。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:8。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:9。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:10。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:11。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:12。在另一个实施方案中,抗her2抗体或抗体片段包含seq id no:2和seq id no:13。在另一个实施方案中,本发明提供了抗her2抗体或抗体片段,其中在根据kabat编号的位置114处以位点特异性方式掺入非天然编码氨基酸。
[0069]
在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与tlr激动剂缀合的抗her2抗体或抗体片段,所述tlr激动剂包含根据图1的任何结构的结构,其中所述tlr激动剂通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合,所述tc进一步包含化学治疗剂或免疫治疗剂。在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与选自表3
‑
7的化合物中的任一者的tlr激动剂缀合的抗her2抗体或抗体片段,其中tlr激动剂通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合。在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与选自表3
‑
7的化合物中的任一者的tlr激动剂缀合的抗her2抗体或抗体片段,其中所述tlr激动剂通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合,所述tc进一步包含化学治疗剂或免疫治疗剂。
[0070]
在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与tlr激动剂缀合的抗her2抗体或抗体片段,所述tlr激动剂包含根据图1的任何结构的结构,其中所述tlr激动剂通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合,所述tc进一步包含药物缀合物。在其他实施方案中,药物缀合物是抗体药物缀合物。在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与选自表3
‑
7的化合物中的任一者的tlr激动剂缀合的抗her2抗体或抗体片段,其中tlr激动剂
通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合。在另一个实施方案中,本发明提供了tlr激动剂缀合物(tc),其包含与选自表3
‑
7的化合物中的任一者的tlr激动剂缀合的抗her2抗体或抗体片段,其中tlr激动剂通过与一个或多个掺入抗体或抗体片段中的非天然编码氨基酸共价键合的接头与抗体或抗体片段缀合,所述tc进一步包含药物缀合物。在其他实施方案中,药物缀合物是抗体药物缀合物。在其他实施方案中,tc进一步包含细胞因子或细胞毒素。
[0071]
在另一个实施方案中,本发明提供了治疗患有癌症或疾病或病症或适应症或疾患的受试者或患者的方法,所述方法包括向受试者或患者施用治疗有效量的本发明的组合物或tc。在某些实施方案中,肿瘤或癌症是her2阳性肿瘤或癌症。在某些实施方案中,肿瘤、癌症、适应症、疾病、疾患或病症是her2阳性肿瘤、癌症、适应症、疾病、疾患或病症。在某些实施方案中,肿瘤或癌症选自由以下组成的组:结肠癌、卵巢癌、乳腺癌、黑素瘤、肺癌、胶质母细胞瘤、前列腺癌、膀胱癌、宫颈癌、胰腺癌、肾癌、食道癌、阴道癌、胃癌和白血病。
[0072]
在另一个实施方案中,本发明提供了治疗患有癌症或疾病或病症的受试者或患者的方法,所述方法包括向受试者或患者施用治疗有效量的进一步包含化学治疗剂或免疫治疗剂的本发明组合物或tc。在某些实施方案中,tc与至少一种化学治疗剂共同施用。化学治疗剂可选自由以下组成的组:替莫唑胺、吉西他滨、多柔比星、环磷酰胺、紫杉醇、顺铂、氟嘧啶、紫杉烷、蒽环类、拉帕替尼、卡培他滨、来曲唑、帕妥珠单抗、多西他赛、ifn
‑
α。在本发明的另一个实施方案中,tc与至少一种化学治疗剂共同施用。
[0073]
在另一个实施方案中,本发明提供了治疗患有癌症或疾病或病症的受试者或患者的方法,所述方法包括向受试者或患者施用治疗有效量的进一步包含抗体药物缀合物、细胞毒性剂或检查点抑制剂的本发明组合物或tc。
[0074]
在另一个实施方案中,本发明提供了杀伤细胞的方法,其包括使细胞与本发明的tc接触。在其他实施方案中,所述细胞是肿瘤细胞或癌细胞。在某些实施方案中,肿瘤细胞或癌细胞是结肠癌、卵巢癌、乳腺癌、黑素瘤、肺癌、胶质母细胞瘤、前列腺癌、膀胱癌、宫颈癌、胰腺癌、肾癌、食管癌、阴道癌、胃癌或白血病的癌细胞。在某些实施方案中,肿瘤或癌症是her2阳性肿瘤或癌症。在某些实施方案中,待治疗的肿瘤、癌症、适应症、疾病、疾患或病症是her2阳性肿瘤、癌症、适应症、疾病、疾患或病症。
[0075]
本发明提供了抑制或降低肿瘤或癌症生长的方法,其包括使肿瘤与有效量的本发明tc接触,以刺激患者肿瘤附近的免疫系统。本发明提供了抑制或降低肿瘤或癌症生长的方法,其包括使肿瘤与有效量的本发明的聚乙二醇化tc或tc的稳定二聚体或多聚体接触。在一个实施方案中,tc是非聚乙二醇化或单聚乙二醇化的。在一个实施方案中,tc是二聚乙二醇化的。在一个实施方案中,tc具有与之附接的多于一个和/或不同的tlr激动剂分子。本发明的另一个实施方案提供了使用本发明的tc来调节对肿瘤细胞的免疫应答的方法。
[0076]
在一些实施方案中,本发明提供了使用tc来治疗癌症的方法。在一些实施方案中,本发明的tc可用于治疗或预防癌症相关疾病、疾患和病症,包括与癌症直接或间接相关的病症,例如血管生成和癌前状态,比如发育异常。在一些实施方案中,肿瘤是液体肿瘤或实体肿瘤。在一些实施方案中,待治疗的病症是癌症。癌症可以是但不限于乳腺癌、脑癌、胰腺癌、皮肤癌、肺癌、肝癌、胆囊癌、结肠癌、卵巢癌、前列腺癌、子宫癌、骨癌和血癌(白血病)或与任何这些癌症有关的癌症或疾病或病症。癌是始于上皮细胞的癌症,上皮细胞为覆盖身
体表面、产生激素和组成腺体的细胞。作为非限制性实例,癌包括乳腺癌、胰腺癌、肺癌、结肠癌、结肠直肠癌、直肠癌、肾癌、膀胱癌、胃癌、前列腺癌、肝癌、卵巢癌、脑癌、阴道癌、外阴癌、子宫癌、口腔癌、阴茎癌、睾丸癌、食管癌、皮肤癌、输卵管癌、头颈癌、胃肠间质癌、腺癌、皮肤或眼内黑素瘤、肛区癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、尿道癌、肾盂癌、输尿管癌、子宫内膜癌、宫颈癌、垂体癌、中枢神经系统(cns)赘生物、原发性cns淋巴瘤、脑干胶质瘤和脊柱轴肿瘤(spinal axis tumor)。在一些情况下,癌症是皮肤癌,比如基底细胞癌、鳞状癌、黑素瘤、非黑素瘤或光化性(日光性)角化病。在一些实施方案中,本发明还涉及治疗哺乳动物的急性白血病的方法,其包括向所述哺乳动物施用治疗有效量的本发明的tc。本发明还提供了用于抑制急性白血病母细胞增殖的方法,其包括向患有急性白血病的哺乳动物施用治疗有效剂量的本发明的tc。
[0077]
在另一个实施方案中,本文公开的tc可用于调节免疫应答。免疫应答的调节可包括刺激、激活、增进、增强或上调免疫应答。免疫应答的调节可包括压制、抑制、阻止、降低或下调免疫应答。
[0078]
在另一个实施方案中,本发明提供了药物组合物,其包含治疗有效量本发明的组合物或tc和药学上可接受的载体或赋形剂。
[0079]
在另一个实施方案中,本发明提供了本发明的组合物在制造药剂方面的用途。
[0080]
在另一个实施方案中,本发明提供了包含根据图1的任何一种结构的tlr激动剂的免疫刺激抗体缀合物(isac)。在另一个实施方案中,本发明提供了包含根据表3、4、5、6、7中的任何一种化合物的tlr激动剂的免疫刺激抗体缀合物(isac)。在另一个实施方案中,本发明提供了isac,其中tlr激动剂包含选自下组的化合物:axc
‑
862、axc
‑
863、axc
‑
867、axc
‑
868、axc
‑
869、axc
‑
874、axc
‑
875、axc
‑
876、axc
‑
879、axc
‑
880、axc
‑
882、axc
‑
893、axc
‑
896、axc
‑
897、axc
‑
901、axc
‑
907和axc
‑
910化合物。
[0081]
在另一个实施方案中,本发明提供了具有根据图1的结构的任何一种化合物的盐。在另一个实施方案中,本发明提供了表3、4、5、6、7的任何一种化合物的盐。在另一个实施方案中,本发明提供了根据本发明公开的组合物、化合物和tc的药物组合物或其盐。在其他实施方案中,药物组合物或盐进一步包含药学上可接受的赋形剂。
附图说明
[0082]
图1描绘了适用于发明的tlr激动剂的一般结构。
[0083]
图2描绘了各种tc缀合物的结构。
[0084]
图3描绘了另外的tc缀合物的结构。
[0085]
图4描绘了选定tc缀合物在细胞增殖测定中的生物活性。
[0086]
图5a和5b描绘了各种tlr7激动剂的tlr7活性。
[0087]
图6描绘了与接头附接的各种tlr7激动剂的tlr7活性。
[0088]
图7描绘了另外的tlr7激动剂和与接头附接的tlr7激动剂的tlr7活性。
[0089]
图8描绘了另外的tlr7激动剂和与接头附接的tlr7激动剂的tlr7活性。
[0090]
图9描绘了与非天然氨基酸paf(dl
‑
paf)相比,与接头(药物接头或dl)附接的不同tlr7激动剂的tlr7活性。
[0091]
图10a
‑
10c描绘了具有在氨基酸位置ha114的非天然氨基酸的未缀合抗her2抗体
的hplc色谱图(图10a),以及在氨基酸位置ha114与tlr激动剂axc
‑
875(图10b)和axc
‑
880(图10c)缀合的抗her2抗体的hplc色谱图。
[0092]
图11a
‑
11c比较了在skov3 her2高表达的肿瘤细胞系(图11a);jimt
‑
1her2中等/低表达的肿瘤细胞系(图11b);和a431 her2低表达的肿瘤细胞系(图11c)中与抗her2抗体缀合的各种有效负载接头的肿瘤依赖性isac活性。
[0093]
图12a和12b比较了在skbr3 her2高表达的肿瘤细胞系(图12a)和hcc1806 her2极低表达的肿瘤细胞系(图12b)中与抗her2抗体缀合的另外的有效负载接头的肿瘤依赖性isac活性。
[0094]
图13a和13b比较了在skbr3 her2高表达的肿瘤细胞系(图13a)和hcc1806 her2极低表达的肿瘤细胞系(图13b)中与抗her2抗体缀合的另外的有效负载接头的肿瘤依赖性isac活性。
[0095]
图14a和14b比较了在skbr3 her2高表达的肿瘤细胞系(图14a)和hcc1806 her2极低表达的肿瘤细胞系(图14b)中与抗her2抗体缀合的另外的有效负载接头的肿瘤依赖性isac活性。
[0096]
图15a和15b比较了在skbr3 her2高表达的肿瘤细胞系(图15a)和hcc1806 her2极低表达的肿瘤细胞系(图15b)中与抗her2抗体缀合的三(3)个有效负载接头的肿瘤依赖性isac活性,其显示her2
‑
axc
‑
879具有最佳的isac活性。
具体实施方式
[0097]
本文公开了包含诸如抗体的靶向部分和一种或多种tlr激动剂的tc。tlr激动剂可进一步包含一个或多个接头。本发明的tc可包含与靶向部分中的非天然氨基酸连接的tlr激动剂。还包括制备此类包含掺入到靶向部分多肽中的非天然氨基酸的tc的方法。
[0098]
在某些实施方案中,提供了包含任何所述化合物和药学上可接受的载体、赋形剂或粘合剂的药物组合物。
[0099]
在进一步或替代的实施方案中是检测患者中多肽的存在的方法,所述方法包括施用包含至少一个含杂环的非天然氨基酸的多肽,并且相对于同源的天然存在的氨基酸多肽,所得含杂环的非天然氨基酸多肽调节所述多肽的免疫原性。
[0100]
应当理解,本文描述的方法和组合物不限于本文描述的特定方法、方案、细胞系、构建体和试剂,并且正因为如此,它们可以变化。还应理解,本文使用的术语仅出于描述特定实施方案的目的,并且不意图限制本文所述的方法和组合物的范围,所述范围将仅受所附权利要求书限制。
[0101]
如本文中和所附权利要求中使用的,除非上下文另外明确地指示,单数形式“一个”、“一种”和“该”包括复数指代。
[0102]
除非另外定义,本文中使用的全部技术术语和科学术语具有与本文所述的本发明所属领域的普通技术人员通常所理解的相同的含义。虽然可以在本文所述的本发明的实践或测试中使用类似于或等同于本文所述那些的任何方法、装置和材料,但现在描述了优选的方法、装置和材料。
[0103]
出于描述和公开例如描述于公布中并且可与目前所描述的发明结合使用的构建体以及方法学的目的,本文所提及的所有公布和专利均以引用的方式整体并入本文。本文
所论述的出版物仅仅出于其在本技术的提交日期之前公开而提供。本文中任何内容不应被解释为承认本文所述的发明人无权凭借在先发明或由于任何其他原因而先于这样的公开。
[0104]
术语“基于羟醛的键联”或“基于混合羟醛的键联”是指一种羰基化合物与可以相同也可以不同的另一种羰基化合物的烯醇酯/烯醇的酸催化或碱催化缩合,从而生成β
‑
羟基羰基化合物——羟醛。
[0105]
如本文所用的术语“亲和标记物”是指可逆或不可逆地结合另一个分子的标记物,所述标记物对它进行修饰、破坏它或与它形成化合物。举例来说,亲和标记物包括酶及其底物或抗体及其抗原。
[0106]
术语“烷氧基”、“烷氨基”和“烷硫基”(或硫代烷氧基)以其传统意义加以使用,并且是指分别通过氧原子、氨基或硫原子与分子连接的那些烷基。
[0107]
除非另外说明,否则术语“烷基”本身或作为另一个分子的一部分,表示直链或支链或环状烃基或其组合,其可以是完全饱和的、单或多不饱和的,并且可以包括具有指定碳原子数的二价和多价基团(即c1‑
c
10
表示一至十个碳)。饱和烃基的实例包括但不限于诸如甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、仲丁基、环己基、(环己基)甲基、环丙基甲基,例如正戊基、正己基、正庚基、正辛基等的同源物和异构体等基团。不饱和烷基是具有一个或多个双键或三键的烷基。不饱和烷基的实例包括但不限于乙烯基、2
‑
丙烯基、巴豆基、2
‑
异戊烯基、2
‑
(丁二烯基)、2,4
‑
戊二烯基、3
‑
(1,4
‑
戊二烯基)、乙炔基、1
‑
和3
‑
丙炔基、3
‑
丁炔基,以及更高级的同源物和异构体。除非另有说明,术语“烷基”还意指包括本文更详细定义的那些烷基衍生物,比如“杂烷基”、“卤代烷基”和“同烷基”。
[0108]
术语“亚烷基”本身或作为另一分子的一部分表示来源于烷烃的二价基团,例如(
–
ch2–
)
n
,其中n可以是1至约24。仅举例来说,这样的基团包括但不限于具有10个或更少碳原子的基团,比如结构
–
ch2ch2–
和
–
ch2ch2ch2ch2–
。“低级烷基”或“低级亚烷基”是链较短的烷基或亚烷基,通常具有8个或更少的碳原子。除非另有说明,术语“亚烷基”还旨在包括本文所述为“杂亚烷基”的那些基团。
[0109]
术语“氨基酸”是指天然存在的和非天然的氨基酸,以及以与天然存在的氨基酸相似的方式起作用的氨基酸类似物和氨基酸模拟物。天然编码氨基酸是20种常见氨基酸(丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酰胺、谷氨酸、甘氨酸、组氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸和缬氨酸)以及吡咯赖氨酸和硒代半胱氨酸。氨基酸类似物是指与天然存在的氨基酸具有相同的基本化学结构的化合物,仅举例来说,所述基本化学结构如与氢、羧基、氨基和r基团结合的α
‑
碳。这样的类似物可以具有修饰的r基团(举例来说,正亮氨酸),或者可以具有修饰的肽主链,同时仍然保留与天然存在的氨基酸相同的基本化学结构。氨基酸类似物的非限制性实例包括高丝氨酸、正亮氨酸、甲硫氨酸亚砜、甲硫氨酸甲基锍。
[0110]
氨基酸在本文中可以通过它们的名称、它们通常已知的三字母符号或由iupac
‑
iub生物化学命名委员会推荐的单字母符号来指代。另外,核苷酸可用它们的被普遍接受的单字母代码来指代。
[0111]“氨基末端修饰基团”是指可以与末端胺基附接的任何分子。举例来说,这样的末端胺基可以在聚合分子的末端,其中这样的聚合物分子包括但不限于多肽、多核苷酸和多糖。末端修饰基团包括但不限于各种水溶性聚合物、肽或蛋白质。仅举例来说,末端修饰基
团包括聚乙二醇或血清白蛋白。末端修饰基团可用于修饰聚合分子的治疗特性,包括但不限于延长肽的血清半衰期。
[0112]
关于本文的“抗体”意指由一种或多种多肽组成的蛋白质,所述多肽基本上由抗体基因的全部或部分编码。免疫球蛋白基因包括但不限于κ、λ、α、γ(igg1、igg2、igg3和igg4)、δ、ε和μ恒定区基因,以及许多的免疫球蛋白可变区基因。本文的抗体预期包括全长抗体和抗体片段,并且包括天然存在于任何生物体中的抗体或经工程改造的抗体(例如,变体)。
[0113]
关于“抗体片段”,意指除全长形式之外的任何抗体形式。本文的抗体片段包括作为存在于全长抗体中的较小组分的抗体,以及已经被工程改造的抗体。抗体片段包括但不限于fv、fc、fab和(fab')2、单链fv(scfv)、双抗体、三抗体、四抗体、双功能杂交抗体、cdr1、cdr2、cdr3、cdr的组合、可变区、框架区、恒定区、重链、轻链和可变区以及替代支架非抗体分子、双特异性抗体等(maynard和georgiou,2000,annu.rev.biomed.eng.2:339
‑
76;hudson,1998,curr.opin.biotechnol.9:395
‑
402)。另一种功能性亚结构是单链fv(scfv),其由免疫球蛋白重链和轻链的可变区组成,通过肽接头共价连接(s
‑
zhu等人,1996,cancer research,56,3055
‑
3061)。这些小的(mr 25,000)蛋白质通常保留对单个多肽中的抗原的特异性和亲和力,并且可以为更大的抗原特异性分子提供合宜的结构单元(building block)。除非另有特别说明,否则使用术语“抗体”或“多种抗体”的声明和权利要求书具体包括“抗体片段”和“多个抗体片段”。
[0114]
如本文所用的“抗体
‑
药物缀合物”或“adc”是指与一个或多个生物活性分子共价键合的抗体分子或其片段。生物活性分子可以通过接头、聚合物或其他共价键与抗体缀合。
[0115]
如本文所用的术语“芳族”或“芳基”是指这样的闭环结构,其具有至少一个具有共轭π电子系统的环,并且包括碳环芳基和杂环芳基(或“杂芳基”或“杂芳族”)基团。碳环或杂环芳族基团可包含5至20个环原子。该术语包括共价连接的单环或稠环多环(即,共享相邻碳原子对的环)基团。芳族基团可以是未取代的或取代的。“芳族”或“芳基”基团的非限制性实例包括苯基、1
‑
萘基、2
‑
萘基、4
‑
联苯基、蒽基和菲基(phenanthracenyl)。上述每个芳基和杂芳基环系统的取代基选自本文所述的可接受的取代基的组。
[0116]
为简洁起见,当术语“芳族”或“芳基”与其他术语(包括但不限于芳氧基、芳硫氧基、芳烷基)组合使用时,其包括如上定义的芳基环和杂芳基环。因而,术语“芳烷基”或“烷芳基”预期包括其中芳基与烷基(包括但不限于苄基、苯乙基、吡啶基甲基等)附接的那些基团,包括其中碳原子(包括但不限于亚甲基)已经被例如杂原子(仅举例来说,氧原子)替换的那些烷基。这样的芳基的实例包括但不限于苯氧基甲基、2
‑
吡啶基氧基甲基、3
‑
(1
‑
萘氧基)丙基等。
[0117]
如本文所用的,术语“亚芳基”是指二价芳基。“亚芳基”的非限制性实例包括亚苯基、亚吡啶基、亚嘧啶基和亚噻吩基。亚芳基的取代基选自本文所述的可接受的取代基的组。
[0118]“双官能聚合物”,也被称为“双官能接头”,是指包含两个能够与其他部分特异性地反应以形成共价或非共价键联的官能团的聚合物。所述部分可包括但不限于天然或非天然氨基酸或含有所述天然或非天然氨基酸的肽上的侧基。可连接于双官能接头或双官能聚合物的其他部分可为相同或不同部分。仅举例来说,双官能接头可具有与第一肽上的基团
具有反应性的官能团,以及与第二肽上的基团具有反应性的另一官能团,借此形成包括所述第一肽、所述双官能接头和所述第二肽的缀合物。用于使各种化合物与肽连接的许多程序和接头分子是已知的。参见,例如欧洲专利申请第188,256号;美国专利第4,671,958号、第4,659,839号、第4,414,148号、第4,699,784号;第4,680,338号;和第4,569,789号,将这些专利通过引用完整并入本文。“多官能聚合物”也被称为“多官能接头”,是指包含两个或更多个能够与其他部分反应的官能团的聚合物。这样的部分可包括但不限于在天然或非天然氨基酸或含有这样的天然或非天然氨基酸的肽上的以形成共价或非共价键联的侧基(包括但不限于氨基酸侧基)。双官能聚合物或多官能聚合物可具有任何期望的长度或分子量,并且可被选择为在连接于化合物的一个或多个分子和与之结合的分子或所述化合物之间提供特定的期望的间隔或构象。
[0119]
如本文所用的,术语“生物利用度”是指物质或其活性部分从药物剂型递送以及在作用部位处或在全身循环中变得可用的速率和程度。生物可用度增加是指使物质或它的活性部分从药物剂型递送以及变得在作用部位处或在体循环中可用的速率和程度增加。举例来说,生物利用度增加可指示为,当物质或其活性部分相比于其他物质或活性部分时,其在血液中的浓度增加。评价生物利用度增加的方法是本领域已知的,并且可用于评价任何多肽的生物利用度。
[0120]
当在本文中使用时,术语“生物学活性分子”、“生物活性部分”或“生物活性剂”表示可影响与生物有关的生物系统、途径、分子或相互作用的任何物理或生物化学特性的任何物质,所述生物包括但不限于病毒、细菌、噬菌体、转座子、朊病毒、昆虫、真菌、植物、动物和人。特别地,如本文所用,生物活性分子包括但不限于意图用于诊断、治愈、缓解、治疗或预防人或其他动物的疾病,或另外使人或动物的身体或精神健康状态增强的任何物质。生物活性分子的实例包括但不限于肽、蛋白质、酶、小分子药物、硬药、软药、前药、碳水化合物、无机原子或分子、染料、脂质、核苷、放射性核素、寡核苷酸、毒素、细胞、病毒、脂质体、微粒和胶束。适合于与本文所述的方法和组合物一起使用的生物活性剂的类别包括但不限于药物、前药、放射性核素、成像剂、聚合物、抗生素、杀真菌剂、抗病毒剂、抗炎剂、抗肿瘤剂、心血管剂、抗焦虑剂、激素、生长因子、类固醇剂、源自微生物的毒素等。
[0121]
关于“调节生物活性”,意指增加或降低多肽的反应性,改变多肽的选择性,增强或降低多肽的底物选择性。通过比较非天然多肽的生物活性与天然多肽的生物活性,可进行修饰的生物活性的分析。
[0122]
如本文所用的,术语“生物材料”是指生物来源的材料,包括但不限于从生物反应器和/或重组方法和技术获得的材料。
[0123]
如本文所用的,术语“生物物理探针”是指能检测或监测分子中的结构变化的探针。这样的分子包括但不限于蛋白质,并且“生物物理探针”可用于检测或监测蛋白质与其他大分子的相互作用。生物物理探针的实例包括但不限于自旋标记物、荧光团和光可活化基团。
[0124]
如本文所用,术语“生物合成”是指利用翻译系统(细胞或非细胞)的任何方法,包括使用以下组分中的至少一者:多核苷酸、密码子、trna和核糖体。举例来说,可使用在wo 2002/085923中描述的方法和技术将非天然氨基酸“生物合成掺入”到非天然氨基酸多肽中,将所述专利通过引用完整并入本文。另外,在wo 2002/085923中描述了可以被“生物合
成掺入”到非天然氨基酸多肽中的有用的非天然氨基酸的选择方法,将所述专利通过引用完整并入本文。
[0125]
如本文所用的,术语“生物素类似物”或也称为“生物素模拟物”,是以高亲和力与亲和素和/或链霉亲和素结合的除生物素之外的任何分子。
[0126]
如本文所用的术语“羰基”是指含有选自
‑
c(o)
‑
、
‑
s(o)
‑
、
‑
s(o)2
‑
和
–
c(s)
‑
的部分的基团,包括但不限于含有至少一个酮基、和/或至少一个醛基、和/或至少一个酯基、和/或至少一个羧酸基、和/或至少一个硫酯基的基团。这样的羰基包括酮、醛、羧酸、酯和硫酯。另外,这样的基团可以是线性、支链或环状分子的一部分。
[0127]
术语“羧基末端修饰基团”是指可以与末端羧基附接的任何分子。举例来说,这样的末端羧基可以在聚合分子的末端,其中这样的聚合物分子包括但不限于多肽、多核苷酸和多糖。末端修饰基团包括但不限于各种水溶性聚合物、肽或蛋白质。仅举例来说,末端修饰基团包括聚乙二醇或血清白蛋白。末端修饰基团可用于修饰聚合分子的治疗特性,包括但不限于延长肽的血清半衰期。
[0128]
如本文所用的,术语“化学可裂解基团”,也称为“化学不稳定的”,是指在暴露于酸、碱、氧化剂、还原剂、化学引发剂或自由基引发剂时断裂或裂解的基团。
[0129]
如本文所用的,“共折叠”是指采用至少两个分子的再折叠过程、反应或方法,所述两个分子彼此相互作用并导致解折叠或不正确折叠的分子转化为正确折叠的分子。仅举例来说,“共折叠”采用至少两个多肽,所述两个多肽彼此相互作用并导致解折叠或不正确折叠的多肽转化为天然的正确折叠的多肽。这样的多肽可以含有天然氨基酸和/或至少一个非天然氨基酸。
[0130]
如本文所用的,“缀合物”是指直接地或通过接头与本文所述的化合物或化合物
‑
接头连接的(例如共价连接的)多肽,所述化合物或化合物
‑
接头例如是根据图1的任何一种结构或表3
‑
7的任何一种结构的化合物或盐。“靶向部分”是指相对于其他非靶分子对靶分子具有选择性亲和力的结构。本发明的靶向部分与靶分子结合。靶向部分可包括例如抗体、肽、配体、受体或其结合部分。靶生物分子可以是生物受体或细胞的其他结构,比如肿瘤抗原。如本文所用的,术语“本发明的缀合物”、“靶向部分缀合物”、“靶向缀合物”、“靶向部分
‑
活性分子缀合物”或“tc”是指与存在于细胞或其亚单位上的靶标结合的靶向多肽或其部分、类似物或衍生物,其与生物活性分子、其部分或其类似物(包括但不限于tlr7和/或tlr8激动剂)缀合。如本文所用的,术语“肿瘤靶向部分缀合物”、“肿瘤靶向部分
‑
生物活性分子缀合物”或“btc”是指与存在于肿瘤细胞或其亚单位上的靶标结合的肿瘤向多肽或其部分、类似物或衍生物,其与生物活性分子、其部分或其类似物(包括但不限于tlr7和/或tlr8激动剂)缀合。除非另有说明,术语“本发明的化合物”和“本发明的组合物”用作术语“本发明的缀合物”的替代物。
[0131]
术语“保守修饰的变体”适用于天然和非天然氨基酸以及天然和非天然核酸序列以及它们的组合。就特定核酸序列来说,“经保守修饰变体”是指编码相同或基本上相同的天然和非天然氨基酸序列的那些天然和非天然核酸,或当天然和非天然核酸不编码天然和非天然氨基酸序列时,是指基本上相同的序列。举例来说,由于遗传密码的简并性,所以许多在功能上相同的核酸编码任何给定蛋白质。举例来说,密码子gca、gcc、gcg和gcu全都编码氨基酸丙氨酸。因而,在丙氨酸由密码子指定的每个位置,密码子可以被改变为所述的任
何相应密码子,而不改变编码的多肽。这样的核酸变异是“沉默变异”,其为保守修饰变异的一个种类。因而,举例来说,本文中编码天然或非天然多肽的每个天然或非天然核酸序列也描述了天然或非天然核酸的每种可能的沉默变异。本领域普通技术人员将认识到,天然或非天然核酸中的每个密码子(除通常是甲硫氨酸的唯一密码子的aug和通常是色氨酸的唯一密码子的tgg之外)都可被修饰,以产生功能相同的分子。因此,编码天然和非天然多肽的天然和非天然核酸的每个沉默变异都隐含于每个描述的序列中。
[0132]
关于氨基酸序列,改变、添加或缺失编码序列中的单个天然和非天然氨基酸或小百分比的天然和非天然氨基酸的对核酸、肽、多肽或蛋白质序列的个别取代、缺失或添加是“保守修饰的变体”,其中所述改变导致氨基酸的缺失、氨基酸的添加或天然和非天然氨基酸被化学上相似的氨基酸取代。提供功能相似的天然氨基酸的保守取代表是本领域熟知的。这样的保守修饰的变体附加于并且不排除本文所述的方法和组合物的多态变体、种间同源物和等位基因。
[0133]
提供功能相似的氨基酸的保守取代表是本领域普通技术人员已知的。以下八个组各自含有在彼此之间为保守取代的氨基酸:1)丙氨酸(a)、甘氨酸(g);2)天冬氨酸(d)、谷氨酸(e);3)天冬酰胺(n)、谷氨酰胺(q);4)精氨酸(r)、赖氨酸(k);5)异亮氨酸(i)、亮氨酸(l)、甲硫氨酸(m)、缬氨酸(v);6)苯丙氨酸(f)、酪氨酸(y)、色氨酸(w);7)丝氨酸(s)、苏氨酸(t);以及8)半胱氨酸(c)、甲硫氨酸(m)。(参见,例如creighton,proteins:structures and molecular properties(w h freeman&co.;第2版(1993年12月)。
[0134]
除非另有说明,术语“环烷基”和“杂环烷基”本身或与其他术语组合分别表示“烷基”和“杂烷基”的环状形式。因此,环烷基或杂环烷基包括饱和的、部分不饱和的和完全不饱和的环连接。另外,对于杂环烷基,杂原子可以占据杂环与分子的其余部分附接的位置。杂原子可包括但不限于氧、氮或硫。环烷基的实例包括但不限于环戊基、环己基、1
‑
环己烯基、3
‑
环己烯基、环庚基等。杂环烷基的实例包括但不限于1
‑
(1,2,5,6
‑
四氢吡啶基)、1
‑
哌啶基、2
‑
哌啶基、3
‑
哌啶基、4
‑
吗啉基、3
‑
吗啉基、四氢呋喃
‑2‑
基、四氢呋喃
‑3‑
基、四氢噻吩
‑2‑
基、四氢噻吩
‑3‑
基、1
‑
哌嗪基、2
‑
哌嗪基等。另外,该术语包括多环结构,包括但不限于双环和三环的环结构。类似地,术语“杂环亚烷基”本身或作为另一分子的一部分表示来源于杂环烷基的二价基团,并且术语“环亚烷基”本身或作为另一分子的一部分表示来源于环烷基的二价基团。
[0135]
如本文所用的,术语“环糊精”是指由成环的至少六个至八个葡萄糖分子组成的环状碳水化合物。环的外部含有水溶性基团;在环的中心是能够容纳小分子的相对非极性腔。
[0136]
如本文所用的,术语“细胞毒性的”是指伤害细胞的化合物。
[0137]
如本文所用的,变性剂(“denaturing agent”或“denaturant”)是指将引起聚合物的可逆解折叠的任何化合物或物质。仅举例来说,“变性剂”可以引起蛋白质的可逆解折叠。变性剂的强度将由具体变性剂的性质和浓度决定。举例来说,变性剂包括但不限于离液剂、洗涤剂、有机溶剂、水混溶性溶剂、磷脂或其组合。离液剂的非限制性实例包括但不限于尿素、胍和硫氰酸钠。洗涤剂的非限制性实例可包括但不限于强洗涤剂,比如十二烷基硫酸钠,或聚氧乙烯醚(例如吐温或triton洗涤剂)、十二烷基肌氨酸钠(sarkosyl);温和的非离子洗涤剂(例如,洋地黄皂苷);温和的阳离子洗涤剂,比如n
‑
>2,3
‑‑
(二油氧基)
‑
丙基
‑
n,n,n
‑
三甲铵;温和的离子洗涤剂(例如胆酸钠或脱氧胆酸钠);或两性离子洗涤剂,包括但不限
于磺基甜菜碱(zwittergent)、3
‑
(3
‑
胆酰胺丙基)二甲基氨基
‑1‑
丙烷硫酸盐(chaps)和3
‑
(3
‑
胆酰胺丙基)二甲基氨基
‑2‑
羟基
‑1‑
丙烷磺酸盐(chapso)。有机、水混溶性溶剂的非限制性实例包括但不限于乙腈、低级链烷醇(特别是c2
‑
c4链烷醇如乙醇或异丙醇)、或低级链烷二醇(c2
‑
c4链烷二醇如乙二醇),可用作变性剂。磷脂的非限制性实例包括但不限于天然存在的磷脂,比如磷脂酰乙醇胺、磷脂酰胆碱、磷脂酰丝氨酸和磷脂酰肌醇;或合成的磷脂衍生物或变体,比如二己酰基磷脂酰胆碱或二庚酰基磷脂酰胆碱。
[0138]
如本文所用的,术语“二胺”是指包含至少两个胺官能团的基团/分子,所述胺官能团包括但不限于肼基、脒基、亚胺基、1,1
‑
二胺基、1,2
‑
二胺基、1,3
‑
二胺基和1,4
‑
二胺基。另外,这样的基团可以是线性、支链或环状分子的一部分。
[0139]
如本文所用的,术语“可检测标记物”是指可以使用分析技术观察到的标记物,所述分析技术包括但不限于荧光、化学发光、电子自旋共振、紫外/可见吸收光谱、质谱、核磁共振、磁共振和电化学方法。
[0140]
如本文所用的术语“二羰基”是指含有选自由
‑
c(o)
‑
、
‑
s(o)
‑
、
‑
s(o)2‑
和
–
c(s)
‑
组成的组的至少两个部分的基团,包括但不限于1,2
‑
二羰基、1,3
‑
二羰基和1,4
‑
二羰基;以及含有至少一个酮基、和/或至少一个醛基、和/或至少一个酯基、和/或至少一个羧酸基、和/或至少一个硫酯基的基团。这样的二羰基包括二酮、酮醛、酮酸、酮酯和酮硫酯。另外,这样的基团可以是线性、支链或环状分子的一部分。二羰基中的两个部分可以相同或不同,并且可包括将在两个部分的任一个处产生(仅举例来说)酯、酮、醛、硫酯或酰胺的取代基。
[0141]
如本文所用的,术语“药物”是指用于预防、诊断、减轻、治疗或治愈疾病或病症的任何物质。
[0142]
如本文所用的,术语“有效量”是指所施用的剂或化合物的将在一定程度上减轻被治疗的疾病或病症的一种或多种症状的足够量。结果可以是体征、症状、或病因的减少和/或减轻,或生物系统的任何其他期望的改变。举例来说,所施用的剂或化合物包括但不限于天然氨基酸多肽、非天然氨基酸多肽、经修饰的天然氨基酸多肽或经修饰的非氨基酸多肽。为了预防性、增强性和/或治疗性治疗,可施用含有这样的天然氨基酸多肽、非天然氨基酸多肽、经修饰的天然氨基酸多肽或经修饰的非天然氨基酸多肽的组合物。在任何个别情况下的适当“有效”量都可使用诸如剂量递增研究的技术来确定。
[0143]
术语“增强(enhance/enhancing)”意指在效能或持续时间方面使所需作用增加或延长。举例来说,“增强”治疗剂的作用是指能够在效能或持续时间方面使治疗剂在治疗疾病、病症或疾患期间的作用增加或延长。如本文所用的“增强性有效量”是指足以增强治疗剂在治疗疾病、病症或疾患方面的作用的量。当在患者中使用时,用于这个用途的有效量将取决于疾病、疾患或病症的严重性和病程、先前的疗法、患者的健康状况和对药物的反应以及治疗医师的判断。
[0144]
如本文所用的,术语“真核生物”是指属于系统发育真核生物域(eucarya)的生物,包括但不限于动物(包括但不限于哺乳动物、昆虫、爬行动物、鸟类等)、纤毛虫、植物(包括但不限于单子叶植物、双子叶植物和藻类)、真菌、酵母、鞭毛虫、微孢子虫和原生生物。
[0145]
如本文所用的,术语“脂肪酸”是指具有约c6或更长的烃侧链的羧酸。
[0146]
如本文所用的,术语“荧光团”是指在激发时发射光子并由此发射荧光的分子。
[0147]
如本文所用的,术语“官能团”、“活性部分”、“活化基团”、“离去基团”、“反应性位
点”、“化学反应性基团”和“化学反应性部分”,是指在发生化学反应处的分子部分或单元。这些术语在化学领域中在一定程度上同义,并且在本文中用于指示执行某种功能或活性并且与其他分子反应的分子部分。
[0148]
术语“卤素”包括氟、氯、碘和溴。
[0149]
如本文所用的,术语“卤代酰基”是指含有卤素部分的酰基,包括但不限于
‑
c(o)ch3、
‑
c(o)cf3、
‑
c(o)ch2och3等等。
[0150]
如本文所用的,术语“卤代烷基”是指含有卤素部分的烷基,包括但不限于
‑
cf3和
–
ch2cf3等。
[0151]
如本文所用的,术语“杂烷基”是指由烷基和至少一个杂原子组成的直链或支链或环烃基或其组合,所述杂原子选自由o、n、si和s组成的组,其中氮和硫原子可任选地被氧化,并且氮杂原子可任选地被季铵化。杂原子o、n和s和si可以位于杂烷基的任何内部位置或烷基与分子的其余部分附接的位置。实例包括但不限于,
‑
ch2‑
ch2‑
o
‑
ch3、
‑
ch2‑
ch2‑
nh
‑
ch3、
‑
ch2‑
ch2‑
n(ch3)
‑
ch3、
‑
ch2‑
s
‑
ch2‑
ch3、
‑
ch2‑
ch2,
‑
s(o)
‑
ch3、
‑
ch2‑
ch2‑
s(o)2‑
ch3、
‑
ch=ch
‑
o
‑
ch3、
‑
si(ch3)3、
‑
ch2‑
ch=n
‑
och3和
–
ch=ch
‑
n(ch3)
‑
ch3。另外,至多两个杂原子可以是连续的,比如,举例来说,
‑
ch2‑
nh
‑
och3和
–
ch2‑
o
‑
si(ch3)3。
[0152]
术语“基于杂环的键联”或“杂环键联”是指由二羰基与二胺基反应形成的部分。所得反应产物是杂环,包括杂芳基或杂环烷基。所得杂环基团用作非天然氨基酸或非天然氨基酸多肽和另一个官能团之间的化学连接。在一个实施方案中,杂环键联包括含氮杂环键联,仅举例来说,包括吡唑键联、吡咯键联、吲哚键联、苯二氮(benzodiazepine)键联和吡唑啉酮键联。
[0153]
类似地,术语“杂亚烷基”是指衍生自杂烷基的二价基团,例如但不限于,
‑
ch2‑
ch2‑
s
‑
ch2‑
ch2‑
和
–
ch2‑
s
‑
ch2‑
ch2‑
nh
‑
ch2‑
。对于杂亚烷基,相同或不同的杂原子也可占据链末端的任一个或两个(包括但不限于亚烷氧基、亚烷二氧基(alkylenedioxy)、亚烷基氨基、亚烷基二氨基、氨基氧基亚烷基(aminooxyalkylene)等)。更进一步地,对于亚烷基和杂亚烷基连接基团,连接基团的取向并非由书写连接基团的化学式的方向来暗示。举例来说,式
–
c(o)2r
’‑
代表
–
c(o)2r
’‑
和
–
r’c(o)2‑
两者。
[0154]
如本文所用的,术语“杂芳基”或“杂芳族”是指含有至少一个选自n、o和s的杂原子的芳基;其中氮和硫原子可任选地被氧化,并且氮原子可任选地被季铵化。杂芳基可以是取代或未取代的。杂芳基可以通过杂原子与分子的其余部分附接。杂芳基的非限制性实例包括1
‑
吡咯基、2
‑
吡咯基、3
‑
吡咯基、3
‑
吡唑基、2
‑
咪唑基、4
‑
咪唑基、吡嗪基、2
‑
噁唑基、4
‑
噁唑基、2
‑
苯基
‑4‑
噁唑基、5
‑
噁唑基、3
‑
异噁唑基、4
‑
异噁唑基、5
‑
异噁唑基、2
‑
噻唑基、4
‑
噻唑基、5
‑
噻唑基、2
‑
呋喃基、3
‑
呋喃基、2
‑
噻吩基、3
‑
噻吩基、2
‑
吡啶基、3
‑
吡啶基、4
‑
吡啶基、2
‑
嘧啶基、4
‑
嘧啶基、5
‑
苯并噻唑基、嘌呤基、2
‑
苯并咪唑基、5
‑
吲哚基、1
‑
异喹啉基、5
‑
异喹啉基、2
‑
喹喔啉基、5
‑
喹喔啉基、3
‑
喹啉基和6
‑
喹啉基。
[0155]
如本文所用的,术语“均烷基”是指作为烃基的烷基。
[0156]
如本文所用的,术语“同一的”是指两个或更多个序列或子序列是相同的。此外,如本文所用的术语“大致上同一”是指如使用比较算法或通过手动比对和目视检查所测量,当历经比较窗或指定区域比较以及对准以达成最大对应时,两个或更多个序列具有某一百分比的相同连续单元。仅举例来说,如果连续单元在指定区域上是约60%同一的、约65%同一
的、约70%同一的、约75%同一的、约80%同一的、约85%同一的、约90%同一的或约95%同一的,则两个或更多个序列可以是“基本上同一的”。这样的百分比描述两个或更多个序列的“同一性百分比”。序列的同一性可以存在于在至少约75
‑
100个连续单元长度的区域上,在约50个连续单元长度的区域上,或当未指定时在整个序列上。这个定义还涉及测试序列的互补序列。仅举例来说,历经指定区域,当氨基酸残基是相同的时,两个或更多个多肽序列是同一的,而如果氨基酸残基是约60%同一的、约65%同一的、约70%同一的、约75%同一的、约80%同一的、约85%同一的、约90%同一的或约95%同一的,那么两个或更多个多肽序列是“大致上同一的”。同一性可历经长度是至少约75至约100个氨基酸的区域,历经长度是约50个氨基酸的区域,或当未指定时跨越多肽序列的整个序列而存在。此外,仅举例来说,历经指定区域,当核酸残基是相同的时,两个或更多个多核苷酸序列是同一的,而如果核酸残基是约60%同一的、约65%同一的、约70%同一的、约75%同一的、约80%同一的、约85%同一的、约90%同一的或约95%同一的,那么两个或更多个多核苷酸序列是“大致上同一的”。同一性可以存在于在多核苷酸序列的至少约75个至约100个核酸长度的区域上,在约50个核酸长度的区域上,或当未指定时在整个序列上。
[0157]
为了序列比较,通常将一个序列用作与测试序列进行比较的参考序列。当使用序列比较算法时,将测试序列和参考序列输入到计算机中,必要时指定子序列坐标,并指定序列算法程序参数。可以使用默认程序参数,或者,也可以指定替代参数。然后,基于程序参数,序列比较算法计算出测试序列相对于参考序列的序列同一性百分比。
[0158]
如本文所用的,术语“免疫原性”是指对施用治疗性药物的抗体应答。对治疗性非天然氨基酸多肽的免疫原性可使用用于检测生物流体中的抗非天然氨基酸多肽抗体的定量和定性测定获得。所述测定包括但不限于放射免疫测定(ria)、酶联免疫吸附测定(elisa)、发光免疫测定(lia)和荧光免疫测定(fia)。分析对治疗性非天然氨基酸多肽的免疫原性涉及将在施用治疗性非天然氨基酸多肽后的抗体应答与在施用治疗性天然氨基酸多肽后的抗体应答进行比较。
[0159]
如本文所用的术语“分离”是指使目标组分与非目标组分分离以及将目标组分从非目标组分移除。经分离物质可呈干燥或半干燥状态,或呈包括但不限于水溶液的溶液形式。分离的组分可处于均质状态,或者分离的组分可以是包含另外的药学上可接受的载体和/或赋形剂的药物组合物的一部分。可使用包括但不限于聚丙烯酰胺凝胶电泳或高效液相色谱法的分析化学技术来确定纯度和均质性。另外,当感兴趣的组分被分离,并且是制剂中存在的主要种类时,所述组分在本文中被描述为是基本上纯化的。如本文所用的术语“纯化”可指目标组分是至少85%纯净的、至少90%纯净的、至少95%纯净的、至少99%或更大百分比纯净的。仅举例来说,当核酸或蛋白质不含它在天然状态下与其相伴的细胞组分中的至少一些,或核酸或蛋白质已被浓缩至大于它的体内或体外产生浓度的水平时,所述核酸或蛋白质是“经分离的”。而且,举例来说,当基因与在所述基因侧翼并且编码除感兴趣的基因之外的蛋白质的开放阅读框分开时,则所述基因是分离的。
[0160]
如本文所用的,术语“标记物”是指掺入化合物中并易于被检出的物质,由此可以检测和/或监测其物理分布。
[0161]
如本文所用的,术语“键联”或“接头”是指由接头的官能团与另一分子之间的化学反应形成的键或化学部分。这样的键可包括但不限于共价键联和非共价键,而这样的化学
部分可包括但不限于酯、碳酸酯、亚胺磷酸酯、腙、缩醛、原酸酯、肽键联和寡核苷酸键联。水解稳定键联意指键联在水中是大致上稳定的,并且在有用的ph值下不与水反应,包括但不限于在生理条件下持续延长时期,也许甚至是无限期。水解不稳定或可降解键联意指键联在水中或在包括例如血液的水溶液中可降解。酶不稳定或可降解键联意指键联可由一种或多种酶降解。仅举例来说,peg和相关聚合物可包括在聚合物主链中或在聚合物主链与聚合物分子的一个或多个末端官能团之间的连接基团中的可降解键联。这样的可降解键联包括但不限于通过peg羧酸或活化peg羧酸与生物活性剂上的醇基团的反应形成的酯键联,其中这样的酯基团通常在生理条件下水解以释放所述生物活性剂。其他水解可降解键联包括但不限于碳酸酯键联;由胺和醛的反应产生的亚胺键联;通过使醇与磷酸酯基团反应形成的磷酸酯键联;作为酰肼和醛的反应产物的腙键联;作为醛和醇的反应产物的缩醛键联;作为甲酸酯和醇的反应产物的原酸酯键联;通过包括但不限于在聚合物诸如peg的末端的胺基团和肽的羧基形成的肽键联;以及通过包括但不限于在聚合物的末端的亚磷酰胺基团和寡核苷酸的5'羟基形成的寡核苷酸键联。接头包括但不限于短线性、支链、多臂或树枝状聚合物分子,比如聚合物。在本发明的一些实施方案中,接头可以是支链的。在其他实施方案中,接头可以是双官能接头。在一些实施方案中,接头可以是三官能接头。许多不同的可裂解接头是本领域技术人员已知的。参见美国专利第4,618,492号;第4,542,225和第4,625,014号。从这些连接基团释放剂的机制包括,例如,光不稳定键的照射和酸催化水解。例如,美国专利第4,671,958号包括对包含接头的免疫缀合物的描述,所述接头在体内被患者补体系统的蛋白水解酶在靶位点裂解。根据多肽和与其连接的分子之间的期望的空间关系,可以预先确定或选择接头的长度。鉴于已报道的将各种放射诊断化合物、放射治疗化合物、药物、毒素和其他药剂与抗体附接的大量方法,本领域技术人员将能够确定将给定剂或分子与多肽附接的适合方法。
[0162]
如本文所用的,术语“修饰”是指针对天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽的变化的存在。通过合成后修饰天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽;或通过共翻译或通过翻译后修饰天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽,可以获得这样的变化或修饰。形式“经修饰的或未经修饰的”意指所讨论的天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽是任选地修饰的,即,所讨论的天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽可以是经修饰或未经修饰的。
[0163]
如本文所用的,术语“调节的血清半衰期”是指修饰的生物活性分子相对于它的非修饰形式在循环半衰期方面的正向或负向变化。举例来说,经修饰的生物活性分子包括但不限于天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽。举例来说,通过在施用生物活性分子或修饰的生物活性分子之后的不同时间点采集血液样品并确定每个样品中该分子的浓度来测量血清半衰期。血清浓度与时间的关联允许计算血清半衰期。举例来说,调节的血清半衰期可以是血清半衰期延长,使得能够改进给药方案或避免毒性作用。这样的血清延长可以是至少约两倍、至少约三倍、至少约五倍或至少约十倍。评价任何多肽的血清半衰期延长的方法是技术人员熟知的。
[0164]
如本文所用的,术语“调节的治疗半衰期”是指治疗有效量的修饰的生物活性分子相对于它的非修饰形式在半衰期方面的正向或负向变化。举例来说,经修饰的生物活性分
子包括但不限于天然氨基酸、非天然氨基酸、天然氨基酸多肽或非天然氨基酸多肽。举例来说,治疗半衰期通过在施用之后在各个时间点测量分子的药物代谢动力学和/或药物效应动力学性质来测量。治疗半衰期增加可使得能够达成特定有益给药方案、特定有益总剂量,或避免非所需作用。举例来说,治疗半衰期延长可由效能增加、修饰的分子与它的靶标的结合增加或降低、非修饰分子的另一参数或作用机理的增加或降低、或酶(比如,仅举例来说,蛋白酶)对分子的分解增加或降低所致。评价任何多肽的治疗半衰期延长的方法是技术人员熟知的。
[0165]“非天然氨基酸”是指不是20种常见氨基酸或吡咯赖氨酸(pyrolysine)或硒代半胱氨酸之一的氨基酸。可与术语“非天然氨基酸”同义使用的其他术语是“非天然编码氨基酸”、“非天然氨基酸”、“非天然存在的氨基酸”以及它们的以各种方式以连字符连接和不以连字符连接的形式。术语“非天然氨基酸”包括但不限于通过修饰天然编码氨基酸(包括但不限于20种常见氨基酸或吡咯赖氨酸和硒代半胱氨酸)而天然存在,但它们自身不通过翻译复合物掺入到增长的多肽链中的氨基酸。这样的氨基酸的实例包括但不限于n
‑
乙酰葡糖胺基
‑
l
‑
丝氨酸、n
‑
乙酰葡糖胺基
‑
l
‑
苏氨酸和o
‑
磷酸酪氨酸。另外,术语“非天然氨基酸”包括但不限于非天然存在的并且可合成获得或可通过修饰非天然氨基酸获得的氨基酸。在一些实施方案中,非天然氨基酸包括赖氨酸类似物,例如,n6
‑
叠氮基乙氧基
‑
l
‑
赖氨酸(azk)、n6
‑
炔丙基乙氧基
‑
l
‑
赖氨酸(prak)、bcn
‑
l
‑
赖氨酸、降冰片烯赖氨酸、tco
‑
赖氨酸、甲基四嗪赖氨酸或烯丙氧基羰基赖氨酸。在一些实施方案中,非天然氨基酸包含糖部分。此类氨基酸的实例包括n
‑
乙酰基
‑
l
‑
葡糖胺基
‑
l
‑
丝氨酸、n
‑
乙酰基
‑
l
‑
半乳糖胺基
‑
l
‑
丝氨酸、n
‑
乙酰基
‑
l
‑
葡糖胺基
‑
l
‑
苏氨酸、n
‑
乙酰基
‑
l
‑
葡糖胺基
‑
l
‑
天冬酰胺和o
‑
甘露糖胺基
‑
l
‑
丝氨酸。所述氨基酸的实例还包括其中氨基酸与糖之间的天然存在的n
‑
键联或o
‑
键联由通常不见于自然界中的共价键联
–
包括但不限于烯、肟、硫醚、酰胺等替换的实例。这样的氨基酸的实例还包括通常未见于天然存在的蛋白质中的糖,比如2
‑
脱氧
‑
葡萄糖、2
‑
脱氧半乳糖等。非天然氨基酸的具体实例包括但不限于对乙酰基
‑
l
‑
苯丙氨酸、对炔丙基氧基苯丙氨酸、o
‑
甲基
‑
l
‑
酪氨酸、l
‑3‑
(2
‑
萘基)丙氨酸、3
‑
甲基
‑
苯丙氨酸、o
‑4‑
烯丙基
‑
l
‑
酪氨酸、4
‑
丙基
‑
l
‑
酪氨酸、三
‑
o
‑
乙酰基
‑
glcnacβ
‑
丝氨酸、l
‑
多巴、氟化苯丙氨酸、异丙基
‑
l
‑
苯丙氨酸、对叠氮基
‑
l
‑
苯丙氨酸、对酰基
‑
l
‑
苯丙氨酸、对苯甲酰基
‑
l
‑
苯丙氨酸、l
‑
磷酸丝氨酸、膦酰基丝氨酸、膦酰基酪氨酸、对碘
‑
苯丙氨酸、对溴苯丙氨酸、对氨基
‑
l
‑
苯丙氨酸、对炔丙基氧基
‑
l
‑
苯丙氨酸、4
‑
叠氮基
‑
l
‑
苯丙氨酸、对叠氮基乙氧基苯丙氨酸和对叠氮基甲基
‑
苯丙氨酸等。在一些实施方案中,非天然氨基酸选自由以下组成的组:对乙酰基
‑
苯丙氨酸、4
‑
叠氮基
‑
l
‑
苯丙氨酸、对叠氮乙氧基苯丙氨酸或对叠氮甲基
‑
苯丙氨酸。
[0166]
如本文所用的,术语“核酸”是指单链或双链形式的脱氧核糖核苷酸、脱氧核糖核苷、核糖核苷或核糖核苷酸及其聚合物。仅举例来说,这样的核酸和核酸聚合物包括但不限于(i)天然核苷酸的类似物,其与参考核酸具有相似的结合特性,并且以与天然存在的核苷酸相似的方式被代谢;(ii)寡核苷酸类似物,包括但不限于pna(肽核酸)、用于反义技术中的dna类似物(硫代磷酸酯、氨基磷酸酯等);(iii)其保守修饰的变体(包括但不限于简并密码子取代)和互补序列以及明确指示的序列。举例来说,通过生成其中一个或多个选定的(或全部)密码子的第三位置被混合碱基和/或脱氧肌苷残基取代的序列,可实现简并密码子取代(batzer等人,nucleic acid res.19:5081(1991);ohtsuka等人,j.biol.chem.260:
2605
‑
2608(1985);和rossolini等人,mol.cell.probes 8:91
‑
98(1994))。
[0167]
如本文所用的,术语“氧化剂”是指能够从被氧化的化合物中除去电子的化合物或物质。举例来说,氧化剂包括但不限于氧化型谷胱甘肽、半胱氨酸、胱胺、氧化型二硫苏糖醇、氧化型赤藓糖醇和氧。多种氧化剂适合在本文所述的方法和组合物中使用。
[0168]
如本文所用的,术语“药学上可接受”是指包括但不限于盐、载体或稀释剂的物质,其不消除化合物的生物活性或特性,并且是相对无毒的,即所述物质可施用于个体而不引起不希望的生物作用或以有害的方式与包含它的组合物中的任何组分相互作用。
[0169]
如本文所用的,术语“聚亚烷基二醇”或“聚(烯烃二醇)”是指线性或支链的聚合聚醚多元醇。这样的聚亚烷基二醇包括但不限于聚乙二醇、聚丙二醇、聚丁二醇以及它们的衍生物。其他示例性实施方案列于例如商业供应商目录中,比如shearwater corporation的目录“polyethylene glycol and derivatives for biomedical applications”(2001)。仅举例来说,这样的聚合聚醚多元醇具有在约0.1kda至约100kda之间的平均分子量。举例来说,这样的聚合聚醚多元醇包括但不限于,在约100da至约100,000da之间。聚合物的分子量可介于约100da与约100,000da之间,包括但不限于约100,000da、约95,000da、约90,000da、约85,000da、约80,000da、约75,000da、约70,000da、约65,000da、约60,000da、约55,000da、约50,000da、约45,000da、约40,000da、约35,000da、约30,000da、约25,000da、约20,000da、约15,000da、约10,000da、约9,000da、约8,000da、约7,000da、约6,000da、约5,000da、约4,000da、约3,000da、约2,000da、约1,000da、约900da、约800da、约700da、约600da、约500da、400da、约300da、约200da和约100da。在一些实施方案中,聚合物的分子量介于约100da与约50,000da之间。在一些实施方案中,聚合物的分子量介于约100da与约40,000da之间。在一些实施方案中,聚合物的分子量介于约1,000da与约40,000da之间。在一些实施方案中,聚合物的分子量在约2,000至约50,000da之间。在一些实施方案中,聚合物的分子量介于约5,000da与约40,000da之间。在一些实施方案中,聚合物的分子量介于约10,000da与约40,000da之间。在一些实施方案中,聚(乙二醇)分子是分支聚合物。分支链peg的分子量可在约1,000da与约100,000da之间,包括但不限于约100,000da、约95,000da、约90,000da、约85,000da、约80,000da、约75,000da、约70,000da、约65,000da、约60,000da、约55,000da、约50,000da、约45,000da、约40,000da、约35,000da、约30,000da、约25,000da、约20,000da、约15,000da、约10,000da、约9,000da、约8,000da、约7,000da、约6,000da、约5,000da、约4,000da、约3,000da、约2,000da和约1,000da。在一些实施方案中,分支链peg的分子量介于约1,000da与约50,000da之间。在一些实施方案中,分支链peg的分子量介于约1,000da与约40,000da之间。在一些实施方案中,分支链peg的分子量介于约5,000da与约40,000da之间。在一些实施方案中,分支链peg的分子量介于约5,000da与约20,000da之间。在其他实施方案中,分支链peg的分子量介于约2,000da至约50,000da之间。
[0170]
如本文所用的,术语“聚合物”是指由重复亚单元组成的分子。这样的分子包括但不限于多肽、多核苷酸、或多糖或聚亚烷基二醇。
[0171]
术语“多肽”、“肽”和“蛋白质”在本文中可互换使用,指的是氨基酸残基的聚合物。也就是说,涉及多肽的描述同等适用于对肽的描述和对蛋白质的描述,并且反之亦然。所述术语适用于天然存在的氨基酸聚合物以及其中一个或多个氨基酸残基是非天然氨基酸的氨基酸聚合物。另外,所述“多肽”、“肽”和“蛋白质”包括任何长度的氨基酸链,包括全长蛋
白质,其中氨基酸残基由共价肽键连接。
[0172]
术语“翻译后修饰”是指天然或非天然氨基酸的在这种氨基酸已以翻译方式并入多肽链中之后发生的任何修饰。这样的修饰包括但不限于共翻译体内修饰、共翻译体外修饰(比如在无细胞翻译系统中)、翻译后体内修饰和翻译后体外修饰。
[0173]
如本文所用的,术语“前药”或“药学上可接受的前药”是指体内或体外转化为母体药物的剂,其中,其不消除药物的生物活性或特性,并且是相对无毒的,即所述物质可施用于个体而不引起不希望的生物作用或以有害的方式与包含它的组合物中的任何组分相互作用。前药通常是药物前体,其在施用于受试者以及后续吸收之后通过某一过程比如通过代谢路径的转化而被转化成活性种类或更具活性的种类。一些前药具有存在于前药上的致使它活性较小和/或对药物赋予溶解性或某一其他性质的化学基团。一旦化学基团已被从前药裂解和/或修饰,即产生活性药物。前药在身体内通过酶促或非酶促反应来转化成活性药物。前药可提供改进的生理化学性质,诸如更好的溶解性;增强的递送特征,诸如特异性靶向特定细胞、组织、器官或配体;以及药物的改进治疗价值。这样的前药的益处包括但不限于(i)相比于母体药物,容易施用;(ii)前药通过口服施用可以是生物可利用的,而母体药物则不然;和(iii)相比于母体药物,前药还可在药物组合物中具有改进的溶解度。前药包括活性药物的无药理活性或活性降低的衍生物。前药可被设计为通过操控药物的特性来调节药物或生物活性分子到达期望的作用部位的量,所述特性比如为生理化学特性、生物制药特性或药代动力学特性。在无限制的情况下,前药的实例为非天然氨基酸多肽,其以酯(“前药”)形式施用以有助于跨越细胞膜(在细胞膜中,水溶性对于移动性是有害的)传送,但是,然后,一旦在对水溶性有利的细胞内部时,其即被代谢水解成羧酸,即活性实体。前药可被设计为可逆药物衍生物,以用作增强药物转运至位点特异性组织的调节剂。
[0174]
如本文所用的,术语“预防有效量”是指预防性地应用于患者的含有至少一种非天然氨基酸多肽或至少一种经修饰的非天然氨基酸多肽的组合物的量,其将在一定程度上减轻被治疗疾病、病症或疾患的一种或多种症状。在这样的预防性应用中,这样的量可取决于患者的健康状态、体重等。通过常规实验,包括但不限于剂量递增临床试验,认为这样的预防有效量被认为是在本领域技术范围内。
[0175]
如本文所用的,术语“受保护的”是指“保护基团”或部分的存在,其阻止化学反应性官能团在某些反应条件下的反应。保护基团将根据受保护的化学反应性基团的类型而变化。仅举例来说,(i)如果化学反应性基团是胺或酰肼,则保护基团可以选自叔丁氧基羰基(t
‑
boc)和9
‑
芴基甲氧基羰基(fmoc);(ii)如果化学反应性基团是硫醇,则保护基团可以是邻二硫吡啶;和(iii)如果化学反应性基团是羧酸,比如丁酸或丙酸,或羟基,则保护基团可以是苄基或烷基,比如甲基、乙基或叔丁基。
[0176]
仅举例来说,封闭/保护基团可以选自:
[0177][0178]
另外,保护基团包括但不限于包括光不稳定基团,比如nvoc和menvoc以及本领域已知的其他保护基团。其他保护基团描述于greene和wuts,protective groups in organic synthesis,第3版,john wiley&sons,new york,ny,1999,将其通过引用完整并入本文。
[0179]
术语“重组宿主细胞”,也称为“宿主细胞”,是指包含外源多核苷酸的细胞,其中用于将外源多核苷酸插入到细胞中的方法包括但不限于直接摄取、转导、f
‑
交配或本领域已知的产生重组宿主细胞的其他方法。仅举例来说,这样的外源多核苷酸可以是非整合载体,包括但不限于质粒,或者可以整合到宿主基因组中。
[0180]
如本文所用的,术语“氧化还原活性剂”是指使另一分子氧化或还原的分子,由此氧化还原活性剂被还原或氧化。氧化还原活性剂的实例包括但不限于二茂铁、醌类、ru
2 /3
络合物、co
2 /3
络合物和os
2 /3
络合物。
[0181]
如本文所用的,术语“还原剂”是指能够对被还原的化合物添加电子的化合物或物质。举例来说,还原剂包括但不限于二硫苏糖醇(dtt)、2
‑
巯基乙醇、二硫赤藓糖醇、半胱氨酸、半胱胺(2
‑
氨基乙硫醇)和还原型谷胱甘肽。仅举例来说,这样的还原剂可用于将巯基保持在还原状态,并使分子内或分子间二硫键还原。
[0182]
如本文所用的,“再折叠”描述了将不正确折叠或解折叠状态转化为天然或正确折叠构象的任何过程、反应或方法。仅举例来说,再折叠将含二硫键的多肽从不正确折叠或解折叠状态转化为相对于二硫键的天然或正确折叠构象。这样的含二硫键的多肽可以是天然氨基酸多肽或非天然氨基酸多肽。
[0183]
如本文所用的,术语“安全性”或“安全性概况”是指相对于已施用药物次数的可能与药物施用有关的副作用。举例来说,已多次施用并且仅产生轻微副作用或无副作用的药物被认为具有极好的安全性。用于评价任何多肽的安全性概况的方法是本领域已知的。
[0184]
如本文所用的,短语“选择性杂交”或“特异性杂交”是指当特定核苷酸序列存在于复杂混合物(包括但不限于总细胞或文库dna或rna)中时,一种分子在严格杂交条件下与该
序列结合、双链化或杂交。
[0185]
短语“严格杂交条件”是指dna、rna、pna或其他核酸模拟物或其组合的序列在低离子强度和高温条件下的杂交。举例来说,在严格条件下,探针将与核酸(包括但不限于总细胞或文库dna或rna)的复杂混合物中的它的目标子序列杂交,但不与复杂混合物中的其他序列杂交。严格条件是序列依赖性的并且在不同情况下将是不同的。举例来说,较长的序列在较高的温度下特异性地杂交。严格杂交条件包括但不限于:(i)比特定序列在限定的离子强度和ph下的热熔点(tm)低约5
‑
10℃;(ii)在约ph 7.0至约ph 8.3时的盐浓度为约0.01m至约1.0m,并且对于短探针(包括但不限于约10至约50个核苷酸),温度为至少约30℃,对于长探针(包括但不限于大于50个核苷酸),温度为至少约60℃;(iii)添加去稳定剂,包括但不限于甲酰胺,(iv)50%甲酰胺、5x ssc和1%sds,在42℃孵育,或5x ssc、约1%sds,在65℃孵育,在0.2x ssc中洗涤,以及约0.1%sds,在65℃孵育约5分钟至约120分钟。仅举例来说,选择性或特异性杂交的检测包括但不限于至少是背景的两倍的阳性信号。在tijssen的laboratory techniques in biochemistry and molecular biology
‑‑
hybridization with nucleic probes,“overview of principles of hybridization and the strategy of nucleic acid assays”(1993)中找到关于核酸杂交的广泛指南。
[0186]
如本文所用的,术语“受试者”是指作为治疗、观察或实验对象的动物。仅举例来说,受试者可为但不限于哺乳动物,包括但不限于人。
[0187]
如本文所用的术语“大致上纯化”是指目标组分可大致上或基本上不含在纯化之前通常伴随所述目标组分或与所述目标组分相互作用的其他组分。仅举例来说,当目标组分的制剂含有小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%(以干重计)的污染性组分时,所述目标组分可为“大致上纯化的”。因此,“大致上纯化的”目标组分可具有约70%、约75%、约80%、约85%、约90%、约95%、约96%、约97%、约98%、约99%或更高的纯度水平。仅举例来说,天然氨基酸多肽或非天然氨基酸多肽可从天然细胞纯化,或在重组产生的天然氨基酸多肽或非天然氨基酸多肽的情况下从宿主细胞纯化。举例来说,当制剂含有小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%(以干重计)的污染性物质时,天然氨基酸多肽或非天然氨基酸多肽的所述制剂可为“大致上纯化的”。举例来说,当天然氨基酸多肽或非天然氨基酸多肽由宿主细胞重组产生时,所述天然氨基酸多肽或非天然氨基酸多肽可在细胞干重的约30%、约25%、约20%、约15%、约10%、约5%、约4%、约3%、约2%或约1%或更小下存在。举例来说,当天然氨基酸多肽或非天然氨基酸多肽由宿主细胞重组产生时,所述天然氨基酸多肽或非天然氨基酸多肽可在约5g/l、约4g/l、约3g/l、约2g/l、约1g/l、约750mg/l、约500mg/l、约250mg/l、约100mg/l、约50mg/l、约10mg/l或约1mg/l或更低的细胞干重下存在于培养基中。举例来说,“基本上纯化的”天然氨基酸多肽或非天然氨基酸多肽可具有约30%、约35%、约40%、约45%、约50%、约55%、约60%、约65%、约70%、约75%、约80%、约85%、约90%、约95%、约99%或更高的纯度水平,如通过适当的方法所确定的,所述方法包括但不限于sds/page分析、rp
‑
hplc、sec和毛细管电泳。
[0188]
术语“取代基”也称为“非干扰取代基”,是指可以用来代替分子上另一个基团的基团。这样的基团包括但不限于卤代、c1‑
c
10
烷基、c2‑
c
10
烯基、c2‑
c
10
炔基、c1‑
c
10
烷氧基、c5‑
c
12
芳烷基、c3‑
c
12
环烷基、c4‑
c
12
环烯基、苯基、取代的苯基、甲苯酰基(toluolyl)、二甲苯基、联苯基、c2‑
c
12
烷氧烷基、c5‑
c
12
烷氧芳基、c5‑
c
12
芳氧烷基、c7‑
c
12
氧芳基、c1‑
c6烷基亚磺酰基、c1‑
c
10
烷基磺酰基、
‑
(ch2)
m
‑
o
‑
(c1‑
c
10
烷基),其中m是1
‑
8,芳基、取代的芳基、取代的烷氧基、氟烷基、杂环基、取代的杂环基、硝基烷基、
‑
no2、
‑
cn、
‑
nrc(o)
‑
(c1‑
c
10
烷基)、
‑
c(o)
‑
(c1‑
c
10
烷基)、c2‑
c
10
烷基硫代烷基(alkthioalkyl)、
‑
c(o)o
‑
(c1‑
c
10
烷基)、
‑
oh、
‑
so2、=s、
‑
cooh、
‑
nr2、羰基、
‑
c(o)
‑
(c1‑
c
10
烷基)
‑
cf3、
‑
c(o)
‑
cf3、
‑
c(o)nr2、
‑
(c1‑
c
10
芳基)
‑
s
‑
(c6‑
c
10
芳基)、
‑
c(o)
‑
(c6‑
c
10
芳基)、
‑
(ch2)
m
‑
o
‑
(ch2)
m
‑
o
‑
(c1‑
c
10
烷基),其中m各自是1至8,
‑
c(o)nr2、
‑
c(s)nr2、
‑
so2nr2、
‑
nrc(o)nr2、
‑
nrc(s)nr2,其盐,等等。前述列表中的每个r基团包括但不限于h、烷基或取代的烷基、芳基或取代的芳基或烷芳基。在取代基通过它们的常规化学式加以详细说明的情况下,将其从左到右书写,它们同样涵盖从右到左书写结构所产生的化学上相同的取代基,例如,
‑
ch2o
‑
等同于
–
och2‑
。
[0189]
仅举例来说,烷基和杂烷基的取代基(包括那些称为亚烷基、烯基、杂亚烷基、杂烯基、炔基、环烷基、杂环烷基、环烯基和杂环烯基的基团)包括但不限于:
‑
or、=o、=nr、=n
‑
or、
‑
nr2、
‑
sr、
‑
卤素、
‑
sir3、
‑
oc(o)r、
‑
c(o)r、
‑
co2r、
‑
conr2、
‑
oc(o)nr2、
‑
nrc(o)r、
‑
nrc(o)nr2、
‑
nr(o)2r、
‑
nr
‑
c(nr2)=nr、
‑
s(o)r、
‑
s(o)2r、
‑
s(o)2nr2、
‑
nrso2r、
‑
cn和
–
no2。前述列表中的每个r基团包括但不限于氢、取代或未取代的杂烷基、取代或未取代的芳基,包括但不限于被1
‑
3个卤素取代的芳基、取代或未取代的烷基、烷氧基或硫代烷氧基或芳烷基。当两个r基团与同一个氮原子附接时,它们可与氮原子结合形成5
‑
、6
‑
或7
‑
元环。例如,
‑
nr2预期包括但不限于1
‑
吡咯烷基和4
‑
吗啉基。
[0190]
举例来说,芳基和杂芳基的取代基包括但不限于
‑
or、=o、=nr、=n
‑
or、
‑
nr2、
‑
sr、
‑
卤素、
‑
sir3、
‑
oc(o)r、
‑
c(o)r、
‑
co2r、
‑
conr2、
‑
oc(o)nr2、
‑
nrc(o)r、
‑
nrc(o)nr2、
‑
nr(o)2r、
‑
nr
‑
c(nr2)=nr、
‑
s(o)r、
‑
s(o)2r、
‑
s(o)2nr2、
‑
nrso2r、
‑
cn、
–
no2、
‑
r、
‑
n3、
‑
ch(ph)2、氟(c1‑
c4)烷氧基和氟(c1‑
c4)烷基,其数量范围为从零到芳环系上开放化合价的总数;并且其中前述列表中的每个r基团包括但不限于氢、烷基、杂烷基、芳基和杂芳基。
[0191]
如本文所用的,术语“治疗有效量”是指施用于已罹患疾病、病症或疾患的患者的含有至少一种非天然氨基酸多肽和/或至少一种修饰的非天然氨基酸多肽的组合物的量,其足以治愈或至少部分地阻止或在一定程度上减轻被治疗的疾病、疾患或病症的一种或多种症状。所述组合物的有效性取决于各种状况,包括但不限于疾病、病症或疾患的严重性和过程;先前疗法;患者的健康状况和对药物的响应;以及治疗医师的判断。仅举例来说,通过包括但不限于剂量递增临床试验在内的常规实验,可以确定治疗有效量。
[0192]
如本文所用的,术语“硫代烷氧基”是指通过氧原子与分子连接的含硫烷基。
[0193]
如本文所用的术语“毒性部分”或“毒性基团”是指可引起伤害、障碍或死亡的化合物。毒性部分包括但不限于澳瑞他汀(auristatin)、dna小沟结合剂、dna小沟烷基化剂、烯二炔、莱西托辛(lexitropsin)、倍癌霉素、紫杉烷、嘌呤霉素、tlr激动剂、美登木素生物碱(maytansinoid)、长春花生物碱、afp、mmaf、mmae、aeb、aevb、澳瑞他汀e、紫杉醇、多西他赛、cc
‑
1065、sn
‑
38、托泊替康、吗啉代多柔比星、根霉素、氰基吗啉代多柔比星、tlr激动剂
‑
10、棘霉素、考布他汀(combretatstatin)、卡奇霉素(chalicheamicin)、美登素、dm
‑
1、纺锤菌素、鬼臼毒素(例如依托泊苷、替尼泊苷等)、浆果赤霉素(baccatin)及其衍生物、抗微管蛋白剂(anti
‑
tubulin agent)、念珠藻素(cryptophysin)、考布他汀(combretastatin)、澳瑞
他汀e、长春新碱、长春碱、长春地辛、长春瑞滨、vp
‑
16、喜树碱、埃博霉素a、埃博霉素b、诺考达唑、秋水仙碱、秋水仙胺、雌莫司汀、西马多丁、圆皮海绵内酯(discodermolide)、美登素(maytansine)、软珊瑚醇(eleutherobin)、二氯甲基二乙胺(mechlorethamine)、环磷酰胺、美法仑、卡莫司汀、洛莫司汀、司莫司汀、链脲佐菌素(streptozocin)、氯脲菌素、尿嘧啶氮芥、氮芥(chlormethine)、异环磷酰胺、苯丁酸氮芥、哌泊溴烷、三亚乙基蜜胺、三亚乙基硫代磷酸胺、白消安、达卡巴嗪和替莫唑胺、阿糖胞苷(ytarabine)、胞嘧啶阿拉伯糖苷(cytosine arabinoside)、氟尿嘧啶、氟尿苷、6
‑
硫鸟嘌呤、6
‑
巯基嘌呤、喷司他丁、5
‑
氟尿嘧啶、甲氨蝶呤、10
‑
炔丙基
‑
5,8
‑
二脱氮杂叶酸(10
‑
propargyl
‑
5,8
‑
dideazafolate)、5,8
‑
二脱氮杂四氢叶酸(5,8
‑
dideazatetrahydrofolic acid)、甲酰四氢叶酸、磷酸氟达拉滨、喷司他丁、吉西他滨、ara
‑
c、紫杉醇、多西他赛、脱氧柯福霉素(deoxycoformycin)、丝裂霉素
‑
c、l
‑
天冬酰胺酶、硫唑嘌呤、布喹那、抗生素(例如蒽环类药物(anthracycline)、庆大霉素、头孢噻吩、万古霉素、替拉万星(telavancin)、达托霉素、阿奇霉素、红霉素、罗红霉素(rocithromycin)、呋喃唑酮、阿莫西林、氨苄青霉素、羧苄青霉素、氟氯西林、甲氧西林、青霉素、环丙沙星、莫西沙星、氧氟沙星、多西环素、米诺环素、土霉素、四环素、链霉素、利福布丁(rifabutin)、乙胺丁醇、利福昔明,等)、抗病毒药物(例如阿巴卡韦、阿昔洛韦、安普利近(ampligen)、昔多福韦、地拉韦定(delavirdine)、地达诺新(didanosine)、依法韦仑(efavirenz)、恩替卡韦、膦乙醇(fosfonet)、更昔洛韦、伊巴他滨、异丙肌苷(imunovir)、碘苷、肌苷、洛匹那韦、美替沙腙(methisazone)、奈沙韦(nexavir)、奈韦拉平(nevirapine)、奥司他韦、喷昔洛韦、司他夫定、曲氟尿苷(trifluridine)、特鲁瓦达(truvada)、伐昔洛韦、扎那米韦,等)、盐酸柔红霉素、道诺霉素、红比霉素(rubidomycin)、司比定(cerubidine)、伊达比星(idarubicin)、多柔比星、表柔比星和吗啉代衍生物、吩噁嗪酮双环肽(例如更生霉素)、碱性糖肽(例如博来霉素)、蒽醌糖苷(例如普卡霉素、光辉霉素(mithramycin))、蒽二酮(例如米托蒽醌)、氮丙啶吡咯并吲哚二酮(例如丝裂霉素)、大环免疫抑制剂(例如环孢素(cyclosporine)、fk
‑
506、他克莫司、普乐可复(prograf)、雷帕霉素,等)、诺维本(navelbene)、cpt
‑
11、阿那曲唑(anastrazole)、来曲唑(letrazole)、卡培他滨、雷洛昔芬、环磷酰胺、异环磷酰胺、屈洛昔芬(droloxafine)、别秋水仙碱(allocolchicine)、软海绵素b(halichondrin b)、秋水仙碱、秋水仙碱衍生物、美登素、根霉素(rhizoxin)、紫杉醇、紫杉醇衍生物、多西他赛、硫代秋水仙碱、三苯甲基半胱氨酸(trityl cysterin)、硫酸长春碱、硫酸长春新碱、顺铂、卡铂、羟基脲、n
‑
甲基肼、表鬼臼毒素(epidophyllotoxin)、丙卡巴肼、米托蒽醌、甲酰四氢叶酸和替加氟(tegafur)。“紫杉烷”包括紫杉醇以及任何活性紫杉烷衍生物或前药。
[0194]
如本文所用的,术语“治疗(“treat”、“treating”或“treatment”)”包括减轻、缓和或改善疾病或病症的症状;预防另外的症状;改善或预防症状的潜在代谢原因;抑制疾病或病症,例如阻止疾病或病症的发展;减轻疾病或病症;引起疾病或病症消退;减轻由疾病或病症引起的状况;或终止疾病或病症的症状。术语“治疗(“treat”、“treating”或“treatment”)”包括但不限于防治性治疗和/或治疗性治疗。
[0195]
如本文所用的,术语“水溶性聚合物”是指可溶于水性溶剂的任何聚合物。这样的水溶性聚合物包括但不限于聚乙二醇、聚乙二醇丙醛、其单c1‑
c
10
烷氧基或芳氧基衍生物(描述于美国专利第5,252,714号,将其通过引用并入本文)、单甲氧基
‑
聚乙二醇、聚乙烯吡
咯烷酮、聚乙烯醇、聚氨基酸、二乙烯醚马来酸酐、n
‑
(2
‑
羟丙基)
‑
甲基丙烯酰胺、葡聚糖、葡聚糖衍生物(包括硫酸葡聚糖)、聚丙二醇、聚氧化丙烯/氧化乙烯共聚物、聚氧乙烯化多元醇、肝素、肝素片段、多糖、寡糖、聚糖、纤维素和纤维素衍生物(包括但不限于甲基纤维素和羧甲基纤维素)、血清白蛋白、淀粉和淀粉衍生物、多肽、聚亚烷基二醇及其衍生物、聚亚烷基二醇及其衍生物的共聚物、聚乙烯基乙基醚和α
‑
β
‑
聚[(2
‑
羟乙基)
‑
dl
‑
天冬酰胺等等,或其混合物。仅举例来说,这样的水溶性聚合物与天然氨基酸多肽或非天然多肽偶联可导致包括但不限于以下的变化:水溶性增加;血清半衰期延长或被调节;相对于未经修饰形式,治疗半衰期延长或被调节;生物利用度增加;生物活性被调节;循环时间延长;免疫原性被调节;物理缔合特征被调节,包括但不限于聚集和多聚体形成、受体结合改变、活性调节剂、或其他靶向多肽结合、与一种或多种结合配偶体的结合改变、以及靶向多肽受体二聚化或多聚化的改变。另外,这样的水溶性聚合物可以具有或不具有其自身的生物活性,并且可以用作将靶向多肽附接至其他物质的接头,所述其他物质包括但不限于一种或多种靶向多肽或一种或多种生物活性分子。
[0196]
除非另有说明,采用本领域技术之内的质谱法、nmr、hplc、蛋白质化学、生物化学、重组dna技术和药理学的常规方法。
[0197]
本文提供的化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽、修饰的非天然氨基酸多肽和用于产生前述化合物的试剂)包括同位素标记化合物,其与本文提供的不同的化学式和结构中列举的那些化合物相同,但事实上一个或多个原子被具有不同于通常在自然界发现的原子质量或质量数的原子质量或质量数的原子代替。可掺入到本文化合物中的同位素的实例包括氢、碳、氮、氧、氟和氯的同位素,比如分别为2h、3h、
13
c、
14
c、
15
n、
18
o、
17
o、
35
s、
18
f、
36
cl。本文所述的某些同位素标记的化合物(例如其中掺入放射性同位素比如3h和
14
c的那些化合物)在药物和/或底物组织分布测定法中是有用的。此外,用诸如氘(即2h)的同位素取代可以提供由于代谢稳定性更高而产生的某些治疗优势,例如体内半衰期延长或剂量要求降低。
[0198]
本文的一些化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽以及用于产生前述化合物的试剂)具有不对称碳原子,因此可以作为对映体或非对映体存在。基于其物理化学差异,通过已知的方法,例如通过色谱法和/或分步结晶,可将非对映体混合物分离为它们单独的非对映体。通过与适当的旋光化合物(例如,醇)反应将对映体混合物转化成非对映体混合物,分离非对映体,以及将单独的非对映体转化(例如,水解)成相应的纯对映体,由此可以分离对映体。所有这样的异构体(包括非对映体、对映体及其混合物)被认为是本文所述组合物的一部分。
[0199]
在另外的或进一步的实施方案中,本文所述的化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽,以及用于产生前述化合物的试剂)以前药的形式使用。在另外的或进一步的实施方案中,本文所述的化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽,以及用于产生前述化合物的试剂)在施用于有需要的生物体后被代谢而产生代谢物,然后所述代谢物被用于产生期望的效果,包括期望的治疗效果。在进一步或另外的实施方案中是非天然氨基酸和“经修饰或未经修饰”的非天然氨基酸多肽的活性代谢物。
[0200]
本文所述的方法和制剂包括使用非天然氨基酸、非天然氨基酸多肽和修饰的非天
然氨基酸多肽的n氧化物、结晶形式(也称为多晶型物)或药学上可接受的盐。在某些实施方案中,非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽可以作为互变异构体存在。所有互变异构体都被包括在本文提供的非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽的范围内。另外,本文所述的非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽可以与药学上可接受的溶剂(比如水、乙醇等)以非溶剂化和溶剂化的形式存在。本文提供的非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽的溶剂化形式也被认为在本文中公开。
[0201]
本文的一些化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽以及用于产生前述化合物的试剂)可以以几种互变异构形式存在。所有这样的互变异构体形式都被认为是本文所述组合物的一部分。并且,例如,本文任何化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽以及用于产生前述化合物的试剂)的所有烯醇
‑
酮形式都被认为是本文所述组合物的一部分。
[0202]
本文的一些化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽以及用于产生前述任一化合物的试剂)是酸性的,并且可以与药学上可接受的阳离子形成盐。本文的一些化合物(包括但不限于非天然氨基酸、非天然氨基酸多肽和修饰的非天然氨基酸多肽以及用于产生前述化合物的试剂)可以是碱性的,并且因此可以与药学上可接受的阴离子形成盐。所有这样的盐,包括二盐在内,都在本文所述组合物的范围内,并且它们可以通过常规方法进行制备。例如,可以通过在水性、非水性或部分水性介质中接触酸性和碱性实体来制备盐。通过使用以下技术中的至少一种来回收盐:过滤,用非溶剂沉淀,然后过滤,蒸发溶剂,或者,在水溶液的情况下,冻干。
[0203]
当存在于母体非天然氨基酸多肽中的酸性质子被金属离子例如碱金属离子、碱土金属离子或铝离子取代或与有机碱配位时,可以形成本文公开的非天然氨基酸多肽的药学上可接受的盐。另外,可以使用起始原料或中间体的盐来制备所公开的非天然氨基酸多肽的盐形式。通过本文所述的非天然氨基酸多肽的游离碱形式与药学上可接受的无机酸或有机酸反应,可以将本文所述的非天然氨基酸多肽制成药学上可接受的酸加成盐(其为药学上可接受的盐的一种类型)。可替代地,通过本文所述的非天然氨基酸多肽的游离酸形式与药学上可接受的无机碱或有机碱反应,可以将本文所述的非天然氨基酸多肽制成药学上可接受的碱加成盐(其为药学上可接受的盐的一种类型)。
[0204]
药学上可接受的盐的类型包括但不限于:(1)酸加成盐,与无机酸比如盐酸、氢溴酸、硫酸、硝酸、磷酸等形成;或与有机酸比如乙酸、丙酸、己酸、环戊烷丙酸、乙醇酸、丙酮酸、乳酸、丙二酸、琥珀酸、苹果酸、马来酸、富马酸、酒石酸、柠檬酸、苯甲酸、3
‑
(4
‑
羟基苯甲酰基)苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、1,2
‑
乙二磺酸、2
‑
羟基乙磺酸、苯磺酸、2
‑
萘磺酸、4
‑
甲基双环
‑
[2.2.2]辛
‑2‑
烯
‑1‑
羧酸、葡庚糖酸、4,4'
‑
亚甲基双
‑
(3
‑
羟基
‑2‑
烯
‑1‑
甲酸)、3
‑
苯基丙酸、三甲基乙酸、叔丁基乙酸、十二烷基硫酸、葡萄糖酸、谷氨酸、羟基萘甲酸、水杨酸、硬脂酸、粘康酸等形成;(2)当母体化合物中存在的酸性质子被金属离子例如碱金属离子、碱土金属离子或铝离子替代或与有机碱配位时形成的盐。可接受的有机碱包括乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、n
‑
甲基葡糖胺等。可接受的无机碱包括氢氧化铝、氢氧化钙、氢氧化钾、碳酸钠、氢氧化钠等。
[0205]
使用各种方法,包括但不限于,离子交换色谱法、离子色谱法、毛细管电泳、电感耦
合等离子体、原子吸收光谱法、质谱法或其任何组合,可以分析和鉴定非天然氨基酸多肽的药物可接受盐的相应抗衡离子。另外,使用实施例中描述的技术和方法,可以测试这样的非天然氨基酸多肽的药物可接受盐的治疗活性。
[0206]
应当理解的是,一种盐的提及包括溶剂加成形式或其晶体形式,特别是溶剂合物或多晶型物。溶剂合物含有化学计量和非化学计量的溶剂,并常常在与药学上可接受的溶剂(比如水、乙醇等)一起结晶的过程中形成。当溶剂为水时形成水合物,或者,当溶剂为醇时形成醇化物。多晶型物包括化合物的相同元素组成的不同的晶体堆积排列。多晶型物通常具有不同的x射线衍射图、红外光谱、熔点、密度、硬度、晶形、光学和电学性质、稳定性和溶解度。各种因素比如重结晶溶剂、结晶速率和储存温度可引起单晶形式占优势。
[0207]
使用多种技术,包括但不限于热分析、x射线衍射、光谱学、蒸汽吸附和显微镜检查,可以完成非天然氨基酸多肽的药物可接受盐多晶型物和/或溶剂合物的筛选和表征。热分析方法着手解决热化学降解或热物理过程,包括但不限于多晶型转变,并且这样的方法用于分析多晶型之间的关系、确定重量损失、寻找玻璃化转变温度或用于赋形剂相容性研究。这样的方法包括但不限于差示扫描量热法(dsc)、调制差示扫描量热法(mdcs)、热重分析(tga)和热重
‑
红外分析(tg/ir)。x射线衍射法包括但不限于单晶和粉末衍射仪以及同步加速器源。使用的各种光谱技术包括但不限于拉曼(raman)、ftir、uvis和nmr(液态和固态)。各种显微技术包括但不限于偏振光显微术、具有能量色散x射线分析(edx)的扫描电子显微术(sem)、具有edx的环境扫描电子显微术(在气体或水蒸气气氛中)、ir显微术和拉曼显微术。
[0208]
虽然本文显示并描述了本发明的优选实施方案,但本领域技术人员显而易见的是,这样的实施方案仅作为举例而提供。在不脱离本发明的情况下,众多改变、变化和替代现将为本领域技术人员所想到。应了解本文所述的本发明的实施方案的各种替代方案可用于实施本发明。预期的是,以下权利要求限定本发明的范围,并且由此涵盖这些权利要求的范围内的方法和结构以及它们的等同物。
[0209]
tlr激动剂接头衍生物
[0210]
在一个层面上,本文描述了用于产生和使用包含至少一个非天然氨基酸或具有羰基、二羰基、肟基或羟胺基的经修饰的非天然氨基酸的tc或类似物的靶向多肽的工具(方法、组合物、技术)。这样的包含非天然氨基酸的tc的靶向多肽可含有其他官能性,包括但不限于聚合物;水溶性聚合物;聚乙二醇的衍生物;第二蛋白质或多肽或多肽类似物;抗体或抗体片段;以及它们的任何组合。注意的是,各种以上提及的官能性不意图暗示一种官能性的成员不能被分类为另一官能性的成员。实际上,视特定情况而定,将存在重叠。仅举例来说,水溶性聚合物在范围方面与聚乙二醇衍生物重叠,然而,重叠不是完全的,因此两种官能性均在以上加以引用。
[0211]
一方面是用于选择和设计tlr激动剂接头衍生物以及靶向多肽的方法,以及将使用本文所述的方法、组合物和技术修饰所述靶向多肽。新的tlr激动剂接头衍生物和靶向多肽可被从头设计,仅举例来说,包括被设计为高通量筛选过程(在所述情况下,可设计、合成、表征和/或测试多种多肽)的一部分,或基于研究者的兴趣进行设计。还可基于已知或部分表征的多肽的结构来设计新的tlr接头衍生物和靶向多肽。仅举例来说,tlr激动剂已经是科学界深入研究的主题;可以基于tlr激动剂的结构设计新的化合物。本文单独描述了选
择取代和/或修饰哪些氨基酸的原则。本文还描述了采用哪种修饰的选择,并且可以将其用于满足实验者或最终用户的需要。这样的需要可包括但不限于,操纵多肽的治疗有效性,改进多肽的安全性概况,调整多肽的药代动力学、药理学和/或药效学,比如仅举例来说,增加水溶性、生物利用度,延长血清半衰期,延长治疗半衰期,调节免疫原性,调节生物活性,或延长循环时间。另外,仅举例来说,这样的修饰包括对多肽提供另外的官能性,掺入抗体,以及前述修饰的任何组合。
[0212]
本文还描述了tlr激动剂接头衍生物和靶向多肽,其具有或可被修饰为含有肟基、羰基、二羰基或羟胺基。关于这个方面,包括生产、纯化、表征和使用这样的tlr激动剂接头衍生物和靶向多肽的方法。
[0213]
tlr激动剂接头衍生物或靶向多肽可含有至少一个、至少两个、至少三个、至少四个、至少五个、至少六个、至少七个、至少八个、至少九个、或十个或更多个羰基或二羰基、肟基、羟胺基或它们的保护形式。tlr激动剂接头衍生物或靶向多肽可以是相同的或不同的,例如在包含1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个不同的反应性基团的衍生物中,可以存在1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个不同位点。
[0214]
如本文所述,本公开提供了与另一分子偶联的的靶向多肽,其具有式“靶向多肽
‑
l
‑
m”,其中l是连接基团或化学键,并且m是任何其他分子,包括但不限于另一个靶向多肽。在一些实施方案中,l是体内稳定的。在一些实施方案中,l是体内可水解的。在一些实施方案中,l是体内亚稳态的。
[0215]
使用本领域技术人员已知的标准连接剂和程序,可以通过l将靶向多肽和m连接在一起。在一些方面,靶向多肽和m直接融合,并且l是键。在其他方面,靶向多肽和m通过连接基团l融合。例如,在一些实施方案中,靶向多肽和m通过肽键,任选地通过肽或氨基酸间隔物连接在一起。在一些实施方案中,靶向多肽和m通过化学缀合,任选地通过连接基团(l)连接在一起。在一些实施方案中,l直接与靶向多肽和m中的每一者缀合。
[0216]
通过一种化合物的亲核反应性基团与另一种化合物的亲电反应性基团反应,可以发生化学缀合。在一些实施方案中,当l是键时,通过使靶向多肽上的亲核反应性部分与y上的亲电反应性部分反应,或者通过使靶向多肽上的亲电反应性部分与m上的亲核反应性部分反应,靶向多肽与m缀合。在实施方案中,当l是将靶向多肽和m连接在一起的基团时,通过靶向多肽和/或m上的亲核反应性部分与l上的亲电反应性部分反应,或者通过靶向多肽和/或m上的亲电反应性部分与l上的亲核反应性部分反应,靶向多肽和/或m可以与l缀合。亲核反应性基团的非限制性实例包括氨基、硫醇和羟基。亲电反应性基团的非限制性实例包括羧基、酰氯、酸酐、酯、琥珀酰亚胺酯、烷基卤化物、磺酸酯、马来酰亚胺基、卤代乙酰基和异氰酸酯。在靶向多肽和m通过羧酸与胺反应而缀合在一起的实施方案中,可使用活化剂来形成羧酸的活化酯。
[0217]
羧酸的活化酯可以是,例如,n
‑
羟基琥珀酰亚胺(nhs)、甲苯磺酸酯(tos)、甲磺酸酯、三氟甲磺酸酯、碳二亚胺或六氟磷酸盐。在一些实施方案中,碳二亚胺是1,3
‑
二环己基碳二亚胺(dcc)、1,1
’‑
羰基二咪唑(cdi)、1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳二亚胺盐酸盐(edc)或1,3
‑
二异丙基碳二亚胺(dicd)。在一些实施方案中,六氟磷酸盐选自六氟磷酸苯并三唑
‑1‑
基
‑
氧基
‑
三(二甲基氨基)磷鎓六氟磷酸盐(bop)、苯并三唑
‑1‑
基
‑
氧基三吡咯烷磷
鎓六氟磷酸盐(pybop)、2
‑
(lh
‑7‑
氮杂苯并三唑
‑1‑
基)
‑
1,1,3,3
‑
四甲基脲阳离子六氟磷酸盐(hatu)和o
‑
苯并三唑
‑
n,n,n',n'
‑
四甲基脲阳离子
‑
六氟磷酸盐(hbtu)。
[0218]
在一些实施方案中,靶向多肽包含能够与m或l上的亲电反应性基团缀合的亲核反应性基团(例如赖氨酸、半胱氨酸或丝氨酸侧链的氨基、巯基或羟基)。在一些实施方案中,靶向多肽包含能够与m或l上的亲核反应性基团缀合的亲电反应性基团(例如asp或glu侧链的羧酸酯基团)。在一些实施方案中,将靶向多肽化学修饰为包含能够直接与m或与l缀合的反应基团。在一些实施方案中,靶向多肽在n端或c端被修饰,以包含具有亲核侧链的天然或非天然氨基酸。在示例性实施方案中,靶向多肽的n端或c端氨基酸选自由以下组成的组:赖氨酸、鸟氨酸、丝氨酸、半胱氨酸和同型半胱氨酸。例如,可以将靶向多肽的n端或c端氨基酸修饰为包含赖氨酸残基。在一些实施方案中,靶向多肽在n端或c端氨基酸被修饰为包含具有亲电侧链的天然或非天然氨基酸,例如asp和glu。在一些实施方案中,靶向多肽的内部氨基酸被如前所述具有亲核侧链的天然或非天然氨基酸取代。在示例性实施方案中,被取代的靶向多肽的内部氨基酸选自由以下组成的组:赖氨酸、鸟氨酸、丝氨酸、半胱氨酸和同型半胱氨酸。例如,靶向多肽的内部氨基酸可以被赖氨酸残基取代。在一些实施方案中,靶向多肽的内部氨基酸被具有亲电侧链的天然或非天然氨基酸例如asp和glu取代。
[0219]
在一些实施方案中,m包含能够直接与靶向多肽或与l缀合的反应性基团。在一些实施方案中,m包含能够与靶向多肽或l上的亲电反应性基团缀合的亲核反应性基团(例如胺、硫醇、羟基)。在一些实施方案中,m包含能够与靶向多肽或l上的亲核反应性基团缀合的亲电反应性基团(例如羧基、羧基的活化形式、具有离去基团的化合物)。在一些实施方案中,将m化学修饰为包含能够与靶向多肽或l上的亲电反应性基团缀合的亲核反应性基团。在一些实施方案中,将m化学修饰为包含能够与靶向多肽或l上的亲核反应性基团缀合的亲电反应性基团。
[0220]
在一些实施方案中,通过有机硅烷,例如用戊二醛处理的氨基硅烷;硅烷醇基的羰基二咪唑(cdi)活化;或利用树枝状聚合物,可以进行缀合。多种树枝状聚合物是本领域已知的,包括聚(酰胺胺)(pamam)树枝状聚合物,其通过发散法从氨或乙二胺引发剂核心试剂开始合成;基于三氨基乙烯亚胺核的pamam树枝状聚合物的亚类;径向分层的聚(酰胺胺
‑
有机硅)树枝状聚合物(pamamos),其为由亲水性、亲核性聚酰胺胺(pamam)内部和疏水性有机硅(os)外部组成的反向单分子胶束;聚(丙烯亚胺)(ppi)树枝状聚合物,其通常是具有伯胺作为端基的聚烷基胺,同时树枝状聚合物内部由许多叔三丙烯胺组成;聚丙烯胺(popam)树枝状聚合物;二氨基丁烷(dab)树枝状聚合物;两亲性树枝状聚合物;胶束树枝状聚合物,其为水溶性超支化聚亚苯基的单分子胶束;聚赖氨酸树枝状聚合物;和基于聚苄基醚超支化骨架的树枝状聚合物。
[0221]
在一些实施方案中,可通过烯烃复分解反应进行缀合。在一些实施方案中,m和靶向多肽、m和l、或靶向多肽和l两者都包含能够经历复分解反应的烯烃或炔部分。在一些实施方案中,使用适合的催化剂(例如铜、钌)来加速复分解反应。本领域描述了进行烯烃复分解反应的适合方法。参见,例如schafmeister等人,j.am.chem.soc.122:5891
‑
5892(2000),walensky等人,science 305:1466
‑
1470(2004),和blackwell等人,angew,chem.,int.ed.37:3281
‑
3284(1998)。
[0222]
在一些实施方案中,可以使用点击化学进行缀合。“点击反应”范围广且易于执行,
仅使用容易获得的试剂,并且对氧气和水不敏感。在一些实施方案中,点击反应是在炔基和叠氮基之间形成三唑基的环加成反应。在一些实施方案中,点击反应使用铜或钌催化剂。本领域描述了进行点击反应的适合方法。参见,例如kolb等人,drug discovery today 8:1128(2003);kolb等人,angew.chem.int.ed.40:2004(2001);rostovtsev等人,angew.chem.int.ed.41:2596(2002);tornoe等人,j.org.chem.67:3057(2002);manetsch等人,j.am.chem.soc.126:12809(2004);lewis等人,angew.chem.int.ed.41:1053(2002);speers,j.am.chem.soc.125:4686(2003);chan等人,org.lett.6:2853(2004);zhang等人,j.am.chem.soc.127:15998(2005);和waser等人,j.am.chem.soc.127:8294(2005)。
[0223]
还考虑了通过高亲和力特异性结合配偶体例如链霉亲和素/生物素或亲和素/生物素或凝集素/碳水化合物的间接缀合。
[0224]
在一些实施方案中,用有机衍生剂将靶向多肽和/或m官能化为包含亲核反应性基团或亲电反应性基团。这种衍生剂能够与靶向多肽上的靶向氨基酸的选定侧链或n端或c端残基以及m上的官能团反应。靶向多肽和/或m上的反应性基团包括例如醛基、氨基、酯基、硫醇、α
‑
卤代乙酰基、马来酰亚胺基或肼基。衍生剂包括,例如,马来酰亚胺苯甲酰磺基琥珀酰亚胺酯(通过半胱氨酸残基缀合)、n
‑
羟基琥珀酰亚胺(通过赖氨酸残基)、戊二醛、琥珀酸酐或本领域已知的其他剂。可替代地,靶向多肽和/或m可以通过中间载体比如多糖或多肽载体间接相互连接。多糖载体的实例包括氨基葡聚糖。适合的多肽载体的实例包括聚赖氨酸、聚谷氨酸、聚天冬氨酸、它们的共聚物,以及这些氨基酸和其他氨基酸例如丝氨酸的混合聚合物,从而对所得负载载体赋予期望的溶解性。
[0225]
半胱氨酰残基最通常与a
‑
卤代乙酸酯(和相应的胺)比如氯乙酸或氯乙酰胺反应,得到羧甲基或羧酰胺甲基衍生物。还通过与溴三氟丙酮、α
‑
溴
‑
β
‑
(5
‑
咪唑基(imidozoyl))丙酸、氯乙酰磷酸酯、n
‑
烷基马来酰亚胺、3
‑
硝基
‑2‑
吡啶基二硫化物、甲基
‑2‑
吡啶基二硫化物、对氯汞基苯甲酸盐、2
‑
氯汞基
‑4‑
硝基苯酚或氯
‑7‑
硝基苯并
‑2‑
氧杂
‑
1,3
‑
二唑反应,将半胱氨酰残基衍生化。
[0226]
通过在ph 5.5
‑
7.0下与焦碳酸二乙酯反应将组氨酰残基衍生化,因为这种剂对组氨酸侧链是相对特异的。对溴苯酰基溴也是有用的;该反应优选在ph 6.0的0.1m二甲胂酸钠中进行。
[0227]
使赖氨酰和氨基末端残基与琥珀酸或其他羧酸酐反应。用这些试剂衍生具有逆转赖氨酰残基电荷的作用。用于衍生含α
‑
氨基的残基的其他适合的试剂包括酰亚胺酯,比如甲基吡啶亚胺甲酯、磷酸吡哆醛、吡哆醛、氯硼氢化物、三硝基苯磺酸、o
‑
甲基异脲、2,4
‑
戊二酮,以及转氨酶催化的与乙醛酸酯的反应。
[0228]
通过与一种或几种常规试剂反应来修饰精氨酰残基,其中所述试剂为苯乙二醛、2,3
‑
丁二酮、1,2
‑
环己二酮和茚三酮。精氨酸残基的衍生化要求该反应在碱性条件下进行,因为胍官能团具有高pka。此外,这些试剂可与赖氨酸基团以及精氨酸ε
‑
氨基基团反应。
[0229]
可以对酪氨酰残基进行特异性修饰,特别感兴趣的是通过与芳香重氮化合物或四硝基甲烷反应,将光谱标记物引入到酪氨酰残基中。最常见的是,分别使用n
‑
乙酰基咪唑(n
‑
acetylimidizole)和四硝基甲烷形成o
‑
乙酰酪氨酰种类和3
‑
硝基衍生物。
[0230]
羧基侧基(天冬氨酰基或谷氨酰基)通过与碳二亚胺(r
‑
n=c=n
‑
r’)反应而被选择性修饰,其中r和r’是不同的烷基,比如1
‑
环己基
‑3‑
(2
‑
吗啉基
‑4‑
乙基)碳二亚胺或1
‑
乙
基
‑3‑
(4
‑
氮阳离子
‑
4,4
‑
二甲基戊基)碳二亚胺。此外,天冬氨酰基和谷氨酰基残基通过与铵离子反应转化为天冬酰胺酰基和谷氨酰胺酰基残基。
[0231]
其他修饰包括脯氨酸和赖氨酸的羟基化、丝氨酰或苏氨酰残基的羟基的磷酸化、赖氨酸、精氨酸和组氨酸侧链的α
‑
氨基的甲基化(t.e.creighton,proteins:structure and molecular properties,w.h.freeman&co.,san francisco,第79
‑
86页(1983))、天冬酰胺或谷氨酰胺的脱酰胺、n端胺的乙酰化和/或c端羧酸基团的酰胺化或酯化。
[0232]
另一种类型的共价修饰涉及将糖苷与肽化学偶联或酶促偶联。糖可以附接至(a)精氨酸和组氨酸,(b)游离羧基,(c)游离巯基,比如半胱氨酸的巯基,(d)游离羟基,比如丝氨酸、苏氨酸或羟脯氨酸的羟基,(e)芳香残基,比如酪氨酸或色氨酸的残基,或(f)谷氨酰胺的酰胺基。这些方法描述于wo1987/05330并且见于aplin和wriston,crc crit.rev.biochem.,第259
‑
306页(1981)。
[0233]
在一些实施方案中,l是键。在这些实施方案中,通过使靶向多肽上的亲核反应性部分与m上的亲电反应性部分反应,将靶向多肽和m缀合在一起。在替代实施方案中,通过使靶向多肽上的亲电反应性部分与m上的亲核部分反应,将靶向多肽和m缀合在一起。在示例性实施方案中,l是在靶向多肽上的胺(例如赖氨酸残基的ε
‑
胺)与m上的羧基反应时形成的酰胺键。在替代实施方案中,靶向多肽和/或m是在缀合之前用衍生剂衍生的。
[0234]
在一些实施方案中,l是连接基团。在一些实施方案中,l是双官能接头,并且在与靶向多肽和m缀合之前仅包含两个反应性基团。在靶向多肽和m两者都具有亲电反应性基团的实施方案中,l在与靶向多肽和m缀合之前包含两个相同或两个不同的亲核基团(例如胺、羟基、硫醇)。在靶向多肽和m两者都具有亲核反应性基团的实施方案中,l在与靶向多肽和m缀合之前包含两个相同或两个不同的亲电基团(例如羧基、羧基的活化形式、具有离去基团的化合物)。在靶向多肽或m之一具有亲核反应性基团并且靶向多肽或m的另一者具有亲电反应性基团的实施方案中,l在与靶向多肽和m缀合之前包含一个亲核反应性基团和一个亲电基团。
[0235]
l可以是具有能够与靶向多肽和m中的每一个反应的至少两个反应性基团(在与靶向多肽和m缀合之前)的任何分子。在一些实施方案中,l仅具有两个反应性基团并且是双官能的。l(在与肽缀合之前)可以由式vi表示:
[0236][0237]
其中a和b独立地是亲核或亲电反应性基团。在一些实施方案中,a和b两者都是亲核基团,或两者都是亲电基团。在一些实施方案中,a或b中的一者是亲核基团,并且a或b中的另一者是亲电基团。a和b的非限制性组合在下表1中示出。
[0238]
表1:亲核和亲电基团的非限制性组合
[0239]
[0240]
[0241]
[0242][0243]
在一些实施方案中,a和b可包括适合于烯烃复分解反应的烯烃和/或炔官能团。在一些实施方案中,a和b包括适合点击化学的部分(例如烯烃、炔烃、腈、叠氮化物)。反应性基团(a和b)的其他非限制性实例包括吡啶基二硫醇、芳基叠氮化物、双吖丙啶(diazirine)、碳二亚胺和酰肼。
[0244]
在一些实施方案中,l是疏水的。疏水接头是本领域已知的。参见,例如bioconjugate techniques,g.t.hermanson(academic press,san diego,ca,1996),将其通过引用完整并入本文。本领域已知的适合的疏水连接基团包括,例如,8
‑
羟基辛酸和8
‑
巯基辛酸。在与组合物的肽缀合之前,疏水连接基团包括至少两个反应性基团(a和b),如本文所述并且如下所示:
[0245][0246]
在一些实施方案中,疏水连接基团包含马来酰亚胺基或碘代乙酰基以及羧酸或活化羧酸(例如nhs酯)作为反应性基团。在这些实施方案中,使用或不使用偶联剂,马来酰亚胺基或碘代乙酰基可以与靶向多肽或m上的硫醇部分偶联,并且羧酸或活化羧酸可以与靶向多肽或m上的胺偶联。可以使用本领域技术人员已知的任何偶联剂,例如,dcc、dic、hatu、hbtu、tbtu和本文所述的其他活化剂,将羧酸与游离胺偶联。在具体实施方案中,亲水连接基团包含具有2至100个亚甲基的脂肪链,其中a和b是羧基或其衍生物(例如琥珀酸)。在其他具体实施方案中,l是碘乙酸。
[0247][0248]
在一些实施方案中,连接基团是亲水的,例如像聚亚烷基二醇。在与组合物的肽缀合之前,亲水连接基团包括至少两个反应性基团(a和b),如本文所述并且如下所示:
[0249][0250]
在一些实施方案中,接头是聚乙二醇(peg)。在某些实施方案中,peg具有约100道尔顿至约10,000道尔顿,例如约500道尔顿至约5000道尔顿的分子量。在一些实施方案中,peg具有约10,000道尔顿至约40,000道尔顿的分子量。
[0251]
在一些实施方案中,亲水连接基团包含马来酰亚胺基或碘代乙酰基以及羧酸或活化羧酸(例如nhs酯)作为反应性基团。在这些实施方案中,使用或不使用偶联剂,马来酰亚胺基或碘代乙酰基可以与靶向多肽或m上的硫醇部分偶联,并且羧酸或活化羧酸可以与靶向多肽或m上的胺偶联。可以使用本领域技术人员已知的任何适当的偶联剂,例如,dcc、dic、hatu、hbtu、tbtu和本文所述的其他活化剂,将羧酸与胺偶联。在一些实施方案中,连接基团是马来酰亚胺基
‑
聚合物(20
‑
40kda)
‑
cooh、碘代乙酰基
‑
聚合物(20
‑
40kda)
‑
cooh、马来酰亚胺基
‑
聚合物(20
‑
40kda)
‑
nhs或碘代乙酰基
‑
聚合物(20
‑
40kda)
‑
nhs。
[0252]
在一些实施方案中,连接基团由氨基酸、二肽、三肽或多肽组成,其中氨基酸、二肽、三肽或多肽包含如本文所述的至少两个活化基团。在一些实施方案中,连接基团(l)包含选自由以下组成的组的部分:氨基、醚、硫醚、马来酰亚胺基、二硫化物、酰胺、酯、硫酯、烯烃、环烯烃、炔烃、三唑基(trizoyl)、氨基甲酸酯、碳酸酯、组织蛋白酶b
‑
可裂解的和腙。
[0253]
在一些实施方案中,l包含1个至约60个原子、或1个至30个原子或更长、2个至5个原子、2个至10个原子、5个至10个原子、或10个至20个原子长的链。在一些实施方案中,链原子全部是碳原子。在一些实施方案中,接头主链中的链原子选自c、o、n和s。链原子和接头可以根据它们的预期溶解度(亲水性)加以选择,从而提供更易溶解的缀合物。在一些实施方案中,l提供了通过酶或其他催化剂或在靶组织或器官或细胞中发现的水解条件而被裂解的官能团。在一些实施方案中,l的长度足够长,以降低空间位阻的可能性。
[0254]
在一些实施方案中,l在生物流体比如血液或血液级分中是稳定的。在一些实施方案中,l在血清中稳定持续至少5分钟,例如当在血清中孵育5分钟的时段时,少于25%、20%、15%、10%或5%的缀合物被裂解。在其他实施方案中,l在血清中稳定持续至少10分钟、或20分钟、或25分钟、或30分钟、或60分钟、或90分钟、或120分钟,或3小时、4小时、5小时、6小时、7小时、8小时、9小时、10小时、12小时、15小时、18小时或24小时。在这些实施方案中,l不包含能够经历体内水解的官能团。在一些示例性实施方案中,l在血清中稳定持续至少约72小时。不能经历显著体内水解的官能团的非限制性实例包括酰胺、醚和硫醚。例如,以下化合物不经历显著体内水解:
[0255][0256]
在一些实施方案中,l是体内可水解的。在这些实施方案中,l包含能够经历体内水解的官能团。能够经历体内水解的官能团的非限制性实例包括酯、酸酐和硫酯。例如,以下化合物能够经历体内水解,因为它包含酯基:
[0257][0258]
在一些示例性实施方案中,l是不稳定的,并且在37℃血浆中在3小时内经历大量水解;在6小时内完全水解。在一些示例性实施方案中,l不是不稳定的。
[0259]
在一些实施方案中,l是体内亚稳态的。在这些实施方案中,l包含任选地经过一段时间能够化学或酶促地被体内裂解的官能团(例如,酸不稳定、还原不稳定或酶不稳定的官能团)。在这些实施方案中,l可包含例如腙部分、二硫化物部分或组织蛋白酶可裂解部分。当l是亚稳态时,并且不希望受任何特定理论的束缚,靶向多肽
‑
l
‑
m缀合物在细胞外环境中是稳定的,例如,在上述时间段内在血清中是稳定的,但是在细胞内环境或模拟细胞内环境的条件下是不稳定的,从而它在进入细胞后裂解。在一些实施方案中,当l是亚稳态时,l在血清中稳定持续至少约24小时、25小时、26小时、27小时、28小时、29小时、30小时、31小时、32小时、33小时、34小时、35小时、36小时、42小时或48小时,例如至少约48小时、54小时、60小时、66小时或72小时,或约24
‑
48小时、48
‑
72小时、24
‑
60小时、36
‑
48小时、36
‑
72小时或48
‑
72小时。
[0260]
在另一个实施方案中,本发明的聚合物衍生物包含具有以下结构的聚合物主链:
[0261]
x—ch2ch2o
‑‑
(ch2ch2o)
n
‑‑
ch2ch2–
o
‑
(ch2)
m
‑
w
‑
n=n=n,其中:
[0262][0263]
w是包含1
‑
10个碳原子的脂肪族或芳香族接头部分;
[0264]
n为1至约4000;并且x是如上所述的官能团;m介于1和10之间。
[0265]
可以通过本领域已知和/或本文公开的多种方法制备本发明的含叠氮化物的聚合物衍生物。在下文所示的一种方法中,具有约800da至约100,000da的平均分子量的水溶性聚合物主链与叠氮化物阴离子(其可与许多适合的反离子中的任何反离子配对,包括钠、钾、叔丁基铵等)反应,所述聚合物主链具有与第一官能团键合的第一末端和与适合的离去基团键合的第二末端。离去基团经历亲核置换并被叠氮化物部分代替,从而提供期望的含叠氮化物的聚合物聚合物;
[0266]
x
‑
聚合物
‑
ly n3‑
→
x
‑
聚合物
‑
l n3[0267]
如所展示的,用于本发明的适合的聚合物主链具有式x
‑
聚合物
‑
ly,其中聚合物是聚(乙二醇),x是不与叠氮化物基团反应的官能团,并且y是适合的离去基团。适合的官能团的实例包括但不限于羟基、保护的羟基、缩醛、烯基、胺、氨基氧基、保护的胺、保护的酰肼、保护的硫醇、羧酸、保护的羧酸、马来酰亚胺、二硫代吡啶和乙烯基吡啶以及酮。适合的离去基团的实例包括但不限于氯化物、溴化物、碘化物、甲磺酸酯、三氟乙基磺酸酯(tresylate)和甲苯磺酸酯。
[0268]
在制备本发明的含叠氮化物的聚合物衍生物的另一种方法中,带有叠氮化物官能
性的连接剂与具有约800da至约100,000da平均分子量的水溶性聚合物主链接触,其中所述连接剂带有化学官能性,所述化学官能性将选择性地与聚合物上的化学官能性反应,形成含叠氮化物的聚合物衍生物产物,其中所述叠氮化物通过连接基团与聚合物主链分开。
[0269]
示例性反应方案如下所示:
[0270]
x
‑
聚合物
‑
y n
‑
接头
‑
n=n=n
→
pg
‑
x
‑
聚合物
‑
接头
‑
n=n=n其中:
[0271]
聚合物是聚(乙二醇),并且x是封端基团,比如烷氧基或如上所述的官能团;并且y是不与叠氮化物官能性反应但将与n官能团有效且选择性地反应的官能团。
[0272]
适合的官能团的实例包括,但不限于,如果n是胺,则y是羧酸、碳酸酯或活性酯;如果n是酰肼或氨基氧基部分,则y是酮;如果n是亲核试剂,则y是离去基团。如果需要,可以通过已知的方法完成粗产物的纯化,所述方法包括但不限于产物的沉淀,随后进行色谱法。
[0273]
在聚合物二胺的情况下,更具体的实例如下所示,其中所述胺之一受到保护基团部分比如叔丁基
‑
boc的保护,并且所得的单保护聚合物二胺与带有叠氮化物官能团的连接部分反应:bochn
‑
聚合物
‑‑
nh2 ho2c
‑
(ch2)3‑
n=n=n。
[0274]
在这种情况下,可以使用多种活化剂比如亚硫酰氯或碳二亚胺试剂和n
‑
羟基琥珀酰亚胺或n
‑
羟基苯并三唑将胺基偶联到羧酸基上,从而在单胺聚合物衍生物和带有叠氮化物的接头部分之间产生酰胺键。在酰胺键成功形成之后,所得的n
‑
叔丁基
‑
boc保护的含叠氮化物衍生物可以直接用于修饰生物活性分子,或者可以被进一步加工,以便安装其他有用的官能团。例如,可以通过用强酸处理来水解n
‑
t
‑
boc基团,以生成ω
‑
氨基
‑
聚合物
‑
叠氮化物。所得的胺可用作合成把手(synthetic handle)来安装其他有用的官能性,比如马来酰亚胺基团、活化的二硫化物、活化酯等,以产生有价值的异双官能试剂。
[0275]
当期望将不同的分子与聚合物的每个末端附接时,异双官能衍生物是特别有用的。例如,ω
‑
n
‑
氨基
‑
n
‑
叠氮基聚合物将允许具有诸如醛、酮、活化酯、活化碳酸酯等活化亲电基团的分子与聚合物的一个末端附接,以及具有乙炔基团的分子与聚合物的另一个末端附接。
[0276]
在本发明的另一个实施方案中,a是具有1
‑
10个碳原子的脂肪族接头或具有6
‑
14个碳原子的取代的芳基环。x是不与叠氮基反应的官能团,并且y是适合的离去基团。
[0277]
多个靶向多肽可以通过接头多肽连接,其中所述接头多肽任选地长度为6
‑
14个、7
‑
13个、8
‑
12个、7
‑
11个、9
‑
11个或9个氨基酸。其他接头包括但不限于小聚合物,比如peg,其可以是多臂的,从而允许多个靶向多肽分子连接在一起。通过使用这样的接头或通过每个多肽各自的n端之间的直接化学键合,多个靶向多肽和经修饰的靶向多肽可以按照头对头构型经由它们的n端相互连接。例如,两个靶向多肽可以通过它们的n端氨基或修饰的n端氨基之间的化学键合连接形成二聚体。而且,被设计成包含多个化学官能团以便与每个靶向多肽的n端键合的连接分子可以用于将多个靶向多肽各自在它们对应的n端连接。另外,可以通过除n端氨基酸或c端氨基酸之外的氨基酸之间的键合将多个靶向多肽连接。可用于形成本文所述的靶向多肽的二聚体和多聚体的共价键的实例包括但不限于二硫键或巯基键或硫醇键。另外,可以使用某些酶(比如分选酶)在靶向多肽和接头之间(包括在靶向多肽的n端)形成共价键。
[0278]
接头可具有宽范围的分子量或分子长度。可以使用更大或更小分子量的接头提供期望的在靶向多肽和连接实体之间或在连接实体和其结合配偶体(如果有的话)之间的空
间关系或构象。还可以使用具有更长或更短分子长度的接头提供在靶向多肽和连接实体之间或在连接实体和其结合配偶体之间期望的空间或灵活性。
[0279]
在一些实施方案中,本发明提供了具有哑铃结构的水溶性双官能接头,其包括:a)在聚合物主链的至少第一端上的叠氮化物、炔烃、肼、酰肼、羟胺或含羰基部分;和b)在聚合物主链的第二端上的至少第二官能团。第二官能团可以与第一官能团相同或不同。在一些实施方案中,第二官能团不与第一官能团反应。在一些实施方案中,本发明提供了包含支链分子结构的至少一个臂的水溶性化合物。例如,支链分子结构可以是树枝状的。
[0280]
在示例性实施方案中,聚合物通过接头与靶向多肽或经修饰的靶向多肽连接。例如,接头可包含一个或两个氨基酸,所述氨基酸在一端与聚合物比如白蛋白结合部分结合,并且在另一端与多肽主链上的任何可用位置结合。另外的示例性接头包括亲水接头,比如包含至少5个非氢原子的化学部分,其中30%
‑
50%的这些非氢原子是n或o。可以将聚合物与靶向多肽或经修饰的靶向多肽连接的另外的示例性接头公开于u.s.2012/0295847和wo/2012/168430中,将所述专利每一者通过引用完整并入本文。
[0281]
任选地,多个靶向多肽或经修饰的靶向多肽分子可以通过接头多肽连接,其中所述接头多肽任选地长度为1个、1
‑
2个、1
‑
3个、1
‑
4个、1
‑
5个、1
‑
6个、1
‑
7个、1
‑
8个、1
‑
9个、1
‑
10个、1
‑
11个、1
‑
12个氨基酸,以及更长的长度,其中任选地一个靶向多肽的n端与接头多肽的c端融合,并且接头多肽的n端与另一个靶向多肽的n端融合。在wo/2013/004607中公开了可以利用的进一步的示例性接头多肽,将所述专利通过引用完整并入本文。
[0282]
如本文所用的术语“亲电子基团”、“亲电试剂”等指能够接受电子对以形成共价键的原子或原子团。本文所用的“亲电子基团”包括但不限于含卤化物、羰基和环氧化物的化合物。常见的亲电试剂可以是卤化物,比如硫光气、甘油二氯醇、邻苯二甲酰氯、琥珀酰氯、氯乙酰氯、氯代琥珀酰氯等;酮类,比如氯丙酮、溴丙酮等;醛类,比如乙二醛等;异氰酸酯类,比如六亚甲基二异氰酸酯、甲苯二异氰酸酯、间二甲苯二异氰酸酯、环己基甲烷
‑
4,4
‑
二异氰酸酯等,以及这些化合物的衍生物。
[0283]
如本文所用的术语“亲核基团”、“亲核试剂”等是指具有能够形成共价键的电子对的原子或原子团。这种类型的基团可以是作为阴离子基团进行反应的可电离基团。本文所用的“亲核基团”包括但不限于羟基、伯胺、仲胺、叔胺和硫醇。
[0284]
表2提供了各种起始亲电试剂和亲核试剂,它们可以组合产生期望的官能团。所提供的信息意味着是说明性的,并不限于本文所述的合成技术。
[0285]
表2:共价键联及其前体的实例
[0286]
[0287][0288]
一般而言,碳亲电试剂容易受到包括碳亲核试剂在内的互补亲核试剂的攻击,其中攻击性亲核试剂给碳亲电试剂带来电子对,从而在亲核试剂和碳亲电试剂之间形成新的键。
[0289]
碳亲核试剂的非限制性实例包括但不限于烷基、烯基、芳基和炔基格氏试剂、有机锂、有机锌、烷基
‑
、烯基、芳基
‑
和炔基
‑
锡试剂(有机锡烷)、烷基
‑
、烯基
‑
、芳基
‑
和炔基
‑
硼烷试剂(有机硼烷和有机硼酸盐);这些碳亲核试剂具有在水或极性有机溶剂中动力学稳定
的优点。碳亲核试剂的其他非限制性实例包括磷内鎓盐、烯醇和烯醇化物试剂;这些碳亲核试剂具有的优点是,其相对容易由合成有机化学领域技术人员熟知的前体生成。当碳亲核试剂与碳亲电试剂结合使用时,在碳亲核试剂和碳亲电试剂之间产生新的碳
‑
碳键。
[0290]
适合与碳亲电试剂偶联的非碳亲核试剂的非限制性实例包括但不限于伯胺和仲胺、硫醇、硫醇盐和硫醚、醇、醇盐、叠氮化物、氨基脲等。这些非碳亲核试剂,当与碳亲电试剂结合使用时,通常生成杂原子键联(c
‑
x
‑
c),其中x是杂原子,包括但不限于氧、硫或氮。
[0291]
在一些情况下,本发明中使用的聚合物的一端以羟基或甲氧基终止,即,x是氢或ch3(“甲氧基peg”)。可替代地,聚合物可以反应性基团终止,从而形成双官能聚合物。典型的反应性基团可以包括通常用于与20种常见氨基酸中存在的官能团(包括但不限于马来酰亚胺基团、活化碳酸酯(包括但不限于对硝基苯酯)、活化酯(包括但不限于n
‑
羟基琥珀酰亚胺、对硝基苯酯)和醛)反应的那些反应性基团,以及对20种常见氨基酸为惰性但与互补官能团(包括但不限于叠氮基、炔基)特异性地反应的官能团。注意的是,在上面的式中用y表示的聚合物的另一端,将通过天然存在或非天然编码氨基酸直接或间接地与靶向多肽附接。例如,y可以是与多肽的胺基(包括但不限于赖氨酸的ε胺或n端)连接的酰胺、氨基甲酸酯或脲。可替代地,y可以是与硫醇基(包括但不限于半胱氨酸的硫醇基)连接的马来酰亚胺。可替代地,y可以是与通过20种常见氨基酸不常接近的残基的键联。例如,聚合物上的叠氮基可以与靶向多肽上的炔基反应,形成huisgen[3 2]环加成产物。可替代地,聚合物上的炔基可以与靶向多肽中存在的叠氮基反应,形成类似的产物。在一些实施方案中,在适用的情况下,强亲核试剂(包括但不限于肼、酰肼、羟胺、氨基脲)可以与靶向多肽中存在的醛基或酮基反应,形成腙、肟或氨基脲(在一些情况下可以通过用适当的还原剂处理进一步还原)。可替代地,强亲核试剂可以通过非天然编码氨基酸掺入到靶向多肽中,并用于优先与水溶性聚合物中存在的酮基或醛基反应。
[0292]
可根据实际需要使用聚合物的任何分子量,包括但不限于根据需要约100道尔顿(da)至100,000da或更大(包括但不限于有时0.1
‑
50kda或10
‑
40kda)。聚合物的分子量可具有宽范围,包括但不限于介于约100da与约100,000da或更大之间。聚合物可以介于约100da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da、1,000da、900da、800da、700da、600da、500da、400da、300da、200da和100da。在一些实施方案中,聚合物介于约100da与约50,000da之间。也可以使用支链聚合物,包括但不限于每条链具有1
‑
100kda(包括但不限于1
‑
50kda或5
‑
20kda)范围的分子量的聚合物分子。支链聚合物的每条链的分子量可以是,包括但不限于,介于约1,000da与约100,000da或更大。支链聚合物的每条链的分子量可以介于约1,000da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da和1,000da。在一些实施方案中,支链聚合物的每条链的分子量介于约1,000da与约50,000da之间。在一些实施方案中,支链聚合物的每条链的分子量介于约1,000da与约40,000da之间。在一些实施方案中,支链聚合物的每条链的
分子量介于约5,000da与约40,000da之间。在一些实施方案中,支链聚合物的每条链的分子量介于约5,000da与约20,000da之间。广泛范围的聚合物分子描述于,包括但不限于,shearwater polymers公司目录、nektar therapeutics目录,将其通过引用并入本文。
[0293]
在一些实施方案中,本发明提供了含叠氮化物和乙炔的聚合物衍生物,其包含具有约800da至约100,000da的平均分子量的水溶性聚合物主链。水溶性聚合物的聚合物主链可以是聚乙二醇。然而,应该理解的是,多种水溶性聚合物,包括但不限于聚乙二醇和其他有关聚合物,包括聚(葡聚糖)和聚(丙二醇),也适用于本发明的实践,并且术语peg或聚(乙二醇)的使用旨在涵盖和包括所有这样的分子。术语peg包括但不限于任何形式的聚乙二醇,包括双官能peg、多臂peg、衍生化peg、分叉的peg、支链peg、悬垂的peg(即具有一个或多个悬垂于聚合物主链的官能团的peg或相关聚合物)或其中具有可降解键联的peg。
[0294]
除了这些形式的聚合物之外,还可以用主链中弱的或可降解的键联来制备聚合物。例如,可以通过使聚合物主链中的酯键联经历水解来制备聚合物。如下所示,这种水解导致聚合物裂解成较低分子量的片段:
‑
聚合物
‑
co2‑
聚合物
‑
h2o
→
聚合物
‑
co2h ho
‑
聚合物
‑
[0295]
许多聚合物也适用于本发明。在一些实施方案中,具有2个至约300个末端的水溶性聚合物主链在本发明中是特别有用的。适合的聚合物的实例包括但不限于其它聚(亚烷基二醇),比如聚(丙二醇)(“ppg”)、其共聚物(包括但不限于乙二醇和丙二醇的共聚物)、其三元共聚物、其混合物等。虽然聚合物主链的每条链的分子量可以变化,但它通常在约800da至约100,000da的范围内,通常为约6,000da至约80,000da。聚合物主链的每条链的分子量可以介于约100da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da、1,000da、900da、800da、700da、600da、500da、400da、300da、200da和100da。在一些实施方案中,聚合物主链的每条链的分子量介于约100da与约50,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约100da与约40,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约1,000da与约40,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约5,000da与约40,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约10,000da与约40,000da之间。
[0296]
在本发明的这个实施方案的一个特征中,在水解之前完整的聚合物
‑
缀合物在施用后降解最小,使得可裂解键的水解有效地控制活性靶向多肽释放到血流中的缓慢速率,这与靶向多肽释放到体循环中之前的其酶促降解相反。
[0297]
适当的生理学上可裂解的键联包括但不限于酯、碳酸酯、氨基甲酸酯、硫酸酯、磷酸酯、酰氧基烷基醚、缩醛和缩酮。这样的缀合物应该具有在储存和施用后稳定的生理学上可裂解的键。例如,在制造最终的药物组合物后、在适当的递送媒介物中溶解后(如果采用的话)以及无论通过何种途径施用后,与聚合物连接的靶向多肽或经修饰的靶向多肽应保持其完整性。
[0298]
本发明还包括在us 2017/0182181中公开的用于药物缀合物的细胞内递送的具有可调稳定性的基于磷酸酯的接头,将所述专利通过引用并入本文。基于磷酸酯的接头包括
与接头臂远端共价连接的单磷酸酯、二磷酸酯、三磷酸酯或四磷酸酯基团(磷酸酯基团),所述接头臂从远端到近端方向包含调谐元件、任选的间隔元件和反应性官能团。基于磷酸酯的接头的磷酸酯基团能够与有效负载缀合,并且反应性官能团能够与诸如抗体的细胞特异性靶向配体缀合。基于磷酸酯的接头的一般结构是:磷酸酯基团
‑
调谐元件
‑
任选的间隔元件
‑
官能反应性基团。与有效负载缀合的基于磷酸酯的接头具有以下一般结构:有效负载
‑
磷酸酯基团
‑
调谐元件
‑
任选的间隔元件
‑
官能反应性基团,并且当与靶向配体缀合时具有一般结构:有效负载
‑
磷酸酯基团
‑
调谐元件
‑
任选的间隔元件
‑
靶向配体。相对于细胞内环境(例如溶酶体区室),这些基于磷酸酯的接头在血液中具有分化的和可调的稳定性。磷酸酯基团在细胞内环境中裂解以释放其天然或活性形式的有效负载的速率可受到调谐元件结构的影响,并通过磷酸酯基团的取代以及磷酸酯基团是单磷酸酯、二磷酸酯、三磷酸酯还是四磷酸酯来介导进一步的作用。此外,这些基于磷酸酯的接头提供了构建缀合物比如抗体
‑
药物缀合物的能力,其中与其中使用非本文公开的基于磷酸酯的接头将同样的有效负载与抗体或靶向配体缀合的缀合物相比,缀合物形成聚集体的倾向降低。
[0299]
tlr激动剂接头衍生物的结构和合成:亲电基团和亲核基团
[0300]
具有含有羟胺(也称为氨基氧基)基团的接头的tlr激动剂衍生物允许与多种亲电基团反应形成缀合物(包括但不限于与peg或其他水溶性聚合物)。与肼、酰肼和氨基脲一样,氨基氧基的增强的亲核性使其能够与含有羰基或二羰基的多种分子包括但不限于酮、醛或具有相似化学反应性的其他官能团有效且选择性地反应。参见,例如shao,j.和tam,j.,j.am.chem.soc.117:3893
‑
3899(1995);h.hang和c.bertozzi,acc.chem.res.34(9):727
‑
736(2001)。尽管与肼基反应的结果为相应的腙,然而,氨基氧基与含羰基或二羰基的基团反应通常产生肟,所述含羰基或二羰基的基团诸如举例来说是酮、醛或具有相似化学反应性的其他官能团。在一些实施方案中,具有包含叠氮化物、炔或环炔的接头的tlr激动剂衍生物允许通过环加成反应(例如1,3
‑
偶极环加成、叠氮化物
‑
炔huisgen环加成等)来连接分子。(描述于美国专利第7,807,619号中,在相对于反应的程度上将所述专利通过引用并入本文)。
[0301]
因而,在某些实施方案中,本文描述了具有如下接头的tlr激动剂衍生物,所述接头包含羟胺、醛、受保护的醛、酮、受保护的酮、硫酯、酯、二羰基、肼、脒、亚胺、二胺、酮
‑
胺、酮
‑
炔和烯
‑
二酮羟胺基团、羟胺样基团(具有与羟胺基团相似的反应性,并且在结构上与羟胺基团相似)、掩蔽的羟胺基团(其可容易地转化成羟胺基团)、或受保护的羟胺基团(在脱保护后具有与羟胺基团相似的反应性)。在一些实施方案中,具有接头的tlr激动剂衍生物包含叠氮化物、炔或环炔。
[0302]
这样的tlr激动剂接头衍生物或靶向多肽可以呈盐的形式,或者可以掺入到非天然氨基酸多肽、聚合物、多糖或多核苷酸中,并且任选地经历翻译后修饰。
[0303]
在某些实施方案中,式(i)
‑
(vii)的化合物在弱酸性条件下在水溶液中稳定持续至少1个月。在某些实施方案中,式(i)
‑
(vii)的化合物在弱酸性条件下稳定持续至少2周。在某些实施方案中,式(i)
‑
(vii)的化合物在弱酸性条件下稳定持续至少5天。在某些实施方案中,这样的酸性条件是ph 2至8。
[0304]
本文提供和描述的方法和组合物包括包含具有至少一个羰基或二羰基、肟基、羟胺基或其保护或掩蔽形式的非天然氨基酸的多肽。将至少一个反应性基团引入到tlr激动
剂接头衍生物或靶向多肽中可以允许应用涉及特定化学反应的缀合化学,所述特定化学反应包括但不限于与一种或多种靶向多肽反应,而不与普遍存在的氨基酸反应。一旦掺入,还可以通过利用本文所述或适合于在tlr激动剂接头衍生物或靶向多肽中存在的特定官能团或取代基的化学方法来修饰tc侧链的靶向多肽。
[0305]
本文所述的tlr激动剂接头衍生物和靶向多肽方法和组合物提供了具有多种官能团、取代基或部分的物质与其他物质的缀合物,所述其他物质包括但不限于聚合物;水溶性聚合物;聚乙二醇的衍生物;第二种蛋白质或多肽或多肽类似物;抗体或抗体片段;以及它们的任何组合。
[0306]
在某些实施方案中,本文所述的tlr激动剂接头衍生物、靶向多肽、tc、接头和试剂,包括式(i)
‑
(vii)的化合物,在弱酸性条件下(包括但不限于ph 2
‑
8)在水溶液中是稳定的。在其他实施方案中,这样的化合物在弱酸性条件下稳定持续至少一个月。在其他实施方案中,这样的化合物在弱酸性条件下稳定持续至少2周。在其他实施方案中,这样的化合物在弱酸性条件下稳定持续至少5天。
[0307]
在本文描述的组合物、方法、技术和策略的另一方面,是用于研究或使用任何前述“经修饰或未经修饰”的非天然氨基酸靶向多肽的方法。仅举例来说,这个方面包括治疗、诊断、基于测定法、工业、化妆品、植物生物学、环境、能源生产、消费品和/或军事用途,其将受益于包含“经修饰或未经修饰”的非天然氨基酸多肽或蛋白质的靶向多肽。
[0308]
本发明提供了包含至少一个非天然氨基酸的tc分子。在本发明的某些实施方案中,具有至少一个非天然氨基酸的tc包含至少一个翻译后修饰。在一个实施方案中,所述至少一个翻译后修饰包括利用本领域普通技术人员已知的适合于特定反应性基团的化学方法进行的包含第二反应性基团的分子与至少一种包含第一反应性基团的非天然氨基酸的附接,所述分子包括但不限于标记物、染料、接头、另一种tc多肽、聚合物、水溶性聚合物、聚乙二醇的衍生物、光交联剂、放射性核素、细胞毒性化合物、药物、亲和标记物、光亲和标记物、反应性化合物、树脂、第二种蛋白质或多肽或多肽类似物、抗体或抗体片段、金属螯合剂、辅因子、脂肪酸、碳水化合物、多核苷酸、dna、rna、反义多核苷酸、糖类、环糊精、抑制性核糖核酸、生物材料、纳米颗粒、自旋标记物、荧光团、含金属部分、放射性部分、新官能团、与其他分子共价或非共价相互作用的基团、光笼蔽(photocaged)部分、光化辐射可激发部分、光异构化部分、生物素、生物素衍生物、生物素类似物、掺入重原子的部分、化学可裂解基团、光可裂解基团、延长的侧链、碳连接的糖、氧化还原活性剂、氨基硫代酸、毒性部分、同位素标记部分、生物物理探针、磷光基团、化学发光基团、电子致密基团、磁性基团、嵌入基团、发色团、能量转移剂、生物活性剂、可检测标记物、小分子、量子点、纳米发射器、放射性核素、放射性发射器、中子俘获剂,或上述或任何其他期望的化合物或物质的任何组合。例如,第一反应性基团是炔基部分(包括但不限于非天然氨基酸对炔丙基氧基苯丙氨酸,其中炔丙基有时也被称为乙炔部分),第二反应性基团是叠氮基部分,并且利用了[3 2]环加成化学方法。在另一个实例中,第一反应性基团是叠氮基部分(包括但不限于非天然氨基酸中的对叠氮基
‑
l
‑
苯丙氨酸或如本说明书中有时指的paz),并且第二反应性基团是炔基部分。在本发明的经修饰的tc的某些实施方案中,使用包含至少一个翻译后修饰的至少一个非天然氨基酸(包括但不限于含有酮官能团的非天然氨基酸),其中所述至少一个翻译后修饰包含糖部分。在某些实施方案中,在体内在真核细胞中或在非真核细胞中进行翻译后修饰。接
头、聚合物、水溶性聚合物或其他分子可以将分子与多肽附接。在一个另外的实施方案中,与tc附接的接头足够长以允许形成二聚体。还可以将分子直接与多肽连接。
[0309]
在某些实施方案中,tc蛋白包含通过一个宿主细胞在体内进行的至少一个翻译后修饰,其中所述翻译后修饰通常不是通过另一个宿主细胞类型进行的。在某些实施方案中,所述蛋白包含通过真核细胞在体内进行的至少一个翻译后修饰,其中所述翻译后修饰通常不是通过非真核细胞进行的。翻译后修饰的实例包括但不限于糖基化、乙酰化、酰化、脂质修饰、棕榈酰化、棕榈酸酯添加、磷酸化、糖脂键联修饰等。
[0310]
在一些实施方案中,tc包含一个或多个用于多肽的糖基化、乙酰化、酰化、脂质修饰、棕榈酰化、棕榈酸酯添加、磷酸化或糖脂键联修饰的非天然编码氨基酸。在一些实施方案中,tc包含一个或多个用于多肽的糖基化的非天然编码氨基酸。在一些实施方案中,tc包含一个或多个用于多肽的糖基化、乙酰化、酰化、脂质修饰、棕榈酰化、棕榈酸酯添加、磷酸化或糖脂键联修饰的天然编码氨基酸。在一些实施方案中,tc包含一个或多个用于多肽的糖基化的天然编码氨基酸。
[0311]
在一些实施方案中,tc包含一个或多个增强多肽的糖基化的非天然编码氨基酸添加和/或取代。在一些实施方案中,tc包含一个或多个增强多肽的糖基化的缺失。在一些实施方案中,tc包含一个或多个增强多肽中的不同氨基酸的糖基化的非天然编码氨基酸添加和/或取代。在一些实施方案中,tc包含一个或多个增强多肽中的不同氨基酸的糖基化的缺失。在一些实施方案中,tc包含一个或多个增强多肽中的非天然编码氨基酸的糖基化的非天然编码氨基酸添加和/或取代。在一些实施方案中,tc包含一个或多个增强多肽中的天然编码氨基酸的糖基化的非天然编码氨基酸添加和/或取代。在一些实施方案中,tc包含一个或多个增强多肽中的不同氨基酸的糖基化的天然编码氨基酸添加和/或取代。在一些实施方案中,tc包含一个或多个增强多肽中的天然编码氨基酸的糖基化的非天然编码氨基酸添加和/或取代。在一些实施方案中,tc包含一个或多个增强多肽中的非天然编码氨基酸的糖基化的非天然编码氨基酸添加和/或取代。
[0312]
在一个实施方案中,翻译后修饰包括通过glcnac
‑
天冬酰胺键联将寡糖附接至天冬酰胺(包括但不限于,其中所述寡糖包含(glcnac
‑
man)2‑
man
‑
glcnac
‑
glcnac等)。在另一实施方案中,翻译后修饰包括通过galnac
‑
丝氨酸、galnac
‑
苏氨酸、glcnac
‑
丝氨酸、或glcnac
‑
苏氨酸键联将寡糖(包括但不限于gal
‑
galnac、gal
‑
glcnac等)附接至丝氨酸或苏氨酸。在某些实施方案中,本发明的蛋白质或多肽可包含分泌或定位序列、表位标签、flag标签、多组氨酸标签、gst融合物和/或类似物。分泌信号序列的实例包括但不限于原核分泌信号序列、真核分泌信号序列、针对细菌表达5’优化的真核分泌信号序列、新分泌信号序列、果胶酸裂解酶分泌信号序列、omp a分泌信号序列和噬菌体分泌信号序列。分泌信号序列的实例包括,但不限于,stii(原核)、fd giii和m13(噬菌体)、bgl2(酵母)和源于转座子的信号序列bla。任何这样的序列都可以被修饰以提供关于多肽的期望的结果,包括但不限于用不同的信号序列取代一个信号序列,用不同的前导序列取代一个前导序列等。
[0313]
感兴趣的蛋白质或多肽可以含有至少一个、至少两个、至少三个、至少四个、至少五个、至少六个、至少七个、至少八个、至少九个或十个或更多个非天然氨基酸。非天然氨基酸可以相同或不同,例如,蛋白质中可以有1个、2个、3个、4个、5个、6个、7个、8个、9个、10个或更多个不同的如下位点,它们包含1个、2个、3个、4个、5个、6个、7个、8个、9个、10个或更多
个不同的非天然氨基酸。在某些实施方案中,存在于蛋白质的天然存在形式中的至少一个但少于全部的特定氨基酸被非天然氨基酸取代。
[0314]
本发明提供了基于包含至少一个非天然编码氨基酸的tc的方法和组合物。将至少一个非天然编码氨基酸引入到tc中可以允许应用涉及特定化学反应的缀合化学,所述特定化学反应包括但不限于与一个或多个非天然编码氨基酸反应,而不与普遍存在的20种氨基酸反应。在一些实施方案中,包含非天然编码氨基酸的tc经由非天然编码氨基酸的侧链与水溶性聚合物比如聚乙二醇(peg)或接头连接。本发明提供了一种用peg衍生物或tlr
‑
接头衍生物选择性修饰蛋白质的高效方法,所述方法涉及响应于选择密码子将非遗传编码氨基酸选择性掺入到蛋白质中,所述非遗传编码氨基酸包括但不限于那些含有未发现于20种天然掺入的氨基酸中的官能团或取代基的氨基酸,所述官能团或取代基包括但不限于酮、叠氮化物或乙炔部分;随后用适当反应性的peg衍生物修饰这些氨基酸。一旦掺入,则可以通过利用本领域普通技术人员已知的化学方法修饰氨基酸侧链,使其适合于非天然编码氨基酸中存在的特定官能团或取代基。多种已知的化学方法适用于本发明,用于将水溶性聚合物掺入蛋白质中。这样的方法包括但不限于huisgen[3 2]环加成反应(参见,例如padwa,a.comprehensive organic synthesis,第4卷,(1991)版trost,b.m.,pergamon,oxford,第1069
‑
1109页;以及,huisgen,r.1,3
‑
dipolar cycloaddition chemistry,(1984)版,padwa,a.,wiley,new york,第1
‑
176页),其分别具有,包括但不限于乙炔或叠氮化物衍生物。
[0315]
因为huisgen[3 2]环加成方法涉及环加成而不是亲核取代反应,所以能以极高的选择性修饰蛋白质。通过向反应混合物中加入催化量的cu(i)盐,可以在室温下在水性条件下以优异的区域选择性进行该反应(1,4>1,5)。参见,例如tornoe等人,(2002)j.org.chem.67:3057
‑
3064;以及,rostovtsev等人,(2002)angew.chem.int.ed.41:2596
‑
2599;和wo 03/101972。可以通过[3 2]环加成添加至本发明蛋白质的分子几乎包括具有适合官能团或取代基的任何分子,所述官能团或取代基包括但不限于叠氮基或乙炔衍生物。这些分子可以分别添加至具有乙炔基的非天然氨基酸,包括但不限于对炔丙基氧基苯丙氨酸;或具有叠氮基的非天然氨基酸,包括但不限于对叠氮基
‑
苯丙氨酸。
[0316]
由huisgen[3 2]环加成产生的五元环在还原环境中通常是不可逆的,并且在水性环境中稳定抗水解持续延长的时段。因此,用本发明的活性peg衍生物或tlr
‑
接头衍生物可以在苛刻的水性条件下改变多种物质的物理和化学特性。甚至更重要的是,因为叠氮化物和乙炔部分是相互特异性的(例如,不与20种常见的遗传编码氨基酸中的任何一种反应),所以可以在一个或多个特定位点以极高的选择性修饰蛋白质。
[0317]
本发明还提供了peg衍生物或tlr接头衍生物的水溶性和水解稳定的衍生物以及具有一个或多个乙炔或叠氮化物部分的相关亲水聚合物。含有乙炔部分的peg聚合物衍生物对于与叠氮化物部分的偶联是高度选择性的,所述叠氮化物部分响应于选择密码子已被选择性地引入到蛋白质中。类似地,含有叠氮化物部分的peg聚合物衍生物对于与乙炔部分的偶联是高度选择性的,所述乙炔部分响应于选择密码子已被选择性地引入到蛋白质中。更具体地说,叠氮化物部分包括但不限于烷基叠氮化物、芳基叠氮化物和这些叠氮化物的衍生物。只要保持乙炔特异性反应性,烷基和芳基叠氮化物的衍生物可以包括其他取代基。乙炔部分包括烷基和芳基乙炔以及各自的衍生物。只要保持叠氮化物特异性反应性,烷基
和芳基乙炔的衍生物可以包括其他取代基。
[0318]
本发明提供了具有多种官能团、取代基或部分的物质与其他物质的缀合物,所述其他物质包括但不限于标记物;染料;聚合物;水溶性聚合物;聚乙二醇的衍生物;光交联剂;放射性核素;细胞毒性化合物;药物;亲和标记物;光亲和标记物;反应性化合物;树脂;第二种蛋白质或多肽或多肽类似物;抗体或抗体片段;金属螯合剂;辅因子;脂肪酸;碳水化合物;多核苷酸;dna;rna;反义多核苷酸;糖类;水溶性树枝状聚合物;环糊精;抑制性核糖核酸;生物材料;纳米颗粒;自旋标记物;荧光团;含金属部分;放射性部分;新官能团;与其他分子共价或非共价相互作用的基团;光笼蔽部分;光化辐射可激发部分;光异构化部分;生物素;生物素衍生物;生物素类似物;掺入重原子的部分;化学可裂解基团;光可裂解基团;延长的侧链;碳连接的糖;氧化还原活性剂;氨基硫代酸;毒性部分;同位素标记部分;生物物理探针;磷光基团;化学发光基团;电子致密基团;磁性基团;嵌入基团;发色团;能量转移剂;生物活性剂;可检测标记物;小分子;量子点;纳米发射器;放射性核素;放射性发射器;中子俘获剂,或上述或任何其他期望的化合物或物质的任何组合。本发明还包括具有叠氮化物或乙炔部分的物质与具有相应的乙炔或叠氮化物部分的peg聚合物衍生物的缀合物。例如,含有叠氮化物部分的peg聚合物可以在蛋白质中在含有带有乙炔官能团的非遗传编码氨基酸的位置与生物活性分子偶联。peg和生物活性分子偶联的键联包括但不限于huisgen[3 2]环加成产物。
[0319]
在本领域中很好地确立了peg可以用于修饰生物材料的表面(参见,例如,美国专利6,610,281;mehvar,r.,j.pharm pharm sci.,3(1):125
‑
136(2000),将其通过引用并入本文)。本发明还包括包含具有一个或多个反应性叠氮化物或乙炔位点的表面以及一种或多种本发明的含叠氮化物或含乙炔的聚合物的生物材料,所述聚合物通过huisgen[3 2]环加成键联与所述表面偶联。还可以通过除叠氮化物或乙炔键联之外的键联,比如通过包含羧酸、胺、醇或硫醇部分的键联,将生物材料和其他物质与叠氮化物或乙炔活化的聚合物衍生物偶联,使得叠氮化物或乙炔部分可用于随后的反应。
[0320]
本发明包括合成本发明的含叠氮化物和含乙炔的聚合物的方法。在含叠氮化物的peg衍生物的情况下,叠氮化物可与聚合物的碳原子直接键合。可替代地,通过将在一个末端具有叠氮化物部分的连接剂与常规的活化聚合物附接,使得所得聚合物具有在其末端的叠氮化物部分,从而可以制备含叠氮化物的peg衍生物。在含乙炔的peg衍生物的情况下,乙炔可与聚合物的碳原子直接键合。可替代地,通过将在一个末端具有乙炔部分的连接剂与常规的活化聚合物附接,使得所得聚合物具有在其末端的乙炔部分,从而可以制备含乙炔的peg衍生物。
[0321]
更具体地说,在含叠氮化物的peg衍生物的情况下,具有至少一个活性羟基部分的水溶性聚合物经历这样的反应,从而产生其上具有诸如甲磺酸酯、三氟乙基磺酸酯(tresylate)、甲苯磺酸酯或卤素离去基团的更具反应性的部分的取代聚合物。含有磺酰卤、卤原子和其他离去基团的peg衍生物或tlr
‑
接头衍生物的制备和使用是本领域普通技术人员已知的。然后,所得的取代聚合物经历这样的反应,以便在聚合物的末端将所述更具反应性的部分取代为叠氮化物部分。可替代地,具有至少一个活性亲核或亲电部分的水溶性聚合物经历与在一个末端具有叠氮化物的连接剂的反应,使得在peg聚合物和连接剂之间形成共价键,并且叠氮化物部分定位在聚合物的末端。亲核部分和亲电部分,包括胺、硫
醇、酰肼、肼、醇、羧酸酯、醛、酮、硫酯等,是普通技术人员已知的。
[0322]
更具体地说,在含乙炔的peg衍生物的情况下,具有至少一个活性羟基部分的水溶性聚合物经历这样的反应,从而从含有乙炔部分的前体中置换卤素或其他活化离去基团。可替代地,具有至少一个活性亲核或亲电部分的水溶性聚合物经历与在一个末端具有乙炔的连接剂的反应,使得在peg聚合物和连接剂之间形成共价键,并且乙炔部分定位在聚合物的末端。在有机合成背景下卤素部分、活化离去基团、亲核和亲电部分的使用以及peg衍生物或tlr
‑
接头衍生物的制备和使用是本领域从业人员良好确立的。
[0323]
本发明还提供了一种选择性修饰蛋白质的方法,从而向修饰的蛋白质中添加其他物质,包括但不限于含有叠氮化物或乙炔部分的水溶性聚合物比如peg和peg衍生物或tlr
‑
接头衍生物、接头或另一种tc多肽。含叠氮化物和含乙炔的peg衍生物或tlr接头衍生物可用于修饰表面和分子的性质,其中生物相容性、稳定性、溶解性和无免疫原性是重要的,同时提供了比本领域先前已知的更具选择性的将peg衍生物或tlr接头衍生物与蛋白质附接的手段。
[0324]
与本发明一起使用的一般重组核酸方法
[0325]
在本发明的众多实施方案中,将使用重组方法分离、克隆并且经常改变编码感兴趣的tc的靶向多肽的核酸。这样的实施方案用于,包括但不限于,蛋白质表达或在来源于tc的靶向多肽的变体、衍生物、表达盒或其他序列的产生过程中。在一些实施方案中,编码本发明的多肽的序列与异源启动子可操作连接。
[0326]
可以基于亲本多肽的氨基酸序列合成编码包含非天然编码氨基酸的tc的靶向多肽的核苷酸序列,然后改变所述核苷酸序列,从而实现相关氨基酸残基的引入(即,掺入或取代)或去除(即,缺失或取代)。可以根据常规方法通过定点诱变合宜地修饰核苷酸序列。可替代地,可以通过化学合成(包括但不限于通过使用寡核苷酸合成仪)制备核苷酸序列,其中基于期望的多肽的氨基酸序列来设计寡核苷酸,并且优选地选择在其中将产生重组多肽的宿主细胞中有利的那些密码子。例如,可以通过pcr、连接或连接链反应来合成和组装编码期望多肽的多个部分的若干小寡核苷酸。参见,例如barany等人,proc.natl.acad.sci.88:189
‑
193(1991);美国专利6,521,427,将其通过引用并入本文。
[0327]
本发明利用重组遗传学领域的常规技术。公开本发明中使用的一般方法的基本文本包括sambrook等人,molecular cloning,alaboratory manual(2001年第3版);kriegler,gene transfer and expression:a laboratory manual(1990);和current protocols in molecular biology(ausubel等人编辑,1994年))。
[0328]
本发明还涉及用于通过正交trna/rs对进行非天然氨基酸的体内掺入的真核宿主细胞、非真核宿主细胞和生物。用本发明的多核苷酸或构建体对宿主细胞进行基因工程改造(包括但不限于,转化、转导或转染),所述构建体包括本发明的多核苷酸,包括但不限于本发明的载体,其可以是,例如,克隆载体或表达载体。
[0329]
将靶核酸引入细胞中的若干熟知的方法是可获得的,其中任何一种均可用于本发明。这些方法包括:受体细胞与含有dna的细菌原生质体的融合、电穿孔、微粒轰击以及用病毒载体感染(下文进一步论述)等。可使用细菌细胞来扩增含有本发明dna构建体的质粒的数量。使细菌生长至对数期,并且可通过本领域已知的多种方法分离细菌内的质粒(参见,例如,sambrook)。另外,用于纯化来自细菌的质粒的试剂盒可商购获得(参见,例如,
easyprep
tm
、flexiprep
tm
,两者都来自pharmacia biotech;来自stratagene的strataclean
tm
;以及来自qiagen的qiaprep
tm
)。然后将分离和纯化的质粒进一步操作以产生其他质粒,用于转染细胞或掺入相关载体来感染生物体。典型的载体含有转录和翻译终止子、转录和翻译起始序列以及用于调节特定靶核酸表达的启动子。载体任选地包含含有至少一个独立终止子序列的通用表达盒、允许所述盒在真核生物或原核生物或两者中复制的序列(包括但不限于穿梭载体)以及用于原核和真核系统的选择标志物。载体适合于在原核生物、真核生物或两者中复制和整合。参见,gillam&smith,gene 8:81(1979);roberts等人,nature,328:731(1987);schneider,e.等人,protein expr.purif.6(1):10
‑
14,1995;ausubel,sambrook,berger(全部同上)。例如,atcc提供了可用于克隆的细菌和噬菌体的目录,例如由atcc公布的gherna等人(编辑)的atcc细菌和噬菌体目录(1992)。在watson等人(1992)的recombinant dna second edition scientific american books(ny)中也发现了用于测序、克隆和分子生物学的其他方面的另外的基本程序以及基本理论考虑。另外,基本上任何核酸(以及几乎任何标记的核酸,无论是标准核酸还是非标准核酸)都可从多种商业来源定制订购或标准订购,所述来源比如midland certified reagent company(midland,tx,可在万维网mcrc.com获得)、the great american gene company(ramona,ca,可在万维网genco.com获得)、expressgen inc.(chicago,il,可在万维网expressgen.com获得)、operon technologies inc.(alameda,ca)和许多其他来源。
[0330]
选择密码子
[0331]
本发明的选择密码子扩展了蛋白质生物合成机器的遗传密码子框架。例如,选择密码子包括但不限于独特的三碱基密码子;无义密码子,比如终止密码子,包括但不限于琥珀密码子(uag)、赭石密码子或蛋白石密码子(uga);非天然密码子;四个或更多个碱基的密码子;稀有密码子等。对于本领域普通技术人员显而易见的是,可以引入到期望的基因或多核苷酸中的选择密码子的数目范围很广,包括但不限于,在编码tc的至少一部分的单个多核苷酸中有一个或多个、两个或更多个、三个或更多个、4个、5个、6个、7个、8个、9个、10个或更多个。
[0332]
在一个实施方案中,所述方法涉及选择密码子的使用,所述选择密码子是终止密码子,用于一个或多个非天然氨基酸的体内掺入。例如,产生了识别终止密码子(包括但不限于uag)的o
‑
trna,并通过具有期望的非天然氨基酸的o
‑
rs将其氨酰化。这种o
‑
trna不被天然存在的宿主的氨酰trna合成酶识别。可以使用常规的定点诱变在感兴趣的多肽中感兴趣的位点处引入终止密码子,包括但不限于tag。参见,例如sayers,j.r.等人(1988),5
’‑3’
exonucleases in phosphorothioate
‑
based oligonucleotide
‑
directed mutagenesis.nucleic acids res,16:791
‑
802。当o
‑
rs、o
‑
trna和编码感兴趣的多肽的核酸在体内组合时,响应于uag密码子掺入非天然氨基酸,从而产生含有在指定位置处的非天然氨基酸的多肽。
[0333]
可以进行非天然氨基酸的体内掺入而不显著扰乱真核宿主细胞。例如,因为uag密码子的抑制效率取决于o
‑
trna(包括但不限于琥珀抑制基因trna)和真核释放因子(包括但不限于erf)(其结合至终止密码子并启动生长的肽从核糖体释放)之间的竞争,可以通过(包括但不限于)提高o
‑
trna和/或抑制trna的表达水平来调节所述抑制效率。
[0334]
也可以用稀有密码子编码非天然氨基酸。例如,当体外蛋白质合成反应中的精氨
酸浓度降低时,已证明稀有精氨酸密码子agg有效地通过用丙氨酸酰化的合成trna将ala插入。参见,例如ma等人,biochemistry,32:7939(1993)。在这种情况下,合成trna与在大肠杆菌中作为次要种类存在的天然存在的trnaarg竞争。一些生物不使用所有的三联体密码子。藤黄微球菌(micrococcus luteus)中的未指定密码子aga已被用于在体外转录/翻译提取物中插入氨基酸。参见,例如kowal和oliver,nucl.acid.res.,25:4685(1997)。可以生成本发明的组分,从而在体内使用这些稀有密码子。
[0335]
选择密码子还包括延长密码子,包括但不限于四个或更多个碱基的密码子,比如四个、五个、六个或更多个碱基的密码子。四碱基密码子的实例包括但不限于agga、cuag、uaga、cccu等。五碱基密码子的实例包括但不限于aggac、ccccu、cccuc、cuaga、cuacu、uaggc等。本发明的特征包括使用基于移码抑制的延长密码子。四个或更多个碱基密码子可以将包括但不限于一个或多个非天然氨基酸插入到同一蛋白质中。例如,在突变的o
‑
trna(包括但不限于,具有反密码子环例如具有至少8
‑
10个nt的反密码子环的特殊的移码抑制trna)存在下,四个或更多个碱基密码子被阅读为单个氨基酸。在其他实施方案中,反密码子环可以解码,包括但不限于,至少四碱基密码子、至少五碱基密码子或至少六碱基密码子或更多碱基的密码子。由于存在256个可能的四碱基密码子,因此可使用四碱基或更多碱基的密码子在同一细胞中编码多个非天然氨基酸。参见,anderson等人,(2002)exploring the limits of codon and anticodon size,chemistry and biology,9:237
‑
244;magliery,(2001)expanding the genetic code:selection of efficient suppressors of four
‑
base codons and identification of“shifty”four
‑
base codons with a library approach in escherichia coli,j.mol.biol.307:755
‑
769。
[0336]
例如,已经使用四碱基密码子,使用体外生物合成方法将非天然氨基酸掺入到蛋白质中。参见,例如ma等人,(1993)biochemistry,32:7939;和hohsaka等人,(1999)j.am.chem.soc.,121:34。使用cggg和aggu,通过两种化学酰化的移码抑制trna在体外将2
‑
萘基丙氨酸和赖氨酸的nbd衍生物同时掺入链霉亲和素中。参见,例如hohsaka等人,(1999)j.am.chem.soc.,121:12194。在体内研究中,moore等人检验了具有ncua反密码子的trnaleu衍生物抑制uagn密码子(n可以是u、a、g或c)的能力,并且发现四联体uaga可被具有ucua反密码子的trnaleu解码,效率为13%至26%,其中在0框或
–
1框中几乎没有解码。参见,moore等人,(2000)j.mol.biol.,298:195。在一个实施方案中,基于稀有密码子或无义密码子的延长密码子可用于本发明中,其可减少其他不需要位点处的错义通读和移码抑制。
[0337]
对于给定系统,选择密码子还可以包括天然三碱基密码子之一,其中内源系统不使用(或很少使用)天然碱基密码子。例如,这包括缺乏识别天然三碱基密码子的trna的系统,和/或其中三碱基密码子是稀有密码子的系统。
[0338]
选择密码子任选地包括非天然碱基对。这些非天然碱基对进一步扩展了现有的遗传字母表。一个额外的碱基对将三联体密码子的数目从64个增加到125个。第三碱基对的特性包括:稳定和选择性的碱基配对,通过聚合酶以高保真度有效酶促掺入到dna中,以及新生非天然碱基对的合成之后的有效继续引物延伸。可适用于方法和组合物的非天然碱基对的描述包括,例如hirao等人,(2002)an unnatural base pair for incorporating amino acid analogues into protein,nature biotechnology,20:177
‑
182。还参见,例如wu,y.
等人,(2002)j.am.chem.soc.124:14626
‑
14630。其他有关出版物在下文列出。
[0339]
对于体内使用,非天然核苷是可透过膜的,并被磷酸化形成相应的三磷酸。另外,增加的遗传信息是稳定的,并且不被细胞酶破坏。benner和其他人以前的努力利用了与规范沃森
‑
克里克对中不同的氢键合模式,其中最值得注意的实例是iso
‑
c:iso
‑
g对。参见,例如switzer等人,(1989)j.am.chem.soc.,111:8322;和piccirilli等人,(1990)nature,343:33;kool,(2000)curr.opin.chem.biol.,4:602。这些碱基通常在一定程度上与天然碱基错配,并且不能酶促复制。kool和同事证明了碱基之间的疏水堆积相互作用能代替氢键来驱动碱基对的形成。参见,kool,(2000)curr.opin.chem.biol.,4:602;以及guckian和kool,(1998)angew.chem.int.ed.engl.,36,2825。为了开发满足上述所有要求的非天然碱基对,schultz、romesberg及其同事系统地合成和研究了一系列非天然疏水碱基。发现pics:pics自身对相比于天然碱基对更稳定,并且可通过大肠杆菌dna聚合酶i的klenow片段(kf)有效地掺入到dna中。参见,例如mcminn等人,(1999)j.am.chem.soc.,121:11585
‑
6;和ogawa等人,(2000)j.am.chem.soc.,122:3274。kf能够以对于生物功能足够的效率和选择性合成3mn:3mn自身对。参见,例如ogawa等人,(2000)j.am.chem.soc.,122:8803。然而,两个碱基都用作进一步复制的链终止子。最近进化了突变体dna聚合酶,其可用于复制pics自身对。另外,可以复制7ai自身对。参见,例如tae等人,(2001)j.am.chem.soc.,123:7439。还开发了一种新型的金属碱基(metallobase)对,dipic:py,它在结合cu(ii)后形成稳定的对。参见,meggers等人,(2000)j.am.chem.soc.,122:10714。因为延长密码子和非天然密码子本质上与天然密码子正交,所以本发明的方法可利用这个特性生成针对它们的正交trna。
[0340]
还可使用翻译旁路系统将非天然氨基酸掺入期望的多肽中。在翻译旁路系统中,大序列被掺入基因中,但未被翻译成蛋白质。所述序列含有用作线索的结构,用于诱导核糖体跳过该序列,并恢复在该插入下游的翻译。
[0341]
编码感兴趣的蛋白质比如tc的靶向多肽的核酸分子可容易地被突变,以在多肽的任何期望的位置处引入半胱氨酸。半胱氨酸广泛用于将活性分子、水溶性聚合物、蛋白质或多种其他分子引入到感兴趣的蛋白质上。适合于将半胱氨酸掺入多肽的期望位置的方法是本领域普通技术人员已知的,例如美国专利第6,608,183号中描述的方法(将其通过引用并入本文)以及标准诱变技术。
[0342]
iii.非天然编码氨基酸
[0343]
很多种非天然编码氨基酸适合在本发明中使用。可以将任何数目的非天然编码氨基酸引入到tc中。一般而言,引入的非天然编码氨基酸对20种常见的遗传编码氨基酸(即丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酰胺、谷氨酸、甘氨酸、组氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸和缬氨酸)基本上是化学惰性的。在一些实施方案中,非天然编码氨基酸包含有效地和选择性地与未发现于20种常见氨基酸中的官能团反应以形成稳定缀合物的侧链官能团(包括但不限于叠氮基、酮基、醛基和氨基氧基)。例如,包含含有叠氮基官能团的非天然编码氨基酸的tc的靶向多肽可以与聚合物(包括但不限于聚(乙二醇),或者,可替代地,含有炔部分的第二多肽或接头)反应,由于叠氮化物和炔官能团的选择性反应而形成huisgen[3 2]环加成产物,从而形成稳定的缀合物。
[0344]
α
‑
氨基酸的通用结构如下所示(式i):
[0345]
i
[0346][0347]
非天然编码氨基酸通常是具有以上列出的化学式的任何结构并且可以适用于本发明,其中r基团是除二十种天然氨基酸中使用的取代基之外的任何取代基。因为本发明的非天然编码氨基酸通常仅在侧链结构方面不同于天然氨基酸,所以非天然编码氨基酸以它们在天然存在的多肽中形成的相同方式与其他氨基酸(包括但不限于天然或非天然编码的)形成酰胺键。然而,非天然编码氨基酸具有将它们与天然氨基酸区分开的侧链基团。例如,r任选地包括烷基
‑
、芳基
‑
、酰基
‑
、酮
‑
、叠氮基
‑
、羟基
‑
、肼、氰基
‑
、卤代
‑
、酰肼、烯基、炔基、醚、硫醇、硒基
‑
、磺酰基
‑
、硼酸盐、硼酸酯、磷酸基、膦酰基、膦、杂环、烯酮、亚胺、醛、酯、硫代酸、羟胺、氨基等或其任何组合。可以适用于本发明的感兴趣的其他非天然氨基酸包括但不限于包含可光活化交联剂的氨基酸、自旋标记的氨基酸、荧光氨基酸、金属结合氨基酸、含金属氨基酸、放射性氨基酸、具有新颖官能团的氨基酸、与其他分子共价或非共价相互作用的氨基酸、光笼蔽氨基酸和/或光异构化氨基酸、包含生物素或生物素类似物的氨基酸、糖基化基酸比如糖取代的丝氨酸、其他碳水化合物修饰的氨基酸、含酮氨基酸、包含聚乙二醇或聚醚的氨基酸、重原子取代的氨基酸、化学可裂解和/或光可裂解的氨基酸、与天然氨基酸相比具有延长侧链(包括但不限于,包括但不限于大于约5个或大于约10个碳原子的聚醚或长链烃)的氨基酸、碳连接的含糖氨基酸、氧化还原活性氨基酸、含氨基硫代酸的氨基酸以及包含一个或多个毒性部分的氨基酸。
[0348]
可以适用于本发明并且可用于与水溶性聚合物反应的示例性非天然编码氨基酸包括但不限于具有羰基、氨基氧基、肼、酰肼、氨基脲、叠氮化物和炔反应性基团的那些。在一些实施方案中,非天然编码氨基酸包含糖部分。此类氨基酸的实例包括n
‑
乙酰基
‑
l
‑
葡糖胺基
‑
l
‑
丝氨酸、n
‑
乙酰基
‑
l
‑
半乳糖胺基
‑
l
‑
丝氨酸、n
‑
乙酰基
‑
l
‑
葡糖胺基
‑
l
‑
苏氨酸、n
‑
乙酰基
‑
l
‑
葡糖胺基
‑
l
‑
天冬酰胺和o
‑
甘露糖胺基
‑
l
‑
丝氨酸。这样的氨基酸的实例还包括其中氨基酸与糖之间的天然存在的n
‑
键联或o
‑
键联被通常未发现于自然界中的共价键联(包括但不限于烯、肟、硫醚、酰胺等)替换的实例。这样的氨基酸的实例还包括通常未见于天然存在的蛋白质中的糖,比如2
‑
脱氧
‑
葡萄糖、2
‑
脱氧半乳糖等。
[0349]
本文中提供的许多非天然编码氨基酸是可商购的,例如,购自sigma
‑
aldrich(st.louis,mo,usa)、novabiochem(a division of emd biosciences,darmstadt,germany)或peptech(burlington,ma,usa)。任选地如本文所提供的或使用本领域普通技术人员已知的标准方法合成不可商购的那些氨基酸。对于有机合成技术,参见例如,参见例如fessendon和fessendon的organic chemistry(1982年,第二版,willard grant press,boston mass.);march的advanced organic chemistry(第三版,1985年,wiley和sons,new york);以及carey和sundberg的advanced organic chemistry(第三版,部分a和b,1990年,plenum press,new york)。还参见美国专利第7,045,337号和第7,083,970号,将所述文献通过引用并入本文。除了含有新侧链的非天然氨基酸之外,可适用于本发明的非天然氨基酸还任选地包含经修饰的主链结构,包括但不限于,如式ii和iii的结构所示:
[0350]
ii
[0351][0352]
iii
[0353][0354]
其中z通常包括oh、nh2、sh、nh
‑
r
′
或s
‑
r
′
;x和y可以相同或不同,通常包括s或o,并且r和r'(任选地相同或不同)通常选自以上针对具有式i的非天然氨基酸描述的r基团的相同组分列表以及氢。例如,本发明的非天然氨基酸任选地包含如式ii和式iii所示的氨基或羧基中的取代。这种类型的非天然氨基酸包括但不限于α
‑
羟基酸、α
‑
硫代酸、α
‑
氨基硫代甲酸盐,包括但不限于,具有对应于常见的二十种天然氨基酸的侧链或非天然侧链。另外,在α
‑
碳的取代任选地包括但不限于l,d或α
‑
α
‑
二取代的氨基酸,比如d
‑
谷氨酸、d
‑
丙氨酸、d
‑
甲基
‑
o
‑
酪氨酸、氨基丁酸,等。其他结构替代物包括环状氨基酸,比如脯氨酸类似物以及3元、4元、6元、7元、8元和9元环脯氨酸类似物,β和γ氨基酸,比如取代的β
‑
丙氨酸和γ
‑
氨基丁酸。
[0355]
许多非天然氨基酸基于天然氨基酸,比如酪氨酸、谷氨酰胺、苯丙氨酸等,并且适合于在本发明中使用。酪氨酸类似物包括但不限于对位取代的酪氨酸、邻位取代的酪氨酸和间位取代的酪氨酸,其中取代的酪氨酸包含,包括但不限于酮基(包括但不限于乙酰基)、苯甲酰基、氨基、肼、羟胺、硫醇基、羧基、异丙基、甲基、c6‑
c
20
直链或支链烃、饱和或不饱和烃、o
‑
甲基、聚醚基、硝基、炔基等。另外,还考虑了多取代的芳环。可适用于本发明的谷氨酰胺类似物包括但不限于α
‑
羟基衍生物、γ
‑
取代的衍生物、环状衍生物和酰胺取代的谷氨酰胺衍生物。可适用于本发明的示例性苯丙氨酸类似物包括但不限于对位取代的苯丙氨酸、邻位取代的苯丙氨酸和间位取代的苯丙氨酸,其中取代基包含(包括但不限于)羟基、甲氧基、甲基、烯丙基、醛、叠氮基、碘基、溴基、酮基(包括但不限于乙酰基)、苯甲酰基、炔基等。可适用于本发明的非天然氨基酸的具体实例包括但不限于对乙酰基
‑
l
‑
苯丙氨酸、o
‑
甲基
‑
l
‑
酪氨酸、l
‑3‑
(2
‑
萘基)丙氨酸、3
‑
甲基
‑
苯丙氨酸、o
‑4‑
烯丙基
‑
l
‑
酪氨酸、4
‑
丙基
‑
l
‑
酪氨酸、三
‑
o
‑
乙酰基
‑
glcnacβ
‑
丝氨酸、l
‑
多巴、氟化苯丙氨酸、异丙基
‑
l
‑
苯丙氨酸、对叠氮基
‑
l
‑
苯丙氨酸、对酰基
‑
l
‑
苯丙氨酸、对苯甲酰基
‑
l
‑
苯丙氨酸、l
‑
磷酸丝氨酸、膦酰基丝氨酸、膦酰基酪氨酸、对碘
‑
苯丙氨酸、对溴苯丙氨酸、对氨基
‑
l
‑
苯丙氨酸、异丙基
‑
l
‑
苯丙氨酸以及对炔丙基氧基
‑
苯丙氨酸等。在例如标题为“in vivo incorporation of unnatural amino acids”的wo2002/085923中提供了可适用于本发明的多种非天然氨基酸的结构的实例。关于另外的甲硫氨酸类似物,还参见kiick等人,(2002)incorporation of azides into recombinant proteins for chemoselective modification by the staudinger ligation,pnas 99:19
‑
24,将其通过引用并入本文。标题为“compositions containing,methods involving,and uses of non
‑
natural amino acids and polypeptides”的国际
申请第pct/us06/47822号描述了芳香胺部分(包括但不限于对氨基
‑
苯丙氨酸)的还原烷基化和还原胺化,将该申请通过引用并入本文。
[0356]
在本发明的另一个实施方案中,将具有一个或多个非天然编码氨基酸的tc多肽共价修饰。与生物系统的不同官能性正交的选择性化学反应被认为是化学生物学中的重要工具。作为合成化学库的相对新事物,这些生物正交反应激发了用于化合物库合成、蛋白质工程改造、功能蛋白质组学和细胞表面化学重塑的新策略。叠氮化物作为生物缀合的独特化学把手(chemical handle)发挥了突出的作用。已经与膦一起使用staudinger连接对通过代谢引入细胞糖缀合物中的叠氮糖加标签。staudinger连接可以在活体动物中进行,没有生理伤害;尽管如此,staudinger反应并非没有不利因素(liability)。必需的膦类易受空气氧化的影响,并且已证明对它们进行优化以提高水溶性和增加反应速率具有综合挑战性。
[0357]
叠氮基具有生物正交反应性的替代模式:由huisgen描述的与炔的[3 2]环加成反应。在其经典形式中,由于对合理反应速率的高温(或压力)要求,该反应在生物系统中具有有限的适用性。sharpless和同事克服了这个障碍,开发了一种被称为“点击化学”的铜(i)催化形式,其在生理温度和功能丰富的生物环境中容易进行。这一发现使得选择性修饰来自复杂的组织裂解物的病毒颗粒、核酸和蛋白质成为可能。不幸的是,强制铜催化剂对细菌和哺乳动物细胞都是有毒的,因此排除了其中细胞必须保持活力的应用。据报道,通过吸电子取代基活化的炔的无催化剂huisgen环加成在环境温度下发生。然而,这些化合物与生物亲核试剂发生michael反应。
[0358]
在一个实施方案中,提供了包含非天然氨基酸(比如对(炔丙基氧基)
‑
苯丙氨酸)的tc的靶向多肽的组合物。还提供了包含对(炔丙基氧基)
‑
苯丙氨酸的各种组合物,并且其包括但不限于蛋白质和/或细胞。一方面,包含对(炔丙基氧基)
‑
苯丙氨酸非天然氨基酸的组合物进一步包含正交trna。非天然氨基酸可与正交trna键合(包括但不限于共价键合),包括但不限于通过氨基
‑
酰基键与正交trna共价键合、与正交trna的末端核糖的3’oh或2’oh共价键合等。
[0359]
可经由非天然氨基酸掺入到蛋白质中的化学部分提供了蛋白质的多种优点和操作。举例来说,利用许多含肼或羟胺试剂中的任何试剂,酮官能团的独特反应性允许蛋白质的体内和体外选择性修饰。例如,重原子非天然氨基酸可用于对x射线结构数据进行定相。在选择重原子的位置方面,使用非天然氨基酸的位点特异性重原子引入还提供了选择性和灵活性。例如,光反应性非天然氨基酸(包括但不限于具有二苯甲酮和芳基叠氮化物(包括但不限于苯基叠氮化物)侧链的氨基酸)允许蛋白质的有效体内和体外光交联。光反应性非天然氨基酸的实例包括但不限于对叠氮基
‑
苯丙氨酸和对苯甲酰基
‑
苯丙氨酸。然后,可通过随意激发提供光反应性基团的时间控制使具有光反应性非天然氨基酸的蛋白质交联。在一个实例中,(包括但不限于)在使用核磁共振和振动光谱学的情况下,可以用作为局部结构和动力学的探针的同位素标记基团(包括但不限于甲基)取代非天然氨基酸的甲基。例如,炔基或叠氮基官能团允许用分子通过[3 2]环加成反应选择性修饰蛋白质。
[0360]
在氨基末端掺入多肽的非天然氨基酸可以由r基团和第2反应性基团组成,所述r基团是除了在二十种天然氨基酸中使用的取代基之外的任何取代基,所述第二反应性基团不同于通常存在于α
‑
氨基酸中的nh2基团。可以在c端掺入相似的非天然氨基酸,其中第2反
应性基团不同于通常存在于α
‑
氨基酸中的cooh基团。
[0361]
可以选择或设计本发明的非天然氨基酸,以提供另外的在二十种天然氨基酸中不可获得的特征。例如,任选地将非天然氨基酸设计或选择为,例如,修饰它们被掺入其中的蛋白质的生物学特性。例如,通过将非天然氨基酸包含在蛋白质中,可以任选地修饰以下特性:毒性、生物分布、溶解性、稳定性(例如,热、水解、氧化、对酶降解的抗性等)、纯化和加工的便利性、结构特性、光谱特性、化学和/或光化学特性、催化活性、氧化还原电位、半衰期、与其他分子反应的能力(例如共价或非共价)等。
[0362]
在一些实施方案中,本发明提供了通过肟键与水溶性聚合物例如peg连接的tc。许多类型的非天然编码氨基酸适合于形成肟键。这些包括但不限于含有羰基、二羰基或羟胺基的非天然编码氨基酸。这样的氨基酸描述于美国专利公开第2006/0194256号、第2006/0217532号和第2006/0217289号以及标题为“compositions containing,methods involving,and uses of non
‑
natural amino acids and polypeptides”的wo2006/069246,将所述专利通过引用完整并入本文。非天然编码氨基酸还描述于美国专利第7,083,970号和美国专利第7,045,337号中,将所述专利通过引用完整并入本文。
[0363]
本发明的一些实施方案利用在一个或多个位置用对乙酰苯丙氨酸氨基酸取代的tc多肽。在zhang,z.等人,biochemistry 42:6735
‑
6746(2003)中描述了对乙酰基
‑
( /
‑
)
‑
苯丙氨酸和间乙酰基
‑
( /
‑
)
‑
苯丙氨酸的合成,将其通过引用并入本文。本领域普通技术人员可以类似地制备其他含羰基或二羰基的氨基酸。此外,本文包括的非天然氨基酸的非限制性示例性合成在美国专利第7,083,970号中提出,将所述专利通过引用完整并入本文。
[0364]
具有亲电反应性基团的氨基酸允许通过亲核加成反应等多种反应来连接分子。这样的亲电反应性基团包括羰基(包括酮基和二羰基)、羰基样基团(其具有与羰基(包括酮基和二羰基)相似的反应性并且在结构上与羰基相似)、掩蔽的羰基(其可以容易地转化为羰基(包括酮基和二羰基))、或受保护的羰基(其在脱保护后具有与羰基(包括酮基和二羰基)相似的反应性)。这样的氨基酸包括具有式(iv)的结构的氨基酸:
[0365][0366]
其中:
[0367]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0368]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c
(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0369]
j是j是
[0370]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0371]
每个r”独立地是氢、烷基、取代的烷基或保护基团,或者当存在多于一个的r”基团时,两个r”任选地形成杂环烷基;
[0372]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0373]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0374]
r3和r4各自独立地是氢、卤素、低级烷基或取代的低级烷基,或者r3和r4或两个r3基团任选地形成环烷基或杂环烷基;
[0375]
或者,
–
a
‑
b
‑
j
‑
r基团一起形成包含至少一个羰基(包括二羰基)、受保护的羰基(包括受保护的二羰基)或掩蔽的羰基(包括掩蔽的二羰基)的双环或三环环烷基或杂环烷基;
[0376]
或者,
–
j
‑
r基团一起形成包含至少一个羰基(包括二羰基)、受保护的羰基(包括受保护的二羰基)或掩蔽的羰基(包括掩蔽的二羰基)的单环或双环环烷基或杂环烷基;
[0377]
条件是当a是亚苯基并且每个r3是h时,b存在;并且当a是
–
(ch2)4‑
并且每个r3是h时,b不是
–
nhc(o)(ch2ch2)
‑
;并且当a和b不存在并且每个r3是h时,r不是甲基。
[0378]
另外,包括具有式(v)的结构:
[0379][0380]
其中:
[0381]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0382]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0383]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0384]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0385]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0386]
条件是当a是亚苯基时,b存在;并且当a是
–
(ch2)4‑
时,b不是
–
nhc(o)(ch2ch2)
‑
;并且当a和b不存在时,r不是甲基。
[0387]
另外,包括具有式(vi)结构的氨基酸:
[0388][0389]
其中:
[0390]
b是选自由以下基团组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0391]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0392]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0393]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0394]
每个r
a
独立地选自由以下组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基。
[0395]
另外,包括下列氨基酸:
[0396][0397]
其中这样的化合物任选地是氨基保护基团、羧基保护的或其盐。另外,可以将任何下列非天然氨基酸掺入非天然氨基酸多肽中。
[0398]
另外,包括下列具有式(vii)的结构的氨基酸:
[0399][0400]
其中
[0401]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0402]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0403]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0404]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0405]
每个r
a
独立地选自由以下基团组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基;并且n为0至8;
[0406]
条件是当a是
–
(ch2)4‑
时,b不是
–
nhc(o)(ch2ch2)
‑
。
[0407]
另外,包括下列氨基酸:
[0408][0409]
其中此类化合物是任选地氨基保护的、任选地羧基保护的、任选地氨基保护的和羧基保护的,或其盐。另外,可以将这些非天然氨基酸和任何下列非天然氨基酸掺入非天然氨基酸多肽中。
[0410]
另外,包括下列具有式(viii)的结构的氨基酸:
[0411][0412]
其中a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0413]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0414]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0415]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸。
[0416]
另外,包括下列具有式(ix)结构的氨基酸:
[0417][0418]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0419]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0420]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0421]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0422]
其中每个r
a
独立地选自由以下组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基。
[0423]
另外,包括下列氨基酸:
[0424][0425]
其中这样的化合物是任选地氨基保护的、任选地羧基保护
的、任选地氨基保护的和羧基保护的,或其盐。另外,可以将这些非天然氨基酸和任何下列非天然氨基酸掺入非天然氨基酸多肽中。
[0426]
另外,包括下列具有式(x)的结构的氨基酸:
[0427][0428]
其中b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0429]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0430]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0431]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0432]
每个r
a
独立地选自由以下组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基;并且n为0至8。
[0433]
另外,包括下列氨基酸:
[0434][0435]
其中这样的化合物是任选地氨基保护的、任选地羧基保护的、任选地氨基保护的和羧基保护的,或其盐。另外,可以将这些非天然氨基酸和任何下列非天然氨基酸掺入非天然氨基酸多肽中。
[0436]
除了单羰基结构之外,本文描述的非天然氨基酸可以包括诸如二羰基、二羰基样基团、掩蔽的二羰基和受保护的二羰基的基团。
[0437]
例如,包括下列具有式(xi)的结构的氨基酸:
[0438][0439]
其中a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0440]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0441]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0442]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0443]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸。
[0444]
另外,包括下列具有式(xii)的结构的氨基酸:
[0445][0446]
b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n
(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0447]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0448]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0449]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0450]
其中每个r
a
独立地选自由以下组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基。
[0451]
另外,包括下列氨基酸:
[0452]
其中这样的化合物是任选地氨基保护的、任选地羧基保护的、任选地氨基保护的和羧基保护的,或其盐。另外,可以将这些非天然氨基酸和任何下列非天然氨基酸掺入非天然氨基酸多肽中。
[0453]
另外,包括下列具有式(xiii)的结构的氨基酸:
[0454][0455]
其中b是任选的,并且当存在时是选自由以下组成的组的接头:低级亚烷基、取代的低级亚烷基、低级亚烯基、取代的低级亚烯基、低级杂亚烷基、取代的低级杂亚烷基、
‑
o
‑
、
‑
o
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s
‑
、
‑
s
‑
(亚烷基或取代的亚烷基)
‑
、
‑
s(o)
k
‑
(其中k是1、2或3)、
‑
s(o)
k
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)
‑
、
‑
c(o)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(s)
‑
、
‑
c(s)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)
‑
、
‑
nr
’‑
(亚烷基或取代的亚烷基)
‑
、
‑
c(o)n(r’)
‑
、
‑
con(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
csn(r’)
‑
、
‑
csn(r’)
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)co
‑
(亚烷基或取代的亚烷基)
‑
、
‑
n(r’)c(o)o
‑
、
‑
s(o)
k
n(r’)
‑
、
‑
n(r’)c(o)n(r’)
‑
、
‑
n(r’)c(s)n(r’)
‑
、
‑
n(r’)s(o)
k
n(r’)
‑
、
‑
n(r’)
‑
n=、
‑
c(r’)=n
‑
、
‑
c(r’)=n
‑
n(r’)
‑
、
‑
c(r’)=n
‑
n=、
‑
c(r’)2‑
n=n
‑
和
‑
c(r’)2‑
n(r’)
‑
n(r’)
‑
,其中每个r’独立地是h、烷基或取代的烷基;
[0456]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0457]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0458]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0459]
每个r
a
独立地选自由以下组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基;并且n为0至8。
[0460]
另外,包括下列氨基酸:
[0461][0462]
其中此类化合物是任选地氨基保护的、任选地羧基保护的、任选地氨基保护的和羧基保护的,或其盐。另外,可以将这些非天然氨基酸和任何下列非天然氨基酸掺入非天然氨基酸多肽中。
[0463]
另外,包括下列具有式(xiv)的结构的氨基酸:
[0464][0465]
其中:
[0466]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0467]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0468]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0469]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0470]
x1是c、s或s(o);并且l是亚烷基、取代的亚烷基、n(r’)(亚烷基)或n(r’)(取代的亚烷基),其中r’是h、烷基、取代的烷基、环烷基或取代的环烷基。
[0471]
另外,包括下列具有式(xiv
‑
a)的结构的氨基酸:
[0472][0473]
其中:
[0474]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0475]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0476]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0477]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0478]
l是亚烷基、取代的亚烷基、n(r’)(亚烷基)或n(r’)(取代的亚烷基),其中r’是h、烷基、取代的烷基、环烷基或取代的环烷基。
[0479]
另外,包括下列具有式(xiv
‑
b)的结构的氨基酸:
[0480][0481]
其中:
[0482]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0483]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0484]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0485]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0486]
l是亚烷基、取代的亚烷基、n(r’)(亚烷基)或n(r’)(取代的亚烷基),其中r’是h、烷基、取代的烷基、环烷基或取代的环烷基。
[0487]
另外,包括下列具有式(xv)的结构的氨基酸:
[0488][0489]
其中:
[0490]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0491]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0492]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0493]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0494]
x1是c、s或s(o);n是0、1、2、3、4或5;并且在每个cr8r9基团上的每个r8和r9独立地选自由h、烷氧基、烷基胺、卤素、烷基、芳基组成的组,或者任何r8和r9可以一起形成=o或环烷基,或者任何相邻的r8基团可以一起形成环烷基。
[0495]
另外,包括下列具有式(xv
‑
a)的结构的氨基酸:
[0496][0497]
其中:
[0498]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0499]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0500]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0501]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0502]
n是0、1、2、3、4或5;并且在每个cr8r9基团上的每个r8和r9独立地选自由h、烷氧基、烷基胺、卤素、烷基、芳基组成的组,或者任何r8和r9可以一起形成=o或环烷基,或者任何相邻的r8基团可以一起形成环烷基。
[0503]
另外,包括下列具有式(xv
‑
b)的结构的氨基酸:
[0504][0505]
其中:
[0506]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0507]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0508]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0509]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0510]
n是0、1、2、3、4或5;并且在每个cr8r9基团上的每个r8和r9独立地选自由h、烷氧基、烷基胺、卤素、烷基、芳基组成的组,或者任何r8和r9可以一起形成=o或环烷基,或者任何相邻的r8基团可以一起形成环烷基。
[0511]
另外,包括下列具有式(xvi)的结构的氨基酸:
[0512][0513]
其中:
[0514]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0515]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0516]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0517]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0518]
x1是c、s或s(o);并且l是亚烷基、取代的亚烷基、n(r’)(亚烷基)或n(r’)(取代的亚烷基),其中r’是h、烷基、取代的烷基、环烷基或取代的环烷基。
[0519]
另外,包括下列具有式(xvi
‑
a)的结构的氨基酸:
[0520][0521]
其中:
[0522]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0523]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0524]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0525]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0526]
l是亚烷基、取代的亚烷基、n(r’)(亚烷基)或n(r’)(取代的亚烷基),其中r’是h、烷基、取代的烷基、环烷基或取代的环烷基。
[0527]
另外,包括下列具有式(xvi
‑
b)的结构的氨基酸:
[0528][0529]
其中:
[0530]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0531]
r是h、烷基、取代的烷基、环烷基或取代的环烷基;
[0532]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0533]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0534]
l是亚烷基、取代的亚烷基、n(r’)(亚烷基)或n(r’)(取代的亚烷基),其中r’是h、烷基、取代的烷基、环烷基或取代的环烷基。
[0535]
另外,包括具有式(xvii)的结构的氨基酸:
[0536][0537]
其中:
[0538]
a是任选的,并且当存在时,是低级亚烷基、取代的低级亚烷基、低级环亚烷基、取代的低级环亚烷基、低级亚烯基、取代的低级亚烯基、亚炔基、低级杂亚烷基、取代的杂亚烷基、低级杂环亚烷基、取代的低级杂环亚烷基、亚芳基、取代的亚芳基、杂亚芳基、取代的杂亚芳基、亚烷芳基、取代的亚烷芳基、亚芳烷基或取代的亚芳烷基;
[0539]
m是
‑
c(r3)
‑
、、
[0540]
其中(a)指示与a基团键合,并且(b)指示与对应的羰基键合,r3和r4独立地选自h、卤素、烷基、取代的烷基、环烷基或取代的环烷基,或者r3和r4或两个r3基团或两个r4基团任选地形成环烷基或杂环烷基;
[0541]
r是h、卤素、烷基、取代的烷基、环烷基或取代的环烷基;
[0542]
t3是键、c(r)(r)、o或s,并且r是h、卤素、烷基、取代的烷基、环烷基或取代的环烷基;
[0543]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0544]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸。
[0545]
另外,包括具有式(xviii)的结构的氨基酸:
[0546][0547]
其中:
[0548]
m是
‑
c(r3)
‑
、、
[0549]
其中(a)指示与a基团键合,并且(b)指示与对应的羰基键合,r3和r4独立地选自h、卤素、烷基、取代的烷基、环烷基或取代的环烷基,或者r3和r4或两个r3基团或两个r4基团任选地形成环烷基或杂环烷基;
[0550]
r是h、卤素、烷基、取代的烷基、环烷基或取代的环烷基;
[0551]
t3是键、c(r)(r)、o或s,并且r是h、卤素、烷基、取代的烷基、环烷基或取代的环烷基;
[0552]
r1是任选的,并且当存在时,是h、氨基保护基团、树脂、氨基酸、多肽或多核苷酸;并且
[0553]
r2是任选的,并且当存在时,是oh、酯保护基团、树脂、氨基酸、多肽或多核苷酸;
[0554]
每个r
a
独立地选自由以下组成的组:h、卤素、烷基、取代的烷基、
‑
n(r’)2,
‑
c(o)
k
r’(其中k是1、2或3)、
‑
c(o)n(r’)2、
‑
or’和
‑
s(o)
k
r’,其中每个r’独立地是h、烷基或取代的烷基。
[0555]
另外,包括具有式(xix)的结构的氨基酸:
[0556]
[0557]
其中:
[0558]
r是h、卤素、烷基、取代的烷基、环烷基或取代的环烷基;并且
[0559]
t3是o或s。
[0560]
另外,包括具有式(xx)的结构的氨基酸:
[0561][0562]
其中:
[0563]
r是h、卤素、烷基、取代的烷基、环烷基或取代的环烷基。
[0564]
另外,包括下列具有式(xxi)的结构的氨基酸:
[0565][0566]
在一些实施方案中,将包含非天然氨基酸的多肽化学修饰,以生成反应性羰基或二羰基官能团。例如,可以从具有相邻的氨基和羟基的官能性生成可用于缀合反应的醛官能性。当生物活性分子是多肽时,例如,可以使用n端丝氨酸或苏氨酸(其可以正常存在或可以通过化学或酶消化而暴露)在温和的氧化裂解条件下使用高碘酸盐生成醛官能性。参见,例如gaertner等人,bioconjug.chem.3:262
‑
268(1992);geoghegan,k.&stroh,j.,bioconjug.chem.3:138
‑
146(1992);gaertner等人,j.biol.chem.269:7224
‑
7230(1994)。然而,本领域已知的方法仅限于在肽或蛋白质的n端的氨基酸。
[0567]
在本发明中,带有相邻羟基和氨基的非天然氨基酸可以作为“掩蔽的”醛官能团掺入到多肽中。例如,5
‑
羟赖氨酸带有与ε胺相邻的羟基。用于生成醛的反应条件通常涉及在温和条件下添加摩尔过量的偏高碘酸钠,以避免在多肽内的其他位点处的氧化。氧化反应的ph通常为约7.0。典型的反应涉及向多肽的缓冲溶液中添加约1.5摩尔过量的偏高碘酸钠,接着在黑暗中孵育约10分钟。参见,例如美国专利第6,423,685号。
[0568]
羰基或二羰基官能性可以在温和条件下在水溶液中与含羟胺的试剂选择性地反应,形成在生理条件下稳定的相应肟键联。参见,例如jencks,w.p.,j.am.chem.soc.81,475
‑
481(1959);shao,j.和tam,j.p.,j.am.chem.soc.117:3893
‑
3899(1995)。此外,羰基或二羰基的独特反应性允许在其他氨基酸侧链存在下的选择性修饰。参见,例如cornish,v.w.等人,j.am.chem.soc.118:8150
‑
8151(1996);geoghegan,k.f.&stroh,j.g.,bioconjug.chem.3:138
‑
146(1992);mahal,l.k.等人,science 276:1125
‑
1128(1997)。
[0569]
a.羰基反应性基团
[0570]
具有羰基反应性基团的氨基酸尤其允许经由亲核加成或羟醛缩合反应来连接分
子(包括但不限于peg或其他水溶性分子)的多种反应。
[0571]
示例性含羰基氨基酸可以表示如下:
[0572][0573]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基或取代的芳基;r2是h、烷基、芳基、取代的烷基和取代的芳基;r3是h、氨基酸、多肽或氨基末端修饰基团,并且r4是h、氨基酸、多肽或羧基末端修饰基团。在一些实施方案中,n是1,r1是苯基,并且r2是简单的烷基(即甲基、乙基或丙基),并且酮部分位于相对于烷基侧链的对位。在一些实施方案中,n是1,r1是苯基,并且r2是简单的烷基(即甲基、乙基或丙基),并且酮部分位于相对于烷基侧链的间位。
[0574]
在zhang,z.等人,biochemistry 42:6735
‑
6746(2003)中描述了对乙酰基
‑
( /
‑
)
‑
苯丙氨酸和间乙酰基
‑
( /
‑
)
‑
苯丙氨酸的合成,将其通过引用并入本文。本领域普通技术人员可类似地制备其他含羰基的氨基酸。
[0575]
在一些实施方案中,将包含非天然编码氨基酸的多肽化学修饰,以生成反应性羰基官能团。例如,可以从具有相邻的氨基和羟基的官能性生成可用于缀合反应的醛官能性。当生物活性分子是多肽时,例如,可以使用n端丝氨酸或苏氨酸(其可以正常存在或可以通过化学或酶消化而暴露)在温和的氧化裂解条件下使用高碘酸盐生成醛官能性。参见,例如,gaertner等人,bioconjug.chem.3:262
‑
268(1992);geoghegan,k.&stroh,j.,bioconjug.chem.3:138
‑
146(1992);gaertner等人,j.biol.chem.269:7224
‑
7230(1994)。然而,本领域已知的方法仅限于在肽或蛋白质的n端的氨基酸。
[0576]
在本发明中,带有相邻羟基和氨基的非天然编码氨基酸可以作为“掩蔽的”醛官能团掺入到多肽中。例如,5
‑
羟赖氨酸带有与ε胺相邻的羟基。用于生成醛的反应条件通常涉及在温和条件下添加摩尔过量的偏高碘酸钠,以避免在多肽内的其他位点处的氧化。氧化反应的ph通常为约7.0。典型的反应涉及向多肽的缓冲溶液中添加约1.5摩尔过量的偏高碘酸钠,接着在黑暗中孵育约10分钟。参见,例如美国专利第6,423,685号,将其通过引用并入本文。
[0577]
羰基官能性可以在温和条件下在水溶液中与含肼、酰肼、羟胺或氨基脲的试剂选择性地反应,分别形成在生理条件下稳定的相应的腙、肟或缩氨基脲键联。参见,例如jencks,w.p.,j.am.chem.soc.81,475
‑
481(1959);shao,j.和tam,j.p.,j.am.chem.soc.117:3893
‑
3899(1995)。而且,羰基的独特反应性允许在其他氨基酸侧链存在下的选择性修饰。参见,例如,cornish,v.w.,等人,j.am.chem.soc.118:8150
‑
8151(1996);geoghegan,k.f.&stroh,j.g.,bioconjug.chem.3:138
‑
146(1992);mahal,l.k.等人,science 276:1125
‑
1128(1997)。
[0578]
b.肼、酰肼或氨基脲反应性基团
[0579]
含有亲核基团(比如肼、酰肼或氨基脲)的非天然编码氨基酸允许与多种亲电子基团反应形成缀合物(包括但不限于与peg或其他水溶性聚合物反应)。
[0580]
示例性含肼、酰肼或氨基脲的氨基酸可以表示如下:
[0581][0582]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基、或取代的芳基或不存在;x是o、n、或s或不存在;r2是h、氨基酸、多肽或氨基末端修饰基团,并且r3是h、氨基酸、多肽或羧基末端修饰基团。
[0583]
在一些实施方案中,n是4,r1不存在,并且x是n。在一些实施方案中,n是2,r1不存在,并且x不存在。在一些实施方案中,n是1,r1是苯基,x是o,并且氧原子位于芳基环上脂族(alphatic)基团的对位。
[0584]
含有酰肼、肼和氨基脲的氨基酸可从商业来源获得。例如,l
‑
谷氨酸
‑ꢀ‑
酰肼可从sigma chemical(st.louis,mo)获得。本领域普通技术人员可制备不可商购获得的其他氨基酸。参见,例如美国专利第6,281,211号,将其通过引用并入本文。
[0585]
含有带有酰肼、肼或氨基脲官能团的非天然编码氨基酸的多肽可以与含有醛或具有相似化学反应性的其他官能团的多种分子有效且选择性地反应。参见,例如shao,j.和tam,j.,j.am.chem.soc.117:3893
‑
3899(1995)。与20种常见氨基酸上存在的亲核基团(包括但不限于丝氨酸或苏氨酸的羟基或赖氨酸的氨基和n端)相比,酰肼、肼和氨基脲官能团的独特反应性使得它们对醛、酮和其他亲电子基团具有显著更高的反应性。
[0586]
c.含氨基氧基的氨基酸
[0587]
含有氨基氧基(也称为羟胺)基团的非天然编码氨基酸允许与多种亲电子基团反应形成缀合物(包括但不限于与peg或其他水溶性聚合物反应)。与肼、酰肼和氨基脲一样,氨基氧基的增强的亲核性使其能够与含有醛或具有相似化学反应性的其他官能团的多种分子有效且选择性地反应。参见,例如shao,j.和tam,j.,j.am.chem.soc.117:3893
‑
3899(1995);h.hang和c.bertozzi,acc.chem.res.34:727
‑
736(2001)。尽管与肼基团反应的结果为相应的腙,但氨基氧基与含羰基的基团(比如酮)的反应通常产生肟。
[0588]
示例性含氨基氧基的氨基酸可以表示如下:
[0589][0590]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基、或取代的芳基或不存在;x是o、n、s或不存在;m为0
‑
10;y=c(o)或不存在;r2是h、氨基酸、多肽或氨基末端修饰基团,并且r3是h、氨基酸、多肽或羧基末端修饰基团。在一些实施方案中,n是1,r1是苯基,x是o,m是1,并且y存在。在一些实施方案中,n是2,r1和x不存在,m是0,并且y不存在。
[0591]
可由可容易获得的氨基酸前体(高丝氨酸、丝氨酸和苏氨酸)制备含氨基氧基的氨基酸。参见,例如m.carrasco和r.brown,j.org.chem.68:8853
‑
8858(2003)。已经从天然来源分离某些含氨基氧基的氨基酸,比如l
‑2‑
氨基
‑4‑
(氨基氧基)丁酸(rosenthal,g.,life sci.60:1635
‑
1641(1997)。本领域普通技术人员可制备其他含氨基氧基的氨基酸。
[0592]
d.叠氮化物和炔反应性基团
[0593]
叠氮化物和炔官能团的独特反应性使它们对于多肽和其他生物分子的选择性修
饰极其有用。有机叠氮化物(特别是脂肪族叠氮化物)和炔通常对常见的反应性化学条件是稳定的。具体而言,叠氮化物和炔官能团两者对天然存在的多肽中发现的20种常见氨基酸的侧链(即r基团)都是惰性的。然而,当使叠氮基与炔基靠近时,它们的“弹簧负载(spring
‑
loaded)”性质显露,并且它们经由huisgen[3 2]环加成反应选择性且有效地反应,以生成相应的三唑。参见,例如,chin j.等人,science 301:964
‑
7(2003);wang,q.等人,j.am.chem.soc.125,3192
‑
3193(2003);chin,j.w.等人,j.am.chem.soc.124:9026
‑
9027(2002)。
[0594]
因为huisgen环加成反应涉及选择性环加成反应(参见,例如,padwa,a.,c
omprehensive o
rganic s
ynthesis
,第4卷,(trost,b.m.编辑,1991),第1069
‑
1109页;huisgen,r.1,3
‑
d
ipolar c
ycloaddition c
hemistry
,(padwa,a.编辑,1984),第1
‑
176页)而不是亲核取代,所以带有含叠氮化物和炔侧链的非天然编码氨基酸的掺入允许所得多肽在非天然编码氨基酸的位置处被选择性修饰。涉及含叠氮化物或含炔的tc的环加成反应可在室温下在水性条件下通过在催化量的用于将cu(ii)原位还原为cu(i)的还原剂存在下添加cu(ii)(包括但不限于催化量的cuso4的形式)来进行。参见,例如,wang,q.等人,j.am.chem.soc.125,3192
‑
3193(2003);tornoe,c.w.等人,j.org.chem.67:3057
‑
3064(2002);rostovtsev等人,angew.chem.int.编辑41:2596
‑
2599(2002)。示例性还原剂包括(包括但不限于)抗坏血酸盐、金属铜、奎宁(quinine)、氢醌、维生素k、谷胱甘肽、半胱氨酸、fe
2
、co
2
以及外加电势。
[0595]
在一些情况下,在期望叠氮化物和炔之间的huisgen[3 2]环加成反应时,tc包含含有炔部分的非天然编码氨基酸,并且有待与所述氨基酸附接的水溶性聚合物包含叠氮化物部分。或者,也可进行逆向反应(即,在氨基酸上存在叠氮化物部分,而在水溶性聚合物上存在炔部分)。
[0596]
叠氮化物官能团也可与含有芳基酯的水溶性聚合物选择性地反应,并且经芳基膦部分适当地官能化从而生成酰胺键联。芳基膦基团使叠氮化物原位还原,并且所得胺随后与近端酯键联有效反应从而生成相应的酰胺。参见,例如e.saxon和c.bertozzi,science 287,2007
‑
2010(2000)。含叠氮化物的氨基酸可以是烷基叠氮化物(包括但不限于2
‑
氨基
‑6‑
叠氮基
‑1‑
己酸)或芳基叠氮化物(对叠氮基
‑
苯丙氨酸)。
[0597]
含有芳基酯和膦部分的示例性水溶性聚合物可以表示如下:
[0598]
其中x可以是o、n、s或不存在,ph是苯基,w是水溶性聚合物,并且r可以是h、烷基、芳基、取代的烷基和取代的芳基。示例性r基团包括但不限于
‑
ch2、
‑
c(ch3)3、
‑
or’、
‑
nr’r”、
‑
sr’、
‑
卤素、
‑
c(o)r’、
‑
conr’r”、
‑
s(o)2r’、
‑
s(o)2nr’r”、
‑
cn和
–
no2。r’、r”、r
”’
和r
””
各自独立地指氢、取代或未取代的杂烷基、取代或未取代的芳基,包括但不限于被1
‑
3个卤素取代的芳基、取代或未取代的烷基、烷氧基或硫代烷氧基或芳烷基。当本发明的化合物包括多于一个r基团时,例如,当多于一个的这些基团存在时,将每个r基团独立地选择为各个r’、r”、r
’”
和r
””
基团。当r'和r”与同一个氮原子附接时,它们可与氮原子结合形成5
‑
、6
‑
或7
‑
元环。例如,
‑
nr’r”意指包括但不限于1
‑
吡咯烷基和4
‑
吗啉基。根据上述对取代基的讨论,本领域技术人员将理解,术语“烷基”意在包括包含与氢基团以外的基团键合的碳原子的基团,比如卤代烷基(包括但不限于
‑
cf3和
–
ch2cf3)和酰基(包括但不限于
‑
c(o)ch3、
‑
c(o)
cf3、
‑
c(o)ch2och3等)。
[0599]
叠氮化物官能团也可与含有硫酯的水溶性聚合物选择性地反应,并且经芳基膦部分适当地官能化从而生成酰胺键联。芳基膦基团使叠氮化物原位还原,并且所得胺随后与硫酯键联有效反应从而生成相应的酰胺。含有硫酯和膦部分的示例性水溶性聚合物可以表示如下:
[0600]
其中n为1
‑
10;x可以是o、n、s或不存在,ph是苯基,并且w是水溶性聚合物。
[0601]
示例性含炔氨基酸可以表示如下:
[0602][0603]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基、或取代的芳基或不存在;x是o、n、s或不存在;m为0
‑
10,r2是h、氨基酸、多肽或氨基末端修饰基团,并且r3是h、氨基酸、多肽或羧基末端修饰基团。在一些实施方案中,n是1,r1是苯基,x不存在,m是0,并且乙炔部分位于相对于烷基侧链的对位。在一些实施方案中,n是1,r1是苯基,x是o,m是1,并且炔丙基氧基位于相对于烷基侧链的对位(即,o
‑
炔丙基
‑
酪氨酸)。在一些实施方案中,n是1,r1和x不存在,并且m是0(即,炔丙基甘氨酸)。
[0604]
含炔氨基酸可商购获得。例如,从peptech(burlington,ma)可商购获得炔丙基甘氨酸。或者,可以根据标准方法制备含炔氨基酸。例如,可以合成对炔丙基氧基苯丙氨酸,例如,如描述于deiters,a.等人,j.am.chem.soc.125:11782
‑
11783(2003),并且可以合成4
‑
炔基
‑
l
‑
苯丙氨酸,如描述于kayser,b.等人,tetrahedron 53(7):2475
‑
2484(1997)。本领域普通技术人员可制备其他含炔氨基酸。
[0605]
示例性含叠氮化物的氨基酸可以表示如下:
[0606][0607]
其中n为0
‑
10;r1是烷基、芳基、取代的烷基、取代的芳基或不存在;x是o、n、s或不存在;m为0
‑
10;r2是h、氨基酸、多肽或氨基末端修饰基团,并且r3是h、氨基酸、多肽或羧基末端修饰基团。在一些实施方案中,n是1,r1是苯基,x不存在,m是0,并且叠氮化物部分位于烷基侧链的对位。在一些实施方案中,n是0
‑
4,并且r1和x不存在,并且m=0。在一些实施方案中,n是1,r1是苯基,x是o,m是2,并且
‑
叠氮基乙氧基部分位于相对于烷基侧链的对位。
[0608]
含有叠氮化物的氨基酸可从商业来源获得。例如,可从chem
‑
impex international,inc.(wood dale,il)获得4
‑
叠氮基苯丙氨酸。对于那些不可商购获得的含有叠氮化物的氨基酸,可相对容易地使用本领域普通技术人员已知的标准方法来制备叠氮基,所述方法包括但不限于通过置换适合的离去基团(包括但不限于卤化物、甲磺酸根、甲苯磺酸根)或通过打开经适当保护的内酯。参见,例如march的advanced organic chemistry(第三版,1985,wiley and sons,new york)。
[0609]
e.氨基硫醇反应性基团
[0610]
β
‑
取代的氨基硫醇官能团的独特反应性使得它们对通过形成噻唑烷来选择性修饰含有醛基的多肽和其他生物分子极其有用。参见,例如j.shao和j.tam,j.am.chem.soc.1995,117(14)3893
‑
3899。在一些实施方案中,可以将β
‑
取代的氨基硫醇氨基酸掺入到tc多肽中,然后与包含醛官能团的水溶性聚合物反应。在一些实施方案中,水溶性聚合物、药物缀合物或其他有效负载可以通过形成噻唑烷而与包含β
‑
取代的氨基硫醇氨基酸的tc的靶向多肽偶联。
[0611]
f.另外的反应性基团
[0612]
可以掺入到本发明的tc多肽中的另外的反应性基团和非天然编码氨基酸,包括但不限于对氨基
‑
苯丙氨酸,描述于以下专利申请中,将所有这些专利申请通过引用完整并入本文:美国专利公开第2006/0194256号、美国专利公开第2006/0217532号、美国专利公开第2006/0217289号、美国临时专利第60/755,338号;美国临时专利第60/755,711号;美国临时专利第60/755,018号;国际专利申请号pct/us06/49397;wo 2006/069246;美国临时专利第60/743,041;美国临时专利第60/743,040号;国际专利申请第pct/us06/47822号;美国临时专利第60/882,819号;美国临时专利第60/882,500号;和美国临时专利第60/870,594号。这些申请还讨论了可能存在于peg或其他聚合物上的反应性基团,包括但不限于用于缀合的羟胺(氨基氧基)基团。
[0613]
tc多肽中的非天然氨基酸的定位
[0614]
本文所述的方法和组合物包括将一个或多个非天然氨基酸掺入靶向多肽中来制备本发明的tc。可在一个或多个不破坏靶向多肽的活性的特定位置处掺入一个或多个非天然氨基酸。这可通过进行“保守”取代来实现,包括但不限于用非天然或天然疏水性氨基酸取代疏水性氨基酸,用非天然或天然大体积氨基酸取代大体积氨基酸,用非天然或天然亲水性氨基酸取代亲水性氨基酸,和/或将非天然氨基酸插入并非活性所需的位置中。
[0615]
可采用多种生物化学和结构方法来选择在tc的靶向多肽内用非天然氨基酸进行取代的期望位点。在一些实施方案中,非天然氨基酸连接在tlr激动剂衍生物的c端。在其他实施方案中,非天然氨基酸连接在tlr激动剂衍生物的n端。tc的靶向多肽的任何位置适合于选择来掺入非天然氨基酸,并且可以基于合理设计进行选择,或针对任何特定的期望目的或不为所述目的进行随机选择。对期望位点的选择可基于产生具有任何期望特性或活性的非天然氨基酸多肽(其可被进一步修饰或保持未经修饰),包括但不限于受体结合调节剂、受体活性调节剂、与结合配偶体结合的调节剂、结合配偶体活性调节剂、结合配偶体构象调节剂;二聚体或多聚体形成;相比于天然分子,活性或特性无变化;或操纵多肽的任何物理或化学特性,比如溶解性、聚集或稳定性。或者,鉴定为对生物活性至关重要的位点也可为用非天然氨基酸进行取代的良好候选,同样取决于对于多肽所寻求的所需活性。另一替代方案将在于简单地用非天然氨基酸在多肽链上的每个位置中进行连续取代,并且观察对多肽的活性的影响。用于选择用于用非天然氨基酸进行取代以进入任何多肽中的位置的任何手段、技术或方法都适于在本文所述的方法、技术和组合物中使用。
[0616]
还可考查多肽的含有缺失的天然存在的突变体的结构和活性以确定蛋白质的可能耐受用非天然氨基酸进行取代的区域。一旦已经消除对于用非天然氨基酸取代可能不耐受的残基,可使用包括但不限于相关多肽以及任何相关配体或结合蛋白的三维结构的方法来检查所提出的取代在各个剩余位置处的影响。许多多肽的x射线晶体学和nmr结构可在蛋
白质数据库(pdb,www.rcsb.org)中获得,所述蛋白质数据库是一种含有大分子蛋白质和核酸的三维结构数据的集中式数据库,可用于鉴定可用非天然氨基酸取代的氨基酸位置。另外,如果三维结构数据不可用,那么可制作研究多肽的二级和三级结构的模型。因此,可易于获得可被非天然氨基酸取代的氨基酸位置的身份。
[0617]
非天然氨基酸的示例性掺入位点包括但不限于从潜在受体结合区排除的那些,或者,用于与结合蛋白或配体结合的区域可被完全地或部分地暴露于溶剂,与附近残基具有最小或不具有氢键合相互作用,可最小程度地暴露于附近反应性残基,和/或可以在高度柔性区域中,如通过特定多肽与它的相关受体、配体或结合蛋白一起的三维晶体结构所预测。
[0618]
广泛多种非天然氨基酸可取代多肽中的给定位置,或掺入到所述位置中。举例来说,基于对多肽与它的相关配体、受体和/或结合蛋白一起的三维晶体结构的检查、对保守取代的优先选择,可选择用于掺入的特定非天然氨基酸。
[0619]
在一个实施方案中,本文所述的方法包括将非天然氨基酸掺入tc的靶向多肽中,其中tc的靶向多肽包含第一反应性基团;以及使tc的靶向多肽与包含第二反应性基团的分子(包括但不限于第二蛋白质或多肽或多肽类似物;抗体或抗体片段;以及它们的任何组合)接触。在某些实施方案中,第一反应性基团是羟胺部分,并且第二反应性基团是羰基或二羰基部分,借此形成肟键联。在某些实施方案中,第一反应性基团是羰基或二羰基部分,并且第二反应性基团是羟胺部分,借此形成肟键联。在某些实施方案中,第一反应性基团是羰基或二羰基部分,并且第二反应性基团是肟部分,借此发生肟交换反应。在某些实施方案中,第一反应性基团是肟部分,并且第二反应性基团是羰基或二羰基部分,借此发生肟交换反应。
[0620]
在一些情况下,tc的靶向多肽中掺入的非天然氨基酸将与多肽内的其他添加、取代或缺失组合以影响其他化学、物理、药理学和/或生物性状。在一些情况下,其他添加、取代或缺失可使多肽的稳定性(包括但不限于对蛋白水解降解的抗性)增加,或使多肽对它的适当受体、配体和/或结合蛋白的亲和力增加。在一些情况下,其他添加、取代或缺失可使多肽的溶解性增加(包括但不限于在大肠杆菌或其他宿主细胞中表达时)。在一些实施方案中,选择用天然编码或非天然氨基酸取代的位点,此外还有为了使多肽在大肠杆菌或其他重组宿主细胞中表达后溶解性增加的目的用于掺入非天然氨基酸的另一位点。在一些实施方案中,多肽包含另一添加、取代或缺失,其调节对相关配体、结合蛋白和/或受体的亲和力,调节(包括但不限于增加或降低)受体二聚化,使受体二聚体稳定,调节循环半衰期,调节释放或生物利用度,促进纯化,或改进或改变特定的施用途径。类似地,非天然氨基酸多肽可包含化学或酶裂解序列、蛋白酶裂解序列、反应性基团、抗体结合结构域(包括但不限于flag或多聚组氨酸(poly
‑
his))或其他基于亲和力的序列(包括但不限于flag、多聚组氨酸、gst等)或使多肽的检测(包括但不限于gfp)、纯化、穿过组织或细胞膜的转运、前药释放或活化、尺寸减小或其他性状的改进的连接分子(包括但不限于生物素)。
[0621]
作为靶向部分的示例的抗her2抗体
[0622]
本文描述的方法、组合物、策略和技术不限于靶向部分多肽或蛋白质的特定类型、类别或家族。实际上,几乎任何靶向部分多肽都可以被设计或修饰为包括至少一个“经修饰或未经修饰的”非天然氨基酸,其含有本文所述的tc的靶向多肽。仅举例来说,靶向部分多肽可与选自由以下组成的组的治疗性蛋白质同源:α
‑
1抗胰蛋白酶、血管抑素、抗溶血因子、
抗体、抗体片段、单克隆抗体(例如贝伐单抗、西妥昔单抗、帕尼单抗、英夫利昔单抗、阿达木单抗、巴利昔单抗、达利珠单抗、奥马珠单抗、优特克单抗、依那西普、吉妥珠单抗、阿仑单抗、利妥昔单抗、曲妥珠单抗、尼妥珠单抗、帕利珠单抗和阿昔单抗)、载脂蛋白、脱辅基蛋白、心房利钠因子、心房利钠多肽、心房肽、c
‑
x
‑
c趋化因子、t39765、nap
‑
2、ena
‑
78、gro
‑
a、gro
‑
b、gro
‑
c、ip
‑
10、gcp
‑
2、nap
‑
4、sdf
‑
1、pf4、mig、降钙素、c
‑
kit配体、cc趋化因子、单核细胞趋化蛋白
‑
1、单核细胞趋化蛋白
‑
2、单核细胞趋化蛋白
‑
3、单核细胞炎性蛋白
‑
1α、单核细胞炎性蛋白
‑
iβ、rantes、1309、r83915、r91733、hcc1、t58847、d31065、t64262、cd40、cd40配体、c
‑
kit配体、胶原蛋白、集落刺激因子(csf)、补体因子5a、补体抑制剂、补体受体1、细胞因子、上皮中性粒细胞活化肽
‑
78、mip
‑
16、mcp
‑
1、表皮生长因子(egf)、上皮中性粒细胞活化肽、促红细胞生成素(epo)、脱落毒素、因子ix、因子vii、因子viii、因子x、成纤维细胞生长因子(fgf)、纤维蛋白原、纤连蛋白、四螺旋束蛋白、g
‑
csf、glp
‑
1、gm
‑
csf、葡糖脑苷脂酶、促性腺激素、生长因子、生长因子受体、生长激素释放因子、刺猬(hedgehog)蛋白、血红蛋白、肝细胞生长因子(hgf)、水蛭素、人生长激素(hgh)、人血清白蛋白、icam
‑
1、icam
‑
1受体、lfa
‑
1、lfa
‑
1受体、胰岛素、胰岛素样生长因子(igf)、igf
‑
i、igf
‑
ii、干扰素(ifn)、ifn
‑
α、ifn
‑
β、ifn
‑
γ、白细胞介素(il)、il
‑
1、il
‑
2、il
‑
3、il
‑
4、il
‑
5、il
‑
6、il
‑
7、il
‑
8、il
‑
9、il
‑
10、il
‑
11、il
‑
12、角质细胞生长因子(kgf)、乳铁蛋白、白血病抑制因子、萤光素酶、神经秩蛋白、中性粒细胞抑制因子(nif)、抑瘤素m、成骨蛋白、癌基因产物、旁降钙素(paracitonin)、甲状旁腺激素(pth)、pd
‑
ecgf、pdgf、肽激素、多效生长因子(pleiotropin)、蛋白a、蛋白g、致热外毒素a、致热外毒素b、致热外毒素c、肽yy(pyy)、松弛素、肾素、scf、小生物合成蛋白、可溶性补体受体i、可溶性i
‑
cam 1、可溶性白细胞介素受体、可溶性tnf受体、生长调节素、生长抑素、生长激素(somatotropin)、链激酶、超抗原、金黄色葡萄球菌肠毒素、sea、seb、sec1、sec2、sec3、sed、see、类固醇激素受体、超氧化物歧化酶、毒性休克综合征毒素、胸腺素α1、组织纤溶酶原激活物、肿瘤生长因子(tgf)、肿瘤坏死因子、肿瘤坏死因子α、肿瘤坏死因子β、肿瘤坏死因子受体(tnfr)、vla
‑
4蛋白、vcam
‑
1蛋白、血管内皮生长因子(vegf)、尿激酶、mos、ras、raf、met、p53、tat、fos、myc、jun、myb、rel、雌激素受体、孕激素受体、睾酮受体、醛固酮受体、ldl受体和皮质酮等。
[0623]
在一个实施方案中是治疗过表达her
‑
2的实体瘤的方法,所述实体瘤选自由以下组成的组:乳腺癌、小细胞肺癌、卵巢癌、子宫内膜癌、膀胱癌、头颈癌、前列腺癌、胃癌、宫颈癌、子宫癌、食道癌和结肠癌。在另一个实施方案中,实体瘤是乳腺癌。在一个另外的实施方案中,实体瘤是卵巢癌。
[0624]
因此,提供曲妥珠单抗的以下描述仅用于说明性目的,并且仅举例来说,不是对本文所述的方法、组合物、策略和技术范围的限制。此外,在本技术中提及曲妥珠单抗旨在使用通用术语作为任何抗体的实例。因而,应当理解,本文关于曲妥珠单抗描述的修饰和化学同样可以应用于任何抗体或单克隆抗体,包括本文具体列出的那些。
[0625]
曲妥珠单抗是一种人源化单克隆抗体,其与her2/neu受体的胞外段的结构域iv结合。her2基因(也称为her2/neu和erbb2基因)在20%
‑
30%的早期乳腺癌中扩增,使得它过表达。而且,在癌症中,her2可能在有丝分裂原没有到达并与任何受体结合的情况下发送信号,使其活性过度。
[0626]
her2延伸穿过细胞膜,将信号从细胞外传递到细胞内。在健康人体内,称为有丝分
裂原的信号传导化合物到达细胞膜,并与her受体家族其他成员的外部结合。这些结合的受体然后与her2连接(二聚化),将其激活。her2然后向细胞内部发送信号。信号穿过不同的生化途径。这包括pi3k/akt途径和mapk途径。这些信号促进细胞的侵袭、存活和血管生长(血管生成)。
[0627]
用曲妥珠单抗处理的细胞在细胞周期的g1期中经历停滞,因此增殖降低。已经表明曲妥珠单抗通过下调her2/neu导致受体二聚化的破坏和通过下游pi3k级联的信号传导来诱导其作用的一部分。然后,p27kip1不被磷酸化,并且能够进入细胞核并抑制cdk2活性,从而引起细胞周期停滞。而且,曲妥珠单抗通过诱导抗血管生成因子和抑制促血管生成因子来抑制血管生成。人们认为,对癌症中观察到的不受调节的生长的原因可能是由于her2/neu的蛋白水解裂解导致细胞外结构域的释放所致。已经证明曲妥珠单抗抑制乳腺癌细胞中的her2/neu胞外结构域裂解。
[0628]
在非真核生物和真核生物中表达
[0629]
为了获得克隆的tc多核苷酸的高水平表达,通常将编码本发明的tc多肽的靶向多肽的多核苷酸亚克隆到表达载体中,所述表达载体含有指导转录的强启动子、转录/翻译终止子以及(如果是针对编码蛋白质的核酸的话)用于翻译起始的核糖体结合位点。适合的细菌启动子是本领域普通技术人员已知的并且由例如sambrook等人和ausubel等人描述。
[0630]
用于表达本发明的tc多肽的细菌表达系统可在下列细菌中获得,包括但不限于,大肠杆菌、芽孢杆菌属种、荧光假单胞菌、铜绿假单胞菌、恶臭假单胞菌和沙门菌属(palva等人,gene 22:229
‑
235(1983);mosbach等人,nature 302:543
‑
545(1983))。用于这样的表达系统的试剂盒可商购获得。用于哺乳动物细胞、酵母和昆虫细胞的真核表达系统是本领域普通技术人员已知的,并且也可商购获得。在使用正交trna和氨酰trna合成酶(如上所述)表达本发明的tc多肽的情况下,用于表达的宿主细胞的选择根据是它们使用正交组分的能力。示例性宿主细胞包括革兰氏阳性菌(包括但不限于短芽孢杆菌、枯草芽孢杆菌或链霉菌)和革兰氏阴性菌(大肠杆菌、荧光假单胞菌、铜绿假单胞菌、恶臭假单胞菌),以及酵母和其他真核细胞。如本文所述,可以使用包含o
‑
trna/o
‑
rs对的细胞。
[0631]
本发明的真核宿主细胞或非真核宿主细胞提供了以大有用量合成包含非天然氨基酸的蛋白质的能力。在一方面,组合物任选地包含但不限于至少10微克、至少50微克、至少75微克、至少100微克、至少200微克、至少250毫克、至少500毫克、至少1毫克、至少10毫克、至少100毫克、至少一克或更多的包含非天然氨基酸的蛋白质,或包含使用体内蛋白质生产方法(关于重组蛋白质生产和纯化的细节在本文提供)可实现的量。在另一方面,蛋白质任选地以包括但不限于每升至少10微克蛋白质、每升至少50微克蛋白质、每升至少75微克蛋白质、每升至少100微克蛋白质、每升至少200微克蛋白质、每升至少250微克蛋白质、每升至少500微克蛋白质、每升至少1毫克蛋白质或每升至少10毫克蛋白质或更高的浓度存在于组合物中,其在包括但不限于细胞裂解物、缓冲液、药物缓冲液或其他液体悬浮液中(包括但不限于,以包括但不限于从约1nl至约100l或更大的体积)。在真核细胞中产生包含至少一个非天然氨基酸的大量(包括但不限于,比用其他方法通常可能产生的量更多,包括但不限于,体外翻译)蛋白质是本发明的特征。
[0632]
编码tc多肽的靶向多肽的核苷酸序列可包含或可不包含编码信号肽的序列。当多肽有待从表达它的细胞中分泌出来时,存在信号肽。这样的信号肽可以是任何序列。信号肽
可以是原核的或真核的。coloma,m(1992)j.imm.methods 152:89 104)描述了用于哺乳动物细胞中的信号肽(鼠igκ轻链信号肽)。其他信号肽包括但不限于来自酿酒酵母的α
‑
因子信号肽(美国专利第4,870,008号,将其通过引用并入本文)、小鼠唾液淀粉酶的信号肽(o.hagenbuchle等人,nature 289,1981,第643
‑
646页)、经修饰的羧肽酶信号肽(l.a.valls等人,cell48,1987,第887
‑
897页)、酵母bar1信号肽(wo 87/02670,将其通过引用并入本文)以及酵母天冬氨酸蛋白酶3(yap3)信号肽(参见m.egel
‑
mitani等人,yeast 6,1990,第127
‑
137页)。
[0633]
适合的哺乳动物宿主细胞的实例是本领域普通技术人员已知的。这样的宿主细胞可以是中国仓鼠卵巢(cho)细胞(例如cho
‑
k1;atcc ccl
‑
61)、绿猴细胞(cos)(例如cos 1(atcc crl
‑
1650)、cos7(atcc crl
‑
1651));小鼠细胞(例如ns/o)、幼仓鼠肾(bhk)细胞系(例如atcc crl
‑
1632或atcc ccl
‑
10)和人细胞(例如hek 293(atcc crl
‑
1573)),以及组织培养中的植物细胞。这些细胞系和其他细胞系可从诸如美国典型培养物保藏中心(rockville,md.)的公共库获得。为了提供tc多肽的改进的糖基化,可以将哺乳动物宿主细胞修饰为表达唾液酸转移酶,例如1,6
‑
唾液酸转移酶,例如如美国专利第5,047,335号所述,将其通过引用并入本文。
[0634]
将外源dna引入哺乳动物宿主细胞的方法包括但不限于磷酸钙介导的转染、电穿孔、deae
‑
葡聚糖介导的转染、脂质体介导的转染、病毒载体以及由使用lipofectamin 2000的life technologies ltd(paisley,uk)和使用fugene 6的roche diagnostics corporation(indianapolis,usa)描述的转染方法。这些方法是本领域熟知的,并且描述于ausbel等人(编辑),1996,current protocols in molecular biology,john wiley&sons,new york,usa。哺乳动物细胞的培养可以根据已确立的方法来进行,例如公开于(animal cell biotechnology,methods and protocols,nigel jenkins编辑,1999,human press inc.totowa,n.j.,usa,以及harrison mass.和rae if,general techniques of cell culture,cambridge university press 1997)。
[0635]
i.大肠杆菌、假单胞菌属种和其他原核生物细菌表达技术是本领域普通技术人员已知的。广泛的多种载体可用于细菌宿主。载体可以是单拷贝或低或高多拷贝载体。载体可用于克隆和/或表达。鉴于关于载体的大量文献、许多载体的商业可用性以及甚至描述载体及其限制图和特征的手册,这里不需要广泛讨论。众所周知,载体通常包括允许选择的标志物,这些标志物可以提供细胞毒性剂抗性、原养型或免疫。通常,存在多种提供不同特征的标志物。
[0636]
细菌启动子是能够结合细菌rna聚合酶并启动编码序列(例如结构基因)下游(3’)转录成mrna的任何dna序列。启动子具有通常置于编码序列的5’端近端的转录起始区。这个转录起始区通常包括rna聚合酶结合位点和转录起始位点。细菌启动子也可具有称为操作子的第二结构域,其可与开始rna合成的相邻rna聚合酶结合位点重叠。操作子允许负调控(诱导型)转录,因为基因阻遏蛋白可结合操纵子,从而抑制特定基因的转录。组成型表达可在不存在诸如操作子的负调控元件的情况下发生。另外,可通过基因激活蛋白结合序列来实现正调控,所述基因激活蛋白结合序列(如果存在的话)通常在rna聚合酶结合序列的近端(5')。基因激活蛋白的实例是分解代谢物激活蛋白(cap),其帮助起始lac操纵子在大肠杆菌(e.coli)中的转录(参见,raibaud等人,annu.rev.genet.(1984)18:173)。因此,受调
控的表达可以是正向或负向的,从而增强或减弱转录。
[0637]
术语“细菌宿主”或“细菌宿主细胞”是指可以或已经用作重组载体或其他转移dna的受体的细菌该术语包括已被转染的原始细菌宿主细胞的后代。应当理解,由于意外或有意突变,单个亲本细胞的后代在形态学上或在与原始亲本互补的基因组dna或总dna上可能未必完全相同。与将以相关特性(比如编码tc多肽的核苷酸序列的存在)表征的亲本充分相似的亲本细胞的后代被包括在由这个定义所指的后代中。
[0638]
用于表达tc多肽的适合的宿主细菌的选择是本领域普通技术人员已知的。在选择用于表达的细菌宿主时,适合的宿主可包括那些显示特别是具有良好的包涵体形成能力、低蛋白水解活性和总体稳健性的宿主。细菌宿主通常可从多种来源获得,包括但不限于加利福尼亚大学(伯克利分校)生物物理学及医学物理学系细菌遗传储备中心(bacterial genetic stock center,department of biophysics and medical physics,university of california(berkeley,ca));以及美国典型培养物保藏中心(“atcc”)(manassas,va)。工业/药物发酵通常使用源于k菌株(例如w3110)的细菌或源于b菌株(例如bl21)的细菌。这些菌株特别有用,因为它们的生长参数非常广为人知,并且非常具有鲁棒性。另外,这些菌株是非病原性的,出于安全和环境原因,这在商业上是重要的。适合的大肠杆菌宿主的其他实例包括但不限于菌株bl21、dh10b或其衍生物。在本发明的方法的另一个实施方案中,大肠杆菌宿主为蛋白酶阴性菌株,其包括但不限于omp
‑
和lon
‑
。宿主细胞株可以是假单胞菌属的种,包括但不限于荧光假单胞菌、铜绿假单胞菌和恶臭假单胞菌。已知命名为菌株mb101的荧光假单胞菌生物变型1可用于重组生产,并且可用于治疗性蛋白质生产过程。假单胞菌属表达系统的实例包括可从dow chemical company获得的作为宿主菌株的系统(midland,mi,可在万维网dow.com上获得)。
[0639]
一旦建立了重组宿主细胞株(即,已将表达构建体引入宿主细胞中并分离出具有适当表达构建体的宿主细胞),在适于产生tc多肽的条件下培养所述重组宿主细胞株。将对本领域技术人员显而易见的是,重组宿主细胞株的培养方法将取决于所利用的表达构建体的性质和宿主细胞的身份。通常使用本领域普通技术人员已知的方法培养重组宿主菌株。重组宿主细胞通常在液体培养基中培养,所述液体培养基含有可同化的碳、氮和无机盐来源,并且任选地含有维生素、氨基酸、生长因子和本领域普通技术人员已知的其他蛋白质培养补充剂。用于培养宿主细胞的液体培养基可任选地含有抗生素或抗真菌剂,以防止不良微生物和/或化合物的生长,所述抗生素或抗真菌剂包括但不限于选择含有表达载体的宿主细胞的抗生素。
[0640]
可以分批或连续形式培养重组宿主细胞,其中细胞收获(在tc多肽在细胞内积聚的情况下)或培养物上清液的收获以分批或连续形式进行。对于在原核宿主细胞中的生产,优选分批培养和细胞收获。
[0641]
通常在重组系统中表达之后纯化本发明的tc多肽。可通过本领域已知的多种方法从宿主细胞或培养基纯化tc多肽。在细菌宿主细胞中产生的tc多肽可能是难溶或不溶的(呈包涵体形式)。在本发明的一个实施方案中,利用本文公开的方法以及本领域已知的方法,可容易地在tc多肽中进行氨基酸取代,所述取代被选择为提高重组产生的蛋白质的溶解度的目的。在不溶性蛋白质的情况下,可通过离心从宿主细胞裂解物收集蛋白质,并且随后可进一步将细胞均质化。在难溶蛋白质的情况下,可添加包括但不限于聚乙烯亚胺(pei)
的化合物来诱导部分可溶性蛋白质的沉淀。然后可通过离心方便地收集沉淀的蛋白质。使用本领域普通技术人员已知的多种方法,可将重组宿主细胞破坏或均质化以从细胞内释放包涵体。可使用熟知的技术进行宿主细胞破坏或均质化,所述技术包括但不限于酶簇细胞破坏、超声处理、杜恩斯均质化(dounce homogenization)或高压释放破坏。在本发明方法的一个实施方案中,使用高压释放技术来破坏大肠杆菌宿主细胞,从而释放tc多肽的包涵体。当处理tc多肽的包涵体时,为了使包涵体的产率最大化而不因比如溶解、机械剪切或蛋白水解等因素导致损失,使重复的均质时间最小化可能是有利的。
[0642]
然后可使用本领域已知的许多适合的增溶剂中的任何一种来溶解不溶或沉淀的tc多肽。可用尿素或盐酸胍溶解tc多肽。应该使溶解的tc多肽的体积最小化,使得可使用可方便管理的批量进行大批量生产。这个因素在大规模商业环境中可能是有意义的,在所述环境的情况下,重组宿主可以成千上万升体积的批量生长。另外,当在大规模商业化环境中制造tc多肽时,特别是用于人药物用途时,如果可能的话,应避免可损坏机器和容器或蛋白质产物本身的刺激性化学物质。在本发明的方法中已经表明,可以使用较温和的变性剂尿素来溶解tc多肽包涵体,以代替较为刺激性的变性剂盐酸胍。尿素的使用显著降低了在制造和纯化tc多肽过程中使用的不锈钢设备损坏的风险,同时有效地溶解tc多肽包涵体。
[0643]
在可溶性的tc蛋白的靶向多肽的情况下,tc的靶向多肽可以分泌到周质空间或培养基中。另外,可溶性tc可以存在于宿主细胞的细胞质中。可能希望在进行纯化步骤之前浓缩可溶性tc。可使用本领域普通技术人员已知的标准技术从例如细胞裂解物或培养基浓缩可溶性靶向多肽。另外,可使用本领域普通技术人员已知的标准技术破坏宿主细胞以及从宿主细胞的细胞质或周质空间释放可溶性tc。
[0644]
一般而言,有时需要使表达的多肽变性和还原并且然后使多肽再折叠成优选的构象。例如,可将胍、尿素、dtt、dte和/或伴侣蛋白添加到感兴趣的翻译产物中。使蛋白质还原、变性和复性的方法是本领域普通技术人员已知的(参见,上述参考文献和debinski等人,(1993)j.biol.chem.,268:14065
‑
14070;kreitman和pastan(1993)bioconjug.chem.,4:581
‑
585;和buchner等人,(1992)anal.biochem.,205:263
‑
270)。例如debinski等人描述了胍
‑
dte中包涵体蛋白的变性和还原。可在含有(包括但不限于)氧化型谷胱甘肽和l
‑
精氨酸的氧化还原缓冲剂中使蛋白质再折叠。可使再折叠试剂流动或以其他方式移动以使其与一种或多种多肽或其他表达产物接触,或反之亦然。
[0645]
在进行tc多肽的原核产生的情况下,如此产生的tc可能被错误折叠,并且因此而缺乏生物活性或生物活性降低。可以通过“再折叠”来恢复蛋白质的生物活性。一般而言,通过使用例如一种或多种离液剂(例如尿素和/或胍)和能够还原二硫键的还原剂(例如二硫苏糖醇、dtt或2
‑
巯基乙醇(2
‑
me))溶解(在tc多肽也是不溶的情况下)、解折叠并还原多肽链,来使错误折叠的tc多肽再折叠。在中等浓度的离液剂下,然后添加氧化剂(例如氧、胱氨酸或胱胺),以允许二硫键再形成。可以使用本领域已知的标准方法,比如在美国专利第4,511,502号、第4,511,503号和第4,512,922号中描述的方法,使tc多肽再折叠,将这些专利通过引用并入本文。tc多肽也可以与其他蛋白质共折叠而形成异二聚体或异源多聚体。
[0646]
在再折叠之后,可以进一步纯化tc的靶向多肽。可以使用本领域普通技术人员已知的多种技术完成tc的纯化,所述技术包括疏水性相互作用色谱法、尺寸排阻色谱法、离子交换色谱法、反相高效液相色谱法、亲和色谱法等或其任何组合。另外的纯化也可包括干燥
或沉淀纯化蛋白质的步骤。
[0647]
纯化后,可通过本领域已知的多种方法中的任何方法,包括但不限于,透滤和透析,将tc的靶向多肽交换到不同的缓冲液中并且/或者浓缩。可使以单一纯化蛋白质形式提供的tc经历聚集和沉淀。
[0648]
纯化的tc的靶向多肽可以为至少90%纯(如通过反相高效液相色谱法、rp
‑
hplc、或十二烷基硫酸钠
‑
聚丙烯酰胺凝胶电泳(sds
‑
page)所测量)、或至少95%纯、或至少96%纯、或至少97%纯、或至少98%纯或至少99%纯或更纯。无论tc的靶向多肽的纯度的确切数值如何,所述tc的靶向多肽对于用作药物产品或用于进一步处理,比如与诸如peg的水溶性聚合物缀合来说都是足够纯的。
[0649]
某些tc分子可以在不存在其他活性成分或蛋白质(除赋形剂、载体和稳定剂、血清白蛋白等以外)的情况下用作治疗剂,或者它们可与另一种蛋白质或聚合物复合。
[0650]
此前已表明,通过向蛋白质合成反应加入化学氨酰化的抑制trna,非天然氨基酸可以被位点特异性地体外掺入蛋白质中,其中所述蛋白质合成反应用含有期望的琥珀无义突变的基因编程。使用这些方法,人们可以用紧密结构同源物取代多种常见的二十种氨基酸,例如,利用对特定氨基酸营养缺陷的菌株,用氟苯丙氨酸取代苯丙氨酸。参见,例如noren,c.j.,anthony
‑
cahill,griffith,m.c.,schultz,p.g.a general method for site
‑
specific incorporation of unnatural amino acids into proteins,science,244:182
‑
188(1989);m.w.nowak等人,science268:439
‑
42(1995);bain,j.d.,glabe,c.g.,dix,t.a.,chamberlin,a.r.,diala,e.s.biosynthetic site
‑
specific incorporation of a non
‑
natural amino acid into a polypeptide,j.am chem soc,111:8013
‑
8014(1989);n.budisa等人,faseb j.13:41
‑
51(1999);ellman,j.a.,mendel,d.,anthony
‑
cahill,s.,noren,c.j.,schultz,p.g.biosynthetic method for introducing unnatural amino acids site
‑
specifically into proteins,methods in enz.,第202卷,301
‑
336(1992);以及,mendel,d.,cornish,v.w.&schultz,p.g.site
‑
directed mutagenesis with an expanded genetic code,annu rev biophys.biomol struct.24,435
‑
62(1995)。
[0651]
例如,制备了识别终止密码子uag的抑制trna,并用非天然氨基酸将其化学氨酰化。使用常规的定点诱变在蛋白质基因中感兴趣的位点处引入终止密码子tag。参见,例如sayers,j.r.,schmidt,w.eckstein,f.5'
‑
3'exonucleases in phosphorothioate
‑
based oligno ucleotide
‑
directed mutagensis,nucleic acids res,16(3):791
‑
802(1988)。当酰化抑制trna和突变基因在体外转录/翻译系统中组合时,响应于uag密码子掺入非天然氨基酸,从而产生含有在指定位置处的该氨基酸的蛋白质。使用[3h]
‑
phe的实验和使用α
‑
羟基酸的实验证明,期望的氨基酸仅在由uag密码子指定的位置掺入,并且该氨基酸不掺入蛋白质中的任何其他位点。参见,例如noren等人,上文;kobayashi等人,(2003)nature structural biology 10(6):425
‑
432;以及ellman,j.a.,mendel,d.,schultz,p.g.site
‑
specific incorporation of novel backbone structures into proteins,science,255(5041):197
‑
200(1992)。
[0652]
可以通过任何方法或技术,包括但不限于化学或酶促氨酰化,用期望的氨基酸将trna氨酰化。
[0653]
通过氨酰trna合成酶或其他酶分子,包括但不限于核酶,可以完成氨酰化。术语“核酶”可与“催化rna”互换。cech和coworkers(cech,1987,science,236:1532
‑
1539;mccorkle等人,1987,concepts biochem.64:221
‑
226)证明了可以充当催化剂(核酶)的天然存在的rna的存在。然而,尽管这些天然的rna催化剂仅被证明作用于核糖核酸底物进行裂解和剪接,但是核糖酶人工进化的最新发展已经将催化谱扩展到各种化学反应。研究已经鉴定了可催化其自身(2')3
’‑
末端的氨酰基
‑
rna键的rna分子(illangakekare等人,1995science267:643
‑
647),以及可将氨基酸从一个rna分子转移到另一个rna分子的rna分子(lohse等人,1996,nature 381:442
‑
444)。
[0654]
美国专利申请公开2003/0228593描述了构建核酶的方法及其在用天然编码和非天然编码氨基酸将trna氨酰化中的用途,将该专利申请公开通过引用并入本文。可将trna氨酰化的酶分子(包括但不限于核酶)的底物固定形式可以使氨酰化产物的有效亲和纯化成为可能。适合的底物的实例包括琼脂糖、琼脂糖凝胶和磁珠。用于氨酰化的核酶的底物固定形式的生产和用途描述于chemistry and biology2003,10:1077
‑
1084和美国专利申请公开2003/0228593中,将这些文献通过引用并入本文。
[0655]
为了避免在氨酰化中使用合成酶,化学氨酰化方法包括但不限于由下列文献介绍的那些:hecht和同事(hecht,s.m.acc.chem.res.1992,25,545;heckler,t.g.;roesser,j.r.;xu,c.;chang,p.;hecht,s.m.biochemistry 1988,27,7254;hecht,s.m.;alford,b.l.;kuroda,y.;kitano,s.j.biol.chem.1978,253,4517)以及schultz、chamberlin、dougherty等人(cornish,v.w.;mendel,d.;schultz,p.g.angew.chem.int.ed.engl.1995,34,621;robertson,s.a.;ellman,j.a.;schultz,p.g.j.am.chem.soc.1991,113,2722;noren,c.j.;anthony
‑
cahill,s.j.;griffith,m.c.;schultz,p.g.science 1989,244,182;bain,j.d.;glabe,c.g.;dix,t.a.;chamberlin,a.r.j.am.chem.soc.1989,111,8013;bain,j.d.等人,nature 1992,356,537;gallivan,j.p.;lester,h.a.;dougherty,d.a.chem.biol.1997,4,740;turcatti等人,j.biol.chem.1996,271,19991;nowak,m.w.等人,science,1995,268,439;saks,m.e.等人,j.biol.chem.1996,271,23169;hohsaka,t.等人,j.am.chem.soc.1999,121,34),将这些文献通过引用并入本文。可以使用这样的方法或其他化学氨酰化方法将trna分子氨酰化。
[0656]
生成催化性rna的方法可涉及生成随机核酶序列的独立集合,对集合进行定向进化,筛选具有希望的氨酰化活性的集合,以及选择那些展示出希望的氨酰化活性的核酶的序列。
[0657]
还可以使用重组翻译系统。纯化翻译因子的混合物也已经成功地用于将mrna翻译成蛋白质,此外还有裂解物或补充有纯化翻译因子比如起始因子
‑
1(if
‑
1)、if
‑
2、if
‑
3(α或β)、延伸因子t(ef
‑
tu)或终止因子的裂解物的组合。无细胞系统也可以是偶联的转录/翻译系统,其中将dna引入该系统中,转录成mrna,并且将mrna翻译,如描述于current protocols in molecular biology(f.m.ausubel等人,editors,wiley interscience,1993)中,将所述文献通过引用明确并入本文。在真核转录系统中转录的rna可以是异核rna(hnrna)或5
′‑
端帽(7
‑
甲基鸟苷)和3
′‑
端多poly a尾成熟mrna的形式,这在某些翻译系统中可能是优势。例如,在网织红细胞裂解物系统中以高效率翻译加帽的mrna。
[0658]
与tc多肽偶联的大分子聚合物
[0659]
可以使用本文所述的组合物、方法、技术和策略对本文所述的非天然氨基酸多肽进行各种修饰。这些修饰包括将另外的官能性掺入到多肽的非天然氨基酸组分上,包括但不限于,标记物;染料;聚合物;水溶性聚合物;聚乙二醇的衍生物;光交联剂;放射性核素;细胞毒性化合物;药物;亲和标记物;光亲和标记物;反应性化合物;树脂;第二种蛋白质或多肽或多肽类似物;抗体或抗体片段;金属螯合剂;辅因子;脂肪酸;碳水化合物;多核苷酸;dna;rna;反义多核苷酸;糖类;水溶性树枝状聚合物;环糊精;抑制性核糖核酸;生物材料;纳米颗粒;自旋标记物;荧光团;含金属部分;放射性部分;新官能团;与其他分子共价或非共价相互作用的基团;光笼蔽部分;光化辐射可激发部分;光异构化部分;生物素;生物素衍生物;生物素类似物;掺入重原子的部分;化学可裂解基团;光可裂解基团;延长的侧链;碳连接的糖;氧化还原活性剂;氨基硫代酸;毒性部分;同位素标记部分;生物物理探针;磷光基团;化学发光基团;电子致密基团;磁性基团;嵌入基团;发色团;能量转移剂;生物活性剂;可检测标记物;小分子;量子点;纳米发射器;放射性核素;放射性发射器;中子俘获剂,或上述或任何其他期望的化合物或物质的任何组合。作为本文所述的组合物、方法、技术和策略的说明性的、非限制性的实例,以下描述将集中于向非天然氨基酸多肽中添加大分子聚合物,其中要理解的是,本文所述的组合物、方法、技术和策略也适用于添加其他官能性,包括但不限于上面列出的那些(如有必要,可进行适当的修改,并且本领域技术人员可根据本文的公开内容进行修改)。
[0660]
广泛多种的大分子聚合物和其他分子可以与本发明的tc多肽连接,以调节tc多肽的生物特性和/或对tc分子提供新的生物学特性。这些大分子聚合物可以通过天然编码氨基酸、通过非天然编码氨基酸或天然或非天然氨基酸的任何功能取代基或添加至天然或非天然氨基酸的任何取代基或官能团与tc多肽连接。聚合物的分子量可以是宽范围的,包括但不限于介于约100da与约100,000da之间或更大。聚合物的分子量可以介于约100da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da、1,000da、900da、800da、700da、600da、500da、400da、300da、200da和100da。在一些实施方案中,聚合物的分子量介于约100da与约50,000da之间。在一些实施方案中,聚合物的分子量介于约100da与约40,000da之间。在一些实施方案中,聚合物的分子量介于约1,000da与约40,000da之间。在一些实施方案中,聚合物的分子量介于约5,000da与约40,000da之间。在一些实施方案中,聚合物的分子量介于约10,000da与约40,000da之间。
[0661]
本发明提供了聚合物:蛋白质缀合物的基本均匀的制剂。如本文所用的“基本均匀”意指观察到聚合物:蛋白质缀合物分子大于总蛋白质的一半。聚合物:蛋白质缀合物具有生物活性,并且本文提供的本发明“基本均匀的”聚乙二醇化tc多肽制剂是那些足够均匀以显示均匀制剂的优点的制剂,所述优点例如在批次间药代动力学的可预测性方面易于临床应用。
[0662]
人们也可以选择制备聚合物:蛋白质缀合物分子的混合物,并且本文提供的优点是人们可以选择包括在混合物中的单聚合物:蛋白质缀合物的比例。因而,如果需要,人们可以制备附接有不同数量的聚合物部分(即,二聚体、三聚体、四聚体等)的各种蛋白质的混
合物,将所述缀合物与使用本发明方法制备的单聚合物:蛋白质缀合物组合,并且得到具有预定比例的单聚合物:蛋白质缀合物的混合物。
[0663]
所选择的聚合物可以是水溶性的,使得与它附接的蛋白质在诸如生理环境的水性环境中不发生沉淀。聚合物可以是支链或非支链的。对于终产品制剂的治疗用途,聚合物将是药学上可接受的。
[0664]
聚合物的实例包括但不限于聚烷基醚及其烷氧基封端的类似物(例如聚氧乙二醇、聚氧乙烯/丙二醇及其甲氧基或乙氧基封端的类似物,特别是聚氧乙二醇,后者也被称为聚乙二醇或peg);聚乙烯吡咯烷酮;聚乙烯烷基醚;聚噁唑啉、聚烷基噁唑啉和聚羟基烷基噁唑啉;聚丙烯酰胺、聚烷基丙烯酰胺和聚羟基烷基丙烯酰胺(例如,聚羟基丙基甲基丙烯酰胺及其衍生物);聚羟基烷基丙烯酸酯;聚唾液酸及其类似物;亲水性肽序列;多糖及其衍生物,包括葡聚糖和葡聚糖衍生物,例如羧甲基葡聚糖、硫酸葡聚糖、氨基葡聚糖;纤维素及其衍生物,例如羧甲基纤维素、羟烷基纤维素;甲壳质及其衍生物,例如壳聚糖、琥珀酰壳聚糖、羧甲基甲壳质、羧甲基壳聚糖;透明质酸及其衍生物;淀粉;藻酸盐;硫酸软骨素;白蛋白;普鲁兰多糖和羧甲基普鲁兰多糖;聚氨基酸及其衍生物,例如聚谷氨酸、聚赖氨酸、聚天冬氨酸、聚天冬酰胺;马来酸酐共聚物,比如:苯乙烯马来酸酐共聚物、二乙烯基乙醚马来酸酐共聚物;聚乙烯醇;它们的共聚物;它们的三元共聚物;它们的混合物;以及前述物质的衍生物。
[0665]
聚乙二醇分子与蛋白质分子的比例将会变化,它们在反应混合物中的浓度也将会变化。一般而言,最佳比例(就反应效率而言,因为存在最少过量的未反应蛋白质或聚合物)可由选择的聚乙二醇的分子量和可用反应性基团的数量决定。就分子量而言,通常聚合物的分子量越高,可以与蛋白质附接的聚合物分子的数量越少。类似地,当优化这些参数时,应该考虑聚合物的支化。通常,分子量越高(或分支越多),聚合物:蛋白质比例越高。
[0666]
如本文所用,当考虑peg:tc多肽缀合物时,术语“治疗有效量”是指给予患者期望的益处的量。所述量在个体之间不同,并且取决于多种因素,包括患者的总体身体状况和待治疗病症的根本原因。用于治疗的tc多肽的量给出了可接受的变化速率,并且将期望的反应维持在有益的水平。可由本领域普通技术人员使用可公开获得的材料和程序容易地确定本发明组合物的治疗有效量。
[0667]
水溶性聚合物可以是任何结构形式,包括但不限于线性的、分叉的或支链的。通常,水溶性聚合物是聚(亚烷基二醇),比如聚(乙二醇)(peg),但也可采用其他水溶性聚合物。举例来说,peg用于描述本发明的某些实施方案。
[0668]
peg是熟知的水溶性聚合物,其可商购获得或者可以根据本领域普通技术人员已知的方法通过乙二醇的开环聚合来制备(sandler和karo,polymer synthesis,academic press,new york,第3卷,第138
‑
161页)。术语“peg”广泛使用以涵盖任何聚乙二醇分子,而不考虑大小或在peg末端的修饰,并且可以通过下式表示为与tc多肽连接:
[0669]
xo
‑
(ch2ch2o)
n
‑
ch2ch2‑
y
[0670]
其中n为2至10,000,x是h或末端修饰,包括但不限于c1‑4烷基、保护基团或末端官能团。
[0671]
在一些情况下,本发明中使用的peg的一端以羟基或甲氧基终止,即,x是氢或ch3(“甲氧基peg”)。可替代地,peg可以反应性基团终止,从而形成双官能聚合物。典型的反应
性基团可以包括通常用于与20种常见氨基酸中的官能团(包括但不限于马来酰亚胺基团、活化碳酸酯(包括但不限于对硝基苯酯)、活化酯(包括但不限于n
‑
羟基琥珀酰亚胺、对硝基苯酯)和醛)反应的那些反应性基团,以及对20种常见氨基酸为惰性但与存在于非天然编码氨基酸中的互补官能团(包括但不限于叠氮基、炔基)特异性地反应的官能团。注意的是,在上面的式中用y表示的peg的另一端,将通过天然存在或非天然编码氨基酸直接或间接地与tc靶向多肽附接。例如,y可以是与多肽的胺基(包括但不限于赖氨酸的ε胺或n端)连接的酰胺、氨基甲酸酯或脲。可替代地,y可以是与硫醇基(包括但不限于半胱氨酸的硫醇基)连接的马来酰亚胺。可替代地,y可以是与通过20种常见氨基酸不常接近的残基的键联。例如,peg上的叠氮基可以与tc多肽上的炔基反应,形成huisgen[3 2]环加成产物。可替代地,peg上的炔基可以与非天然编码氨基酸中存在的叠氮基反应,形成类似的产物。在一些实施方案中,在适用的情况下,强亲核试剂(包括但不限于肼、酰肼、羟胺、氨基脲)可以与非天然编码氨基酸中存在的醛基或酮基反应,形成腙、肟或氨基脲(在一些情况下可以通过用适当的还原剂处理来进一步还原)。可替代地,强亲核试剂可以通过非天然编码氨基酸掺入到tc多肽中,并用于优先与水溶性聚合物中存在的酮基或醛基反应。
[0672]
可根据实际需要使用peg的任何分子量,包括但不限于根据需要约100道尔顿(da)至100,000da或更大(包括但不限于有时0.1
‑
50kda或10
‑
40kda)。peg的分子量可以是宽范围的,包括但不限于介于约100da与约100,000da之间或更大。peg可以介于约100da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da、1,000da、900da、800da、700da、600da、500da、400da、300da、200da和100da。在一些实施方案中,peg介于约100da与约50,000da之间。在一些实施方案中,peg介于约100da与约40,000da之间。在一些实施方案中,peg介于约1,000da与约40,000da之间。在一些实施方案中,peg介于约5,000da与约40,000da之间。在一些实施方案中,peg介于约10,000da与约40,000da之间。也可以使用支链peg,包括但不限于每条链具有1
‑
100kda(包括但不限于1
‑
50kda或5
‑
20kda)范围的分子量的peg分子。支链peg的每条链的分子量可以是,包括但不限于,介于约1,000da与约100,000da或更大。支链peg的每条链的分子量可以介于约1,000da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da和1,000da。在一些实施方案中,支链peg的每条链的分子量介于约1,000da与约50,000da之间。在一些实施方案中,支链peg的每条链的分子量介于约1,000da与约40,000da之间。在一些实施方案中,支链peg的每条链的分子量介于约5,000da与约40,000da之间。在一些实施方案中,支链peg的每条链的分子量介于约5,000da与约20,000da之间。多种peg分子描述于,包括但不限于,shearwater polymers公司目录、nektar therapeutics目录,将其通过引用并入本文。
[0673]
通常,peg分子的至少一个末端可用于与非天然编码氨基酸反应。例如,带有与氨基酸侧链反应的炔和叠氮化物部分的peg衍生物或的tlr接头衍生物可用于将peg与本文所
述的非天然编码氨基酸附接。如果非天然编码氨基酸包含叠氮化物,那么peg通常将含有炔部分以实现[3 2]环加成产物的形成,或者含有膦基团的活化peg种类(即酯、碳酸酯)以实现酰胺键联的形成。可替代地,如果非天然编码氨基酸包含炔,那么peg通常将含有叠氮化物部分以实现[3 2]huisgen环加成产物的形成。如果非天然编码氨基酸包含羰基,那么peg通常将包含有效的亲核试剂(包括但不限于酰肼、肼、羟胺或氨基脲官能性),以便分别实现相应的腙、肟和氨基脲键联的形成。在其他替代方案中,可以使用与上述反应性基团相反的方向,即,非天然编码氨基酸中的叠氮化物部分可以与含有炔的peg衍生物反应。
[0674]
在一些实施方案中,具有peg衍生物的tc多肽含有与非天然编码氨基酸侧链上存在的化学官能性反应的化学官能性。
[0675]
在一些实施方案中,本发明提供了含叠氮化物和乙炔的聚合物衍生物,其包含具有约800da至约100,000da的平均分子量的水溶性聚合物主链。水溶性聚合物的聚合物主链可以是聚乙二醇。然而,应该理解的是,多种水溶性聚合物,包括但不限于聚乙二醇和其他有关聚合物,包括聚(葡聚糖)和聚(丙二醇),也适用于本发明的实践,并且术语peg或聚(乙二醇)的使用旨在涵盖和包括所有这样的分子。术语peg包括但不限于任何其形式的聚乙二醇,包括双官能peg、多臂peg、衍生化peg、分叉的peg、支链peg、悬垂的peg(即具有一个或多个悬垂于聚合物主链的官能团的peg或相关聚合物)或其中具有可降解键联的peg。
[0676]
peg通常是透明、无色、无味、可溶于水、对热稳定、对许多化学试剂是惰性的、不水解或变质,并且通常是无毒的。聚乙二醇被认为是生物相容的,也就是说peg能够与活组织或生物体共存而不引起伤害。更具体地说,peg基本上是非免疫原性的,也就是说peg不倾向于在体内产生免疫应答。当与体内具有某种期望功能的分子比如生物活性剂附接时,peg倾向于掩蔽所述生物活性剂,并且可以减少或消除任何免疫应答,使得生物体可耐受所述生物活性剂的存在。peg缀合物倾向于不产生显著免疫应答或引起凝血或其他不良作用。具有式
‑‑
ch2ch2o
‑‑
(ch2ch2o)
n
‑‑
ch2ch2‑‑
的peg适用于本发明,其中n为约3至约4000,通常为约20至约2000。具有约800da至约100,000da分子量的peg在本发明的一些实施方案中作为聚合物主链特别有用。peg的分子量可以是宽范围的,包括但不限于介于约100da与约100,000da之间或更大。peg的分子量可以介于约100da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da、1,000da、900da、800da、700da、600da、500da、400da、300da、200da和100da。在一些实施方案中,peg的分子量介于约100da与约50,000da之间。在一些实施方案中,peg的分子量介于约100da与约40,000da之间。在一些实施方案中,peg的分子量介于约1,000da与约40,000da之间。在一些实施方案中,peg的分子量介于约5,000da与约40,000da之间。在一些实施方案中,peg的分子量介于约10,000da与约40,000da之间。
[0677]
聚合物主链可以是线性的或支链的。支链聚合物主链通常是本领域已知的。通常,支链聚合物具有中心分支核部分和多个与中心分支核连接的线性聚合物链。peg通常以分支形式使用,可以通过向诸如甘油、甘油低聚物、季戊四醇和山梨醇之类的各种多元醇添加环氧乙烷来制备所述分支形式。中心分支部分也可来源于几种氨基酸,比如赖氨酸。支链聚乙二醇可以表示为一般形式r(
‑
peg
‑
oh)
m
,其中r来源于核部分,比如甘油、甘油低聚物或季
戊四醇,并且m表示臂的数量。多臂peg分子,例如描述于美国专利第5,932,462号;第5,643,575号;第5,229,490号;第4,289,872号;美国专利申请2003/0143596;wo 96/21469;和wo 93/21259中的那些,也可以用作聚合物主链,将这些专利的每一篇通过引用完整并入本文。
[0678]
支链peg也可以是由peg(
‑‑
ychz2)
n
表示的分叉peg的形式,其中y是连接基团,并且z是通过限定长度的原子链与ch连接的活化端基。又另一种分支形式悬垂的peg,具有沿着peg主链而不是在peg链的末端的反应性基团,比如羧基。
[0679]
除了这些形式的peg之外,还可以用主链中弱的或可降解的键联来制备聚合物。例如,可以通过使聚合物主链中的酯键联经历水解来制备peg。如下所示,这种水解导致聚合物裂解成较低分子量的片段:
[0680]
‑
peg
‑
co2‑
peg
‑
h2o
→
peg
‑
co2h ho
‑
peg
‑
[0681]
本领域普通技术人员应当理解,术语聚(乙二醇)或peg代表或包括本领域已知的所有形式,包括但不限于本文公开的那些。
[0682]
许多其他聚合物也适用于本发明。在一些实施方案中,具有2个至约300个末端的水溶性聚合物主链在本发明中是特别有用的。适合的聚合物的实例包括但不限于其它聚(亚烷基二醇),比如聚(丙二醇)(“ppg”)、其共聚物(包括但不限于乙二醇和丙二醇的共聚物)、其三元共聚物、其混合物等。虽然聚合物主链的每条链的分子量可以变化,但它通常在约800da至约100,000da的范围内,通常为约6,000da至约80,000da。聚合物主链的每条链的分子量可以介于约100da与约100,000da之间,包括但不限于100,000da、95,000da、90,000da、85,000da、80,000da、75,000da、70,000da、65,000da、60,000da、55,000da、50,000da、45,000da、40,000da、35,000da、30,000da、25,000da、20,000da、15,000da、10,000da、9,000da、8,000da、7,000da、6,000da、5,000da、4,000da、3,000da、2,000da、1,000da、900da、800da、700da、600da、500da、400da、300da、200da和100da。在一些实施方案中,聚合物主链的每条链的分子量介于约100da与约50,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约100da与约40,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约1,000da与约40,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约5,000da与约40,000da之间。在一些实施方案中,聚合物主链的每条链的分子量介于约10,000da与约40,000da之间。
[0683]
本领域普通技术人员将认识到,基本上水溶性的主链的前述清单决不是穷尽的,而仅仅是说明性的,并且具有上述质量的所有聚合物材料都被认为适用于本发明。
[0684]
在本发明的一些实施方案中,聚合物衍生物是“多官能的”,意味着聚合物主链具有用官能团官能化或活化的至少两个末端,并且可能多达约300个末端。多官能聚合物衍生物包括但不限于具有两个末端的线性聚合物,其中与官能团键合的每个末端可以相同或不同。
[0685]
术语“受保护的”是指保护基团或部分的存在,其防止化学反应性官能团在某些反应条件下的反应。保护基团将根据受保护的化学反应性基团的类型而变化。例如,如果化学反应性基团是胺或酰肼,则保护基团可以选自叔丁氧基羰基(t
‑
boc)和9
‑
芴基甲氧基羰基(fmoc)的组。如果化学反应性基团是硫醇,则保护基团可以是邻二硫吡啶。如果化学反应性基团是羧酸,比如丁酸或丙酸,或羟基,则保护基团可以是苄基或烷基,比如甲基、乙基或叔丁基。本领域已知的其他保护基团也可用于本发明中。
[0686]
文献中末端官能团的具体实例包括但不限于n
‑
琥珀酰亚胺基碳酸酯(参见,例如,美国专利第5,281,698号、第5,468,478号)、胺(参见,例如buckmann等人,makromol.chem.182:1379(1981),zalipsky等人,eur.polym.j.19:1177(1983))、酰肼(参见,例如andresz等人,makromol.chem.179:301(1978))、琥珀酰亚胺丙酸酯和琥珀酰亚胺丁酸酯(参见,例如olson等人,见于poly(ethylene glycol)chemistry&biological applications,第170
‑
181页,harris和zalipsky编辑,acs,washington,d.c.,1997;还参见美国专利第5,672,662号)、琥珀酰亚胺琥珀酸酯(参见,例如abuchowski等人,cancer biochem.biophys.7:175(1984)和joppich等人,makromol.chem.180:1381(1979)、琥珀酰亚胺酯(参见,例如美国专利第4,670,417号)、苯并三唑碳酸酯(参见,例如美国专利第5,650,234号)、甘油醚(参见,例如pitha等人,eur.j biochem.94:11(1979),elling等人,biotech.appl.biochem.13:354(1991)、氧羰基咪唑(参见,例如beauchamp等人,anal.biochem.131:25(1983),tondelli等人j.controlled release 1:251(1985))、对硝基苯基碳酸酯(参见,例如veronese等人,appl.biochem.biotech.,11:141(1985);和sartore等人,appl.biochem.biotech.,27:45(1991))、醛(参见,例如harris等人,j.polym.sci.chem.ed.22:341(1984),美国专利第5,824,784号、美国专利第5,252,714号)、马来酰亚胺(参见,例如goodson等人,biotechnology(ny)8:343(1990),romani等人,见于chemistry of peptides and proteins 2:29(1984)),以及kogan,synthetic comm.22:2417(1992))、邻二硫吡啶(参见,例如woghiren等人,bioconj.chem.4:314(1993))、丙烯酰基(acrylol)(参见,例如sawhney等人,macromolecules,26:581(1993))、乙烯砜(参见,例如美国专利第5,900,461号)。将所有上述参考文献和专利通过引用并入本文。
[0687]
含有非天然编码氨基酸比如对叠氮基
‑
l
‑
苯丙氨酸的tc多肽的聚乙二醇化(即,添加任何水溶性聚合物)是通过任何方便的方法进行的。例如,用炔终止的mpeg衍生物将tc多肽聚乙二醇化。简言之,在室温下,在搅拌下将过量的固体mpeg(5000)
‑
o
‑
ch2‑
c≡ch加入到含对叠氮基
‑
l
‑
phe的tc多肽的水溶液中。通常,将该水溶液用缓冲液缓冲,所述缓冲液具有接近进行反应时的ph值(通常约ph 4
‑
10)的pk
a
。用于在ph 7.5时聚乙二醇化的适合缓冲液的实例包括,例如,但不限于,hepes、磷酸盐、硼酸盐、tris
‑
hcl、epps和tes。如有需要,连续地监测和调节ph。通常允许反应持续约1
‑
48小时。
[0688]
随后使反应产物经受疏水性相互作用色谱,以将聚乙二醇化的tc多肽与游离mpeg(5000)
‑
o
‑
ch2‑
c≡ch和聚乙二醇化的tc多肽的任何高分子量复合物分离,所述复合物可在活化未封闭peg时在分子的两端形成,从而将tc多肽分子交联。疏水相互作用色谱过程中的条件是游离mpeg(5000)
‑
o
‑
ch2‑
c≡ch流过柱,而任何交联的聚乙二醇化tc多肽复合物在期望形式之后洗脱,其含有一个与一个或多个peg基团缀合的tc多肽分子。适合的条件取决于交联复合物相对于期望的缀合物的相对大小而变化,并且容易由本领域普通技术人员确定。将含有期望的缀合物的洗脱液通过超滤浓缩,并通过透滤脱盐。
[0689]
可使用上述洗脱方法产生基本上纯化的peg
‑
tc,其中所产生的peg
‑
tc具有至少约30%、至少约35%、至少约40%、至少约45%、至少约50%、至少约55%、至少约60%、至少约65%、至少约70%的纯度水平,具体地说,至少约75%、80%、85%的纯度水平,并且更具体地至少约90%的纯度水平、至少约95%的纯度水平、至少约99%或更高的纯度水平,正如通
过适当的方法比如sds/page分析、rp
‑
hplc、sec和毛细管电泳确定的。如果需要,可以通过本领域普通技术人员已知的一种或多种程序进一步纯化从疏水色谱法获得的聚乙二醇化tc多肽,所述程序包括但不限于亲和色谱法;阴离子或阳离子交换色谱法(使用,包括但不限于deae sepharose);硅胶色谱法;反相hplc;凝胶过滤(使用,包括但不限于sephadex g
‑
75);疏水性相互作用色谱法;尺寸排阻色谱法、金属螯合物色谱法;超滤/渗滤;乙醇沉淀;硫酸铵沉淀;色谱聚焦;置换色谱法;电泳程序(包括但不限于制备型等电聚焦)、差异溶解度(包括但不限于硫酸铵沉淀)或提取。可以通过gpc通过与球状蛋白标准品进行比较来估计表观分子量(preneta,az in p
rotein purification methods
,a
practical approach
(harris&angal编辑,irl press 1989,293
‑
306)。可以通过蛋白水解降解(包括但不限于胰蛋白酶裂解)然后进行质谱分析来评估tc
‑
peg缀合物的纯度。pepinsky rb.等人,j.pharmcol.&exp.ther.297(3):1059
‑
66(2001)。
[0690]
与本发明tc多肽的靶向多肽的氨基酸连接的水溶性聚合物可以被进一步衍生化或取代,而没有限制。
[0691]
含叠氮化物的peg衍生物或tlr接头衍生物
[0692]
在本发明的另一个实施方案中,将tc的靶向多肽用含有叠氮化物部分的peg衍生物修饰,所述叠氮化物部分将与非天然编码氨基酸侧链上存在的炔部分反应。一般而言,peg衍生物或tlr
‑
接头衍生物将具有范围在1
‑
100kda的平均分子量,并且在一些实施方案中为10
‑
40kda。
[0693]
在一些实施方案中,叠氮化物末端peg衍生物将具有以下结构:
[0694]
ro
‑
(ch2ch2o)
n
‑
o
‑
(ch2)
m
‑
n3[0695]
其中r是简单的烷基(甲基、乙基、丙基等),m为2
‑
10,并且n为100
‑
1,000(即平均分子量介于5
‑
40kda之间)。
[0696]
在另一个实施方案中,叠氮化物末端peg衍生物将具有以下结构:
[0697]
ro
‑
(ch2ch2o)
n
‑
o
‑
(ch2)
m
‑
nh
‑
c(o)
‑
(ch2)
p
‑
n3[0698]
其中r是简单的烷基(甲基、乙基、丙基等),m为2
‑
10,p为2
‑
10,并且n是100
‑
1,000(即,平均分子量在5
‑
40kda之间)。
[0699]
在本发明的另一个实施方案中,将包含含炔氨基酸的tc的靶向多肽用含有末端叠氮化物部分的支链peg衍生物修饰,其中所述支链peg的每条链具有10
‑
40kda的分子量,并且可以为5
‑
20kda。例如,在一些实施方案中,叠氮化物末端peg衍生物将具有以下结构:
[0700]
[ro
‑
(ch2ch2o)
n
‑
o
‑
(ch2)2‑
nh
‑
c(o)]2ch(ch2)
m
‑
x
‑
(ch2)
p
n3[0701]
其中r是简单的烷基(甲基、乙基、丙基等),m为2
‑
10,p为2
‑
10,n为100
‑
1,000,x任选地为o、n、s或羰基(c=o),其在每种情况下可以存在或不存在。
[0702]
含炔peg衍生物或tlr接头衍生物
[0703]
在本发明的另一个实施方案中,将tc的靶向多肽用含有炔部分的peg衍生物修饰,所述炔部分将与非天然编码氨基酸侧链上存在的叠氮化物部分反应。
[0704]
在一些实施方案中,炔末端peg衍生物将具有以下结构:
[0705]
ro
‑
(ch2ch2o)
n
‑
o
‑
(ch2)
m
‑
c≡ch
[0706]
其中r是简单的烷基(甲基、乙基、丙基等),m为2
‑
10,并且n为100
‑
1,000(即平均分子量介于5
‑
40kda之间)。
[0707]
在本发明的另一个实施方案中,将包含含炔非天然编码氨基酸的tc的靶向多肽用含有末端叠氮化物或末端炔部分的peg衍生物修饰,所述末端叠氮化物或末端炔部分通过酰胺键联与peg主链连接。
[0708]
在一些实施方案中,炔末端peg衍生物将具有以下结构:
[0709]
ro
‑
(ch2ch2o)
n
‑
o
‑
(ch2)
m
‑
nh
‑
c(o)
‑
(ch2)
p
‑
c≡ch
[0710]
其中r是简单的烷基(甲基、乙基、丙基等),m为2
‑
10,p为2
‑
10,并且n是100
‑
1,000。
[0711]
在本发明的另一个实施方案中,将包括含叠氮化物的氨基酸的tc的靶向多肽用含有末端炔部分的支链peg衍生物修饰,其中所述支链peg的每条链具有10
‑
40kda的分子量,并且可以为5
‑
20kda。例如,在一些实施方案中,炔末端peg衍生物将具有以下结构:
[0712]
[ro
‑
(ch2ch2o)
n
‑
o
‑
(ch2)2‑
nh
‑
c(o)]2ch(ch2)
m
‑
x
‑
(ch2)
p c≡ch
[0713]
其中r是简单的烷基(甲基、乙基、丙基等),m为2
‑
10,p为2
‑
10,n为100
‑
1,000,并且x任选地为o、n、s或羰基(c=o),或不存在。
[0714]
含膦peg衍生物或tlr接头衍生物
[0715]
在本发明的另一个实施方案中,将tc的靶向多肽用含有活化官能团(包括但不限于酯、碳酸酯)的peg衍生物修饰,所述活化官能团进一步包含芳基膦基团,所述芳基膦基团将与非天然编码氨基酸侧链上存在的叠氮化物部分反应。一般而言,peg衍生物或tlr
‑
接头衍生物将具有范围在1
‑
100kda的平均分子量,并且在一些实施方案中为10
‑
40kda。
[0716]
在一些实施方案中,peg衍生物将具有以下结构:
[0717][0718]
其中n为1
‑
10;x可以是o、n、s或不存在,ph是苯基,并且w是水溶性聚合物。
[0719]
在一些实施方案中,peg衍生物将具有以下结构:
[0720][0721]
其中x可以是o、n、s或不存在,ph是苯基,w是水溶性聚合物,并且r可以是h、烷基、芳基、取代的烷基和取代的芳基。示例性r基团包括但不限于
‑
ch2、
‑
c(ch3)3、
‑
or’、
‑
nr’r”、
‑
sr’、
‑
卤素、
‑
c(o)r’、
‑
conr’r”、
‑
s(o)2r’、
‑
s(o)2nr’r”、
‑
cn和
–
no2。r’、r”、r
”’
和r
””
各自独立地指氢、取代或未取代的杂烷基、取代或未取代的芳基,包括但不限于被1
‑
3个卤素取代的芳基、取代或未取代的烷基、烷氧基或硫代烷氧基或芳烷基。当本发明的化合物包括多于一个r基团时,例如,当多于一个的这些基团存在时,将每个r基团独立地选择为各个r’、r”、r
’”
和r
””
基团。当r'和r”与同一个氮原子附接时,它们可与氮原子结合形成5
‑
、6
‑
或7
‑
元环。例如,
‑
nr’r”意指包括但不限于1
‑
吡咯烷基和4
‑
吗啉基。根据上述对取代基的讨论,本领域技术人员将理解,术语“烷基”意在包括包含与氢基团以外的基团键合的碳原子的基团,比如卤代烷基(包括但不限于
‑
cf3和
–
ch2cf3)和酰基(包括但不限于
‑
c(o)ch3、
‑
c(o)cf3、
‑
c(o)ch2och3等)。
[0722]
其他peg衍生物或tlr
‑
接头衍生物和通用缀合技术
[0723]
可与tc多肽连接的其他示例性peg分子以及聚乙二醇化方法包括但不限于,描述
于例如下列专利中的那些:美国专利公开第2004/0001838号;第2002/0052009号;第2003/0162949号;第2004/0013637号;第2003/0228274号;第2003/0220447号;第2003/0158333号;第2003/0143596号;第2003/0114647号;第2003/0105275号;第2003/0105224号;第2003/0023023号;第2002/0156047号;第2002/0099133号;第2002/0086939号;第2002/0082345号;第2002/0072573号;第2002/0052430号;第2002/0040076号;第2002/0037949号;第2002/0002250号;第2001/0056171号;第2001/0044526号;第2001/0021763号;美国专利第6,646,110号;第5,824,778号;第5,476,653号;第5,219,564号;第5,629,384号;第5,736,625号;第4,902,502号;第5,281,698号;第5,122,614号;第5,473,034号;第5,516,673号;第5,382,657号;第6,552,167号;第6,610,281号;第6,515,100号;第6,461,603号;第6,436,386号;第6,214,966号;第5,990,237号;第5,900,461号;第5,739,208号;第5,672,662号;第5,446,090号;第5,808,096号;第5,612,460号;第5,324,844号;第5,252,714号;第6,420,339号;第6,201,072号;第6,451,346号;第6,306,821号;第5,559,213号;第5,747,646号;第5,834,594号;第5,849,860号;第5,980,948号;第6,004,573号;第6,129,912号;wo 97/32607、ep 229,108、ep 402,378、wo 92/16555、wo 94/04193、wo 94/14758、wo 94/17039、wo 94/18247、wo 94/28024、wo 95/00162、wo 95/11924、wo95/13090、wo 95/33490、wo 96/00080、wo 97/18832、wo 98/41562、wo 98/48837、wo 99/32134、wo 99/32139、wo 99/32140、wo 96/40791、wo 98/32466、wo 95/06058、ep 439 508、wo 97/03106、wo 96/21469、wo 95/13312、ep 921 131、wo 98/05363、ep 809 996、wo 96/41813、wo 96/07670、ep 605 963、ep 510 356、ep 400 472、ep 183 503和ep 154 316,将这些专利通过引用并入本文。本文所述的任何peg分子可以以任何形式使用,包括但不限于单链、支链、多臂链、单官能、双官能、多官能或它们的任何组合。
[0724]
另外的聚合物和peg衍生物或tlr
‑
接头衍生物,包括但不限于羟胺(氨基氧基)peg衍生物或tlr
‑
接头衍生物,描述于以下专利申请中:美国专利公开第2006/0194256号、美国专利公开第2006/0217532号、美国专利公开第2006/0217289号、美国临时专利第60/755,338号;美国临时专利第60/755,711号;美国临时专利第60/755,018号;国际专利申请号pct/us06/49397;wo 2006/069246;美国临时专利第60/743,041;美国临时专利第60/743,040号;国际专利申请号pct/us06/47822;美国临时专利第60/882,819号;美国临时专利第60/882,500号;和美国临时专利第60/870,594号,将所有这些专利申请通过引用完整并入本文。
[0725]
tc多肽的糖基化
[0726]
糖基化可以显著地影响多肽比如tc多肽的物理性质(例如溶解度),并且在蛋白质稳定性、分泌和亚细胞定位方面也是重要的。糖基化多肽还可以表现出增强的稳定性,或者可以改善一种或多种药代动力学特性,比如半衰期。另外,溶解度提高能够例如产生比包含非糖基化多肽的制剂更适合于药物施用的制剂。
[0727]
本发明包括掺入一个或多个带有糖残基的非天然编码氨基酸的tc多肽。糖残基可以是天然的(包括但不限于n
‑
乙酰葡糖胺)或非天然的(包括但不限于3
‑
氟代半乳糖)。糖可以通过n
‑
或o
‑
连接的糖苷键联(包括但不限于n
‑
乙酰半乳糖
‑
l
‑
丝氨酸)或非天然键联(包括但不限于肟或相应的c
‑
或s
‑
连接的糖苷)与非天然编码氨基酸连接。
[0728]
可以在体内或体外将糖(包括但不限于糖基)部分添加到tc多肽中。在发明的一些
实施方案中,可以用经氨基氧基衍生化的糖来修饰包含含羰基的非天然编码氨基酸的tc的靶向多肽,以生成通过肟键联连接的相应的糖基化多肽。一旦与非天然编码氨基酸附接,可以通过用糖基转移酶和其他酶处理来进一步将糖精细加工,以生成与tc多肽结合的寡糖。参见,例如h.liu等人,j.am.chem.soc.125:1702
‑
1703(2003)。
[0729]
在本发明的一些实施方案中,将包含含羰基的非天然编码氨基酸的tc多肽直接用被制备为氨基氧基衍生物的具有确定结构的聚糖修饰。本领域普通技术人员将认识到,可以使用包括叠氮化物、炔烃、酰肼、肼和氨基脲在内的其他官能性将糖与非天然编码氨基酸连接。
[0730]
在本发明的一些实施方案中,然后通过(包括但不限于)分别与包括但不限于炔基或叠氮化物衍生物的huisgen[3 2]环加成反应,可以修饰包含叠氮化物或含炔基的非天然编码氨基酸的tc的靶向多肽。这种方法允许以极高的选择性修饰蛋白质。
[0731]
tc二聚体和多聚体
[0732]
本发明还提供了tc和tc类似物的组合,比如同二聚体、异二聚体、同多聚体或异多聚体(即,三聚体、四聚体等),其中含有一个或多个非天然编码氨基酸的tc直接与另一个tc或任何其他非tc多肽的多肽主链结合或通过接头结合。由于与单体相比其分子量增加,tc二聚体或多聚体缀合物可表现出新的或期望的特性,包括但不限于相对于单体tc的不同药理学、药代动力学、药效学、调节的治疗半衰期或调节的血浆半衰期。在一些实施方案中,本发明的tc二聚体将调节tc受体的信号转导。在其他实施方案中,本发明的tc二聚体或多聚体将充当tc受体拮抗剂、激动剂或调节剂。
[0733]
在一些实施方案中,在含二聚体或多聚体的tc中存在的一个或多个tc分子包含与水溶性聚合物连接的非天然编码氨基酸。
[0734]
在一些实施方案中,tc多肽直接连接,包括但不限于经由asn
‑
lys酰胺键联或cys
‑
cys二硫键联。在一些实施方案中,tc多肽和/或连接的非tc分子将包含促进二聚化的不同的非天然编码氨基酸,所述二聚化包括但不限于,第一tc多肽的一个非天然编码氨基酸中的炔和第二分子的第二非天然编码氨基酸中的叠氮化物将经由huisgen[3 2]环加成而缀合。可替代地,tc和/或包含含酮的非天然编码氨基酸的连接的非tc分子可以与包含含羟胺的非天然编码氨基酸的第二多肽缀合,并且所述多肽经由相应的肟的形成而反应。
[0735]
可替代地,两个tc多肽和/或连接的非肽tc分子经由接头连接。可以使用任何杂
‑
或同
‑
双官能接头来连接两个分子和/或连接的非肽tc分子,它们可以具有相同或不同的一级序列。在一些情况下,用于将tc和/或连接的非肽tc分子栓系在一起的接头可以是双官能peg试剂。接头可具有宽范围的分子量或分子长度。可以使用更大或更小分子量的接头来提供在tc和连接实体之间或在tc和其受体之间或在连接实体和其结合配偶体(如果有的话)之间的期望的空间关系或构象。还可以使用具有更长或更短分子长度的接头来提供在tc和连接实体之间或在连接实体和其结合配偶体(如果有的话)之间的期望的空间或灵活性。
[0736]
在一些实施方案中,本发明提供了具有哑铃结构的水溶性双官能接头,其包括:a)在聚合物主链的至少第一端上的叠氮化物、炔烃、肼、酰肼、羟胺或含羰基部分;和b)在聚合物主链的第二端上的至少第二官能团。第二官能团可以与第一官能团相同或不同。在一些实施方案中,第二官能团不与第一官能团反应。在一些实施方案中,本发明提供了包含支链分子结构的至少一个臂的水溶性化合物。例如,支链分子结构可以是树枝状的。
[0737]
在一些实施方案中,本发明提供了包含一个或多个tc多肽的多聚体,所述多聚体通过与水溶性活化聚合物反应而形成,具有以下结构:r
‑
(ch2ch2o)
n
‑
o
‑
(ch2)
m
‑
x,其中n为约5至3,000,m为2
‑
10,x可以是叠氮化物、炔烃、肼、酰肼、氨基氧基、羟胺、乙酰基或含羰基部分,并且r是可以与x相同或不同的封端基团、官能团或离去基团。r可以是例如选自由以下组成的组的官能团:羟基、受保护的羟基、烷氧基、n
‑
羟基琥珀酰亚胺酯、1
‑
苯并三唑酯、n
‑
羟基琥珀酰亚胺碳酸酯、1
‑
苯并三唑基碳酸酯、缩醛、醛、醛水合物、烯基、丙烯酸酯、甲基丙烯酸酯、丙烯酰胺、活性砜、胺、氨基氧基、保护的胺、酰肼、保护的酰肼、保护的硫醇、羧酸、保护的羧酸、异氰酸酯、异硫氰酸酯、马来酰亚胺、乙烯砜、二硫代吡啶、乙烯基吡啶、碘乙酰胺、环氧化物、乙二醛、二酮、甲磺酸酯、甲苯磺酸酯和三氟乙基磺酸酯(tresylate)、烯烃和酮。
[0738]
tc多肽活性以及tc多肽对her2靶标亲和力的测量
[0739]
tc多肽活性可以使用标准或已知的体外或体内测定法来测定。可以通过本领域已知的适合方法分析tc的生物活性。这样的测定法包括但不限于,tc反应基因的激活、受体结合测定法、抗病毒活性测定法、细胞病变效应抑制测定法、抗增殖测定法、免疫调节测定法和监测mhc分子的诱导的测定法。
[0740]
可以分析tc多肽的激活tc敏感信号转导途径的能力。一个实例是干扰素刺激反应元件(isre)测定法。用isre萤光素酶载体(pisre
‑
luc,clontech)瞬时转染组成型表达tc受体的细胞。在转染之后,用tc的靶向多肽处理细胞。测试多个蛋白质浓度,例如0.0001
‑
10ng/ml,以生成剂量
‑
反应曲线。如果tc多肽结合并激活tc受体,那么产生的信号转导级联诱导萤光素酶表达。可以通过多种方式测量发光,例如使用topcount
tm
或fusion
tm
酶标仪和steady
‑
glo
r
萤光素酶测定系统(promega)。
[0741]
可以分析tc多肽与tc受体结合的能力。对于包含非天然氨基酸的非聚乙二醇化或聚乙二醇化tc多肽,可以通过使用biacore
tm
生物传感器(pharmacia)来测量tc对其受体的亲和力。适合的结合测定法包括但不限于biacore测定法(pearce等人,biochemistry 38:81
‑
89(1999))和alphascreen
tm
测定法(perkinelmer)。
[0742]
无论使用哪种方法产生tc多肽,都要进行tc多肽的生物活性测定。一般而言,生物活性测试应该提供对期望结果的分析,比如生物活性的升高或降低(与修饰的tc相比)、不同的生物活性(与修饰的tc相比)、受体或结合配偶体亲和力分析、tc本身或其受体的构象或结构变化(与修饰的tc相比)或血清半衰期分析。效力、功能性体内半衰期和药代动力学参数的测量
[0743]
本发明的一个重要方面是延长的生物半衰期,这是通过构建具有或没有多肽与水溶性聚合物部缀合的tc而获得的。tc血清浓度的施用后降低速率可使得评价对用缀合和非缀合tc多肽及其变体治疗的生物反应是重要的。本发明的缀合和非缀合的tc多肽及其变体也可以在施用(通过例如皮下或静脉内施用)之后具有延长的血清半衰期,使得可能通过例如elisa法或通过初级筛选测定法进行测量。可以使用商业来源的elisa或ria试剂盒,比如invitrogen(carlsbad,ca)。如本文所述进行体内生物半衰期的测量。
[0744]
根据本领域普通技术人员已知的方案,可以确定包含非天然编码氨基酸的tc的靶向多肽的效力和功能性体内半衰期。
[0745]
包含非天然编码氨基酸的tc多肽的药代动力学参数可以在正常的sprague
‑
dawley雄性大鼠(n=每个治疗组5只动物)中进行评价。动物将接受25ug/只大鼠静脉内注射或50ug/只大鼠皮下注射的单剂量,并且将根据预定的时间进程采集大约5
‑
7个血液样品,所述时间进程对于包含不与水溶性聚合物缀合的非天然编码氨基酸的tc多肽,通常覆盖大约6小时,并且对于包含非天然编码氨基酸并且与水溶性聚合物缀合的tc多肽,覆盖大约4天。针对没有非天然编码氨基酸的tc的药代动力学数据可以直接地与针对包含非天然编码氨基酸的tc多肽获得的数据进行比较。
[0746]
施用与药物组合物
[0747]
本发明的多肽或蛋白质(包括但不限于tc、合成酶、包含一个或多个非天然氨基酸的蛋白质等)任选地用于治疗用途,包括但不限于与适合的药物载体组合。例如,这样的组合物包含治疗有效量的化合物和药学上可接受的载体或赋形剂。这样的载体或赋形剂包括但不限于盐水、缓冲盐水、右旋糖、水、甘油、乙醇和/或其组合。将制剂制成为适合于施用方式。一般而言,施用蛋白质的方法是本领域普通技术人员已知的,并且可应用于本发明多肽的施用。组合物可以是水溶性形式,比如作为药学上可接受的盐存在,这意味着包括酸加成盐和碱加成盐两者。
[0748]
根据本领域普通技术人员已知的方法,任选地在一种或多种适当的疾病的体外模型和/或体内动物模型中测试包含一种或多种本发明多肽的治疗组合物,以确认功效、组织代谢以及估计剂量。具体而言,即,在相关测定法中,通过相对于天然氨基酸同源物的本文非天然氨基酸的活性、稳定性或其他适合的度量(包括但不限于,将修饰为包含一个或多个非天然氨基酸的tc的靶向多肽与天然氨基酸tc多肽进行比较,以及将修饰为包含一个或多个非天然氨基酸的tc的靶向多肽与当前可用的tc治疗进行比较),可以初步确定剂量。
[0749]
施用是通过通常用于引入分子使其与血液或组织细胞最终接触的任何途径。以任何适合的方式施用本发明的非天然氨基酸多肽,任选地与一种或多种药学上可接受的载体一起施用。在本发明背景下向患者施用这样的多肽的适合方法是可获得的,并且,尽管不止一种途径可以用于施用特定组合物,但是某一种特定的途径比其他途径常常可以提供更即时和更有效的作用或反应。
[0750]
药学上可接受的载体部分地取决于所施用的特定组合物以及用于施用所述组合物的特定方法。因此,存在多种本发明药物组合物的适合制剂。
[0751]
本发明的tc多肽可以通过适用于蛋白质或肽的任何常规途径施用,这些途径包括但不限于胃肠外,例如注射,包括但不限于皮下或静脉注射或任何其他形式的注射或输注。多肽组合物可通过许多途径施用,包括但不限于口服、静脉内、腹膜内、肌内、透皮、皮下、局部、舌下或直肠方式。还可以经由脂质体施用包含经修饰或未经修饰的非天然氨基酸多肽的组合物。这样的施用途径和适当制剂通常是本领域技术人员已知的。tc多肽可以单独使用或与其他适合的组分比如药物载体组合使用。tc多肽可以与其他剂或治疗剂组合使用。
[0752]
还可以将单独的或与其他适合的成分组合的包含非天然氨基酸的tc多肽制成将经由吸入施用的气溶胶制剂(即,它们可以被“雾化”)。气溶胶制剂可以置于加压的可接受的推进剂中,所述推进剂比如二氯二氟甲烷、丙烷、氮气等。
[0753]
适合于肠胃外施用例如通过关节内(在关节中)、静脉内、肌内、真皮内、腹膜内和皮下途径的制剂包括水性和非水性等渗无菌注射溶液(其可含有抗氧化剂、缓冲剂、抑菌剂和使制剂与预期接受者的血液等渗的溶质)以及水性和非水性无菌悬浮液(其可包含悬浮
剂、增溶剂、增稠剂、稳定剂和防腐剂)。可以将tc的制剂提供在单位剂量或多剂量密封容器比如安瓿和小瓶中。
[0754]
肠胃外施用和静脉内施用是优选的施用方法。具体而言,已经用于天然氨基酸同源物治疗剂的施用途径(包括但不限于通常用于epo、gh、g
‑
csf、gm
‑
csf、ifn例如tc、白细胞介素、抗体、fgf和/或任何其他药物递送蛋白的那些),连同当前使用的制剂,为本发明多肽提供了优选的施用途径和制剂。
[0755]
在本发明的背景下,取决于应用,施用于患者的剂量足以具有在患者中随时间的有益治疗响应或其他适当的活性。剂量取决于特定载体或制剂的功效、以及所采用的非天然氨基酸多肽的活性、稳定性或血清半衰期和患者的状况,以及待治疗患者的体重或表面积。剂量的大小还取决于伴随在特定患者中施用特定载体、制剂等的任何不良副作用的存在、性质和程度。
[0756]
在确定在疾病(包括但不限于中性粒细胞减少、再生障碍性贫血、周期性中性粒细胞减少、特发性中性粒细胞减少、chdiak
‑
higashi综合征、系统性红斑狼疮(sle)、白血病、骨髓增生异常综合征和骨髓纤维化等)的治疗或预防中施用的载体或制剂的有效量时,医师评价循环血浆水平、制剂毒性和疾病进展。
[0757]
例如,向70千克患者施用的剂量,通常在相当于针对相关组合物的改变的活性或血清半衰期进行调整的目前使用的治疗性蛋白质的剂量的范围内。通过任何已知的常规疗法,包括抗体施用、疫苗施用,细胞毒性剂、天然氨基酸多肽、核酸、核苷酸类似物、生物反应调节剂的施用等,本发明的载体或药物制剂可以补充治疗条件。
[0758]
对于施用,以通过相关制剂的ld
‑
50或ed
‑
50和/或各种浓度的非天然氨基酸多肽的任何副作用的观察所确定的速率(包括但不限于适用于患者的体重和总体健康状况)施用本发明制剂。可以通过单剂量或分次剂量完成施用。
[0759]
如果经历制剂输注的患者产生发热、寒战或肌肉疼痛,那么他/她接受适当剂量的阿司匹林、布洛芬、对乙酰氨基酚或其他疼痛/发热控制药物。对经历输注反应(比如发热、肌肉疼痛和寒战)的患者,应在未来输注前30分钟进行阿司匹林、对乙酰氨基酚或(包括但不限于)苯海拉明的前驱给药。哌替啶用于对解热药和抗组胺药未快速作出响应的更严重的寒战和肌肉疼痛。取决于反应的严重性,减慢或停止细胞输注。
[0760]
可以将本发明的tc的靶向多肽的人形式直接施用于哺乳动物受试者。通过通常用于将tc多肽引入受试者的任何途径进行施用。根据本发明实施方案的tc多肽组合物包括适合于口服、直肠、局部、吸入(包括但不限于通过气溶胶)、口腔(包括但不限于舌下)、阴道、肠胃外(包括但不限于皮下、肌内、皮内、关节内、胸膜内、腹膜内、脑内、动脉内或静脉内)、局部(即,皮肤和粘膜表面,包括气道表面)、肺部、眼内、鼻内和透皮施用的那些,不过在任何给定情况下的最适合的途径将取决于被治疗病症的性质和严重性。可以全身或局部施用。化合物的制剂可以提供在单位剂量或多剂量密封容器(比如安瓿和小瓶)中。可以用药学上可接受的载体将本发明的tc多肽制备成单位剂量可注射形式(包括但不限于溶液、悬浮液或乳液)的混合物。还可以通过连续输注(使用,包括但不限于,微泵,比如渗透泵)、单次推注或缓释储库制剂来施用本发明的tc多肽。
[0761]
适合于施用的制剂包括水性和非水性溶液、等渗无菌溶液(其可含有抗氧化剂、缓冲剂、抑菌剂和使制剂等渗的溶质)以及水性和非水性无菌悬浮液(其可包含悬浮剂、增溶
剂、增稠剂、稳定剂和防腐剂)。可由前述类型的无菌粉末、颗粒和片剂制备溶液和悬浮液。
[0762]
冷冻干燥是常用的呈现蛋白质的技术,用于从感兴趣的蛋白质制剂中除去水分。冷冻干燥或冻干是这样的过程,通过该过程,待干燥的材料首先被冷冻,然后通过在真空环境中升华除去冰或冷冻溶剂。预冻干制剂中可包含赋形剂,以增强冷冻干燥过程中的稳定性和/或提高冻干产品储存后的稳定性。pikal,m.biopharm.3(9)26
‑
30(1990)和arakawa等人,pharm.res.8(3):285
‑
291(1991)。
[0763]
药物的喷雾干燥也是本领域普通技术人员已知的。例如,参见broadhead,j.等人,"the spray drying of pharmaceuticals,"drug dev.ind.pharm,18(11&12),1169
‑
1206(1992)。除了小分子药物以外,还将多种生物材料喷雾干燥,这些生物材料包括:酶、血清、血浆、微生物和酵母。喷雾干燥是有用的技术,因为它能够在一步法中将液体药物制剂转化成精细的、无尘的或团聚的粉末。基本技术包括以下四个步骤:a)将进料溶液雾化成喷雾;b)喷雾空气接触;c)喷雾的干燥;和d)从干燥空气分离干燥的产品。美国专利第6,235,710号和第6,001,800号描述了通过喷雾干燥制备重组促红细胞生成素,将这些专利通过引用并入本文。
[0764]
本发明的药物组合物和制剂可包含药学上可接受的载体、赋形剂或稳定剂。药学上可接受的载体部分地取决于所施用的特定组合物以及用于施用所述组合物的特定方法。因此,有多种本发明的药物组合物(包含任选的药学上可接受的载体、赋形剂或稳定剂)的适合制剂(参见,例如remington’s pharmaceutical sciences,第17版,1985))。
[0765]
适合的载体包括但不限于含有琥珀酸盐、磷酸盐、硼酸盐、hepes、柠檬酸盐、组氨酸、咪唑、乙酸盐、碳酸氢盐和其他有机酸的缓冲液;抗氧化剂,包括但不限于抗坏血酸;低分子量多肽,包括但不限于少于约10个残基的多肽;蛋白质,包括但不限于血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,包括但不限于聚乙烯吡咯烷酮;氨基酸,包括但不限于甘氨酸、谷氨酰胺、天冬酰胺、精氨酸、组氨酸或组氨酸衍生物、甲硫氨酸、谷氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括但不限于海藻糖、蔗糖、葡萄糖、甘露糖或糊精;螯合剂,包括但不限于edta和乙二胺四乙酸(edentate)二钠;二价金属离子,包括但不限于锌、钴或铜;糖醇,包括但不限于甘露醇或山梨糖醇;成盐反离子,包括但不限于钠和氯化钠;填料,比如微晶纤维素、乳糖、玉米淀粉和其他淀粉;粘合剂;甜味剂和其他调味剂;着色剂;和/或非离子表面活性剂,包括但不限于tween
tm
(包括但不限于吐温80(聚山梨醇酯80)和吐温20(聚山梨醇酯20)、pluronics
tm
和其他普朗尼克酸,包括但不限于普朗尼克酸f68(泊洛沙姆188)或peg。适合的表面活性剂包括例如但不限于基于聚(氧化乙烯)
‑
聚(氧化丙烯)
‑
聚(氧化乙烯)即(peo
‑
ppo
‑
peo)或聚(氧化丙烯)
‑
聚(氧化乙烯)
‑
聚(氧化丙烯)即(ppo
‑
peo
‑
ppo)或其组合的聚醚。peo
‑
ppo
‑
peo和ppo
‑
peo
‑
ppo可以通过商品名pluronics
tm
、r
‑
pluronics
tm
、tetronics
tm
和r
‑
tetronics
tm
(basf wyandotte corp.,wyandotte,mich.)商购获得,并且在美国专利第4,820,352号中有进一步描述,将该专利通过引用完整并入本文。其他乙烯/聚丙烯嵌段聚合物可以是适合的表面活性剂。可以使用表面活性剂或表面活性剂的组合来稳定乙二醇化的tc,以抵抗一种或多种应力,包括但不限于由搅拌产生的应力。以上中的一些可被称为“填充剂”。有些也可称为“张力调节剂”。为了产品稳定性和抗微生物效果,还可应用抗微生物防腐剂;适合的防腐剂包括但不限于苯甲醇、苯扎氯铵、间甲酚、对羟基苯甲酸甲酯/对羟基苯甲酸丙酯、甲酚和苯酚,或其组合。美国专利第7,144,574
号描述了适合于本发明的药物组合物和制剂以及其他递送制剂的另外的材料,将该专利通过引用并入本文。
[0766]
本发明的tc多肽(包括与诸如peg的水溶性聚合物连接的那些)也可以通过缓释系统或作为缓释系统的一部分进行施用。缓释组合物包括(包括但不限于)呈成形物品形式的半透性聚合物基质,包括但不限于薄膜或微胶囊。缓释基质包括生物相容性材料,比如聚甲基丙烯酸2
‑
羟乙酯(langer等人,j.biomed.mater.res.,15:267
‑
277(1981);langer,chem.tech.,12:98
‑
105(1982)、乙烯乙酸乙烯酯(langer等人,上文)或聚
‑
d
‑
(
‑
)
‑3‑
羟基丁酸(ep 133,988)、聚丙交酯(聚乳酸)(美国专利第3,773,919号;ep 58,481)、聚乙交酯(乙醇酸聚合物)、聚丙交酯
‑
乙交酯共聚物(乳酸和乙醇酸的共聚物)聚酸酐、l
‑
谷氨酸和γ
‑
乙基
‑
l
‑
谷氨酸的共聚物(sidman等人,biopolymers,22,547
‑
556(1983)、聚(原酸)酯、多肽、透明质酸、胶原、硫酸软骨素、羧酸、脂肪酸、磷脂、多糖、核酸、聚氨基酸、氨基酸(比如苯丙氨酸、酪氨酸、异亮氨酸)、多核苷酸、聚乙烯丙烯、聚乙烯吡咯烷酮和硅酮。缓释组合物还包括脂质体包封的化合物。通过本身已知的方法来制备含有所述化合物的脂质体:de 3,218,121;eppstein等人,proc.natl.acad.sci.u.s.a.,82:3688
‑
3692(1985);hwang等人,proc.natl.acad.sci.u.s.a.,77:4030
‑
4034(1980);ep 52,322;ep 36,676;美国专利第4,619,794号;ep143,949;美国专利第5,021,234号;日本专利申请83
‑
118008;美国专利第4,485,045号和第4,544,545号;以及ep 102,324。将引用的所有参考文献和专利通过引用并入本文。
[0767]
可以通过例如描述于下列文献中的方法制备脂质体包封的tc多肽:de 3,218,121;eppstein等人,proc.natl.acad.sci.u.s.a.,82:3688
‑
3692(1985);hwang等人,proc.natl.acad.sci.u.s.a.,77:4030
‑
4034(1980);ep 52,322;ep 36,676;美国专利第4,619,794号;ep 143,949;美国专利第5,021,234号;日本专利申请83
‑
118008;美国专利第4,485,045号和第4,544,545号;以及ep 102,324。脂质体的组成和大小是熟知的,或者能够容易地由本领域普通技术人员凭经验确定。脂质体的一些实例描述于例如,park jw等人,proc.natl.acad.sci.usa92:1327
‑
1331(1995);lasic d和papahadjopoulos d(编辑):m
edical a
pplications of l
iposomes
(1998);drummond dc等人,liposomal drug delivery systems for cancer therapy,见于teicher b(编辑):c
ancer d
rug d
iscovery and d
evelopment
(2002);park jw等人,clin.cancer res.8:1172
‑
1181(2002);nielsen ub等人,biochim.biophys.acta1591(1
‑
3):109
‑
118(2002);mamot c等人,cancer res.63:3154
‑
3161(2003)。将引用的所有参考文献和专利通过引用并入本文。
[0768]
在本发明的背景下,施用于患者的剂量应足以在受试者中随时间引起有益的响应。通常,每剂量肠胃外施用的本发明的tc多肽的总药学有效量在按照患者体重计约0.01μg/kg/天至约100μg/kg,或约0.05mg/kg至约1mg/kg的范围内,不过这取决于治疗决定。在这个实施方案的特定方面,可以按照每天大于4μ/kg至每天约20μg/kg范围的剂量施用缀合物。在另外其他方面,可以按照每天大于4μg/kg至每天约9μg/kg范围的剂量施用缀合物。在另外其他方面,可以按照每天约4μg/kg至每天约12.5μg/kg范围的剂量施用缀合物。在特定的方面,可以按照没有过度毒性的最大耐受剂量或低于最大耐受剂量的剂量施用缀合物。此外,可以施用缀合物一周至少两次,或者可以施用缀合物一周至少三次、一周至少四次、一周至少五次、一周至少六次或一周七次。在特定的方面,当缀合物被施用多于一次时,可
以按照每天每次大于4μg/kg的剂量施用缀合物。具体而言,可以在两周或更长的时段施用缀合物。在某些方面,与参考样品,即,不与本发明缀合物接触的细胞样品相比,表达白细胞介素
‑
10受体的细胞的生长可被抑制至少50%、至少65%、至少75%、至少80%、至少85%、至少90%、至少95%或至少99%。在这个实施方案的特定方面,可以按照每天约5.3μg/kg的剂量、或每天约7.1μg/kg的剂量、或每天约9.4μg/kg的剂量、或每天约12.5μg/kg的剂量施用缀合物。给药频率还受限于治疗决定,并且可能比批准用于人的可商购获得的tc多肽产品更频繁或更不频繁。通常,可以通过上述任何施用途径施用本发明的tc的靶向多肽、聚乙二醇化tc多肽、缀合的tc多肽或聚乙二醇化的缀合tc多肽。
[0769]
本发明的tc的治疗用途
[0770]
本发明的tc可用于治疗多种疾患。本发明还包括治疗对tc、cd8 t细胞刺激和/或tc制剂有响应的处于癌症风险、正患有和/或已经患有癌症的哺乳动物的方法。tc的施用可产生短期效应,即,对观察到的若干临床参数的即时有益效果,并且这可以在施用后12小时或24小时产生,并且在另一方面,还可以产生长期效应,其为有益地减缓肿瘤生长的进展,减小肿瘤大小,和/或升高循环cd8 t细胞水平,并且本发明的tc可以通过本领域技术人员已知的任何手段施用,并且可以有利地经由输注施用,例如通过动脉、腹膜内或静脉注射和/或输注,其剂量足以获得期望的药理作用。
[0771]
tc剂量范围可为每次治疗每kg体重10
‑
200mg或40
‑
80mg tc多肽。例如,施用的tc的剂量可以是约每kg体重20
‑
100mg tc多肽,将其以快速注射和/或以临床必要时段的输注的形式给予,例如持续时间范围为几分钟至几小时,例如长达24小时。如有需要,tc施用可以重复一次或若干次。tc的施用可以与其他药剂比如化学治疗剂的施用相组合。此外,本发明涉及用于预防和/或治疗癌症的方法,其包括向有需要的受试者施用有效量的tc。
[0772]
tc的平均量可以变化,并且特别是应该基于有资格医师的推荐和处方。tc的确切量是一个偏好问题,取决于诸如被治疗病症的确切类型、被治疗患者的状况以及组合物中的其他成分等因素。本发明还提供了治疗有效量的另一种活性剂的施用。基于利用tc的治疗,有待给予的量可以由本领域普通技术人员容易地确定。
[0773]
实施例
[0774]
提供以下实施例来说明、但不限制要求保护的发明。
[0775]
实施例1:合成tlr激动剂的一般方法
[0776]
本实施例提供了用于合成本发明的tlr激动剂的一般方法。
[0777]
全部使用可商购获得的无水溶剂,无需进一步纯化,并且将其在氮气气氛下储存。使用uv光在merck硅胶60f254板上进行tlc和/或用kmno4水溶液染色以便可视化。使用实验程序中详述的条件,在来自teledyne isco的combi flash rf上进行色谱纯化。使用phenomenex gemini
–
nx c18 5μm 50x4.6mm柱在shimadzu系统上进行分析型hplc,所述柱用在含0.05%tfa的在水中乙腈的线性梯度以1ml/min洗脱。(流动相a:在水中的0.05%三氟乙酸;流动相b:在90%乙腈(acn)水溶液中的0.05%三氟乙酸)或waters beh 1.7μm v2.1x50 mm柱。分析方法1:0%b在1min内,0
‑
50%b在11min内,50
‑
100%b在0.5min内,100%b持续1.5min,100
‑
0%b在1min内,0%b持续2min;方法2:10
‑
20%b在1min内,20
‑
70%b在11min内,70
‑
100%b在0.5min内,100%b持续1.5min,100
‑
10%b在1min内,10%b持续2min;方法3:方法2:0
‑
40%b在1min内,40
‑
90%b在11min内,90
‑
100%b在0.5min内,100%b
持续1.5min,100
‑
10%b在1min内,10%b持续2min;方法4:5%b在0.3min内,5%至100%b从0.3至1.5min。100%b从1.5min至1.8min,从0min至1.8min流速从0.8ml/min至1.1min/min。
[0778]
取决于规模,使用gemini
–
nx c18 5μm 100x30mm、150x30mm或250x50mm柱,在shimadzu系统上进行制备型hplc。将质谱(ms)记录在shimadzu lcms
‑
2020系统上,并使用shimadzu labsolutions软件处理数据。使用与6230accurate
‑
mass tofms系统偶联的agilent 1260infinity binary lc,进行hr
‑
esi
‑
tof分析。在500mhz bruker nmr光谱仪上收集nmr光谱数据。以ppm报告化学位移(δ),并参考氘代溶剂信号。耦合常数(j)以赫兹(hz)报告。将自旋多重度描述为:s(单峰)、br(宽峰)、d(二重峰)、dd(二重峰的二重峰)、t(三重峰)、q(四重峰)或m(多重峰)。合并单体抗体,0.22μm过滤,并在≤65℃储存,直到进一步使用。
[0779]
实施例2:包含以下结构
–
核心1的tlr激动剂(图1)的合成:
[0780]
核心1
[0781][0782]
在一些实施方案中,x是ch或nh;
[0783]
r2和r3各自连接形成c4至c8环烷基或独立地是
‑
h、c1至c
12
烷基、含硝基烷基、芳香环或
–
c(nh)nh2;
[0784]
r4是c1至c
12
烷基、c1至c
12
取代的烷基、c4至c8环烷基、c4至c8取代的环烷基、芳香环、取代的芳香环、芳杂环、取代的芳杂环、
‑
onh2末端c1至c
12
烷基,或不存在。在一些实施方案中,z1=c1至c6烷基、c3至c8环烷基或c3至c8含硝基的杂环。
[0785]
如以下方案中所公开合成具有核心1结构的tlr激动剂。
[0786][0787]2‑
(2
‑
(3
‑
硝基喹啉
‑4‑
基氨基)乙氧基)乙基氨基甲酸叔丁酯(1):将4
‑
氯
‑3‑
硝基
喹啉(1750mg,8.39mmol)溶解在dcm(30ml)中,用游离胺(1800mg,8.55mmol)处理,然后用tea(2.29ml,17.3mmol)处理。将反应在室温下保持18h,然后用h2o(20ml)、盐水(10ml)洗涤,经mgso4干燥,并且真空浓缩。获得为黄色固体的目标化合物(1)(3130mg,99%),ms m/z 399(m na)
。
[0788]2‑
(2
‑
(3
‑
氨基喹啉
‑4‑
基氨基)乙氧基)乙基氨基甲酸叔丁酯(2):将硝基化合物(1)(3.12g,8.29mmol)溶解在thf(100ml)和水(80ml)中。加入一份锌(13.55g,207.2mmol),然后加入nh4cl(13.3g,248.6mmol)。将悬浮液在室温下剧烈搅拌1h(hplc)。在过滤之后,将滤饼用thf(20mlx2)洗涤。向滤液中加入nacl,直到水相饱和为止。收集液相,并且分离thf层。用thf/ea(50ml/50ml)萃取水层。将有机层合并,经mgso4干燥,并且浓缩,以获得用于下一步的残渣(2)(3.1g,>100%)。ms m/z 347(m h)
。
[0789]2‑
(2
‑
(2
‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基氨基甲酸叔丁酯(3):将胺化合物(2)(3.1g,粗产物,<8.95mmol)和原戊酸三乙酯(3.1ml,13.5mmol)悬浮在甲苯(200ml)中,并加热至110℃。然后加入吡啶hcl(55mg,0.48mmol)。将反应加热4h。将混合物在室温下保持48h。倾析液体,并且将剩余的固体/残渣用甲苯(20mlx2)搅拌,与液体合并,浓缩。将残渣溶解在dcm中,通过柱色谱法纯化(dcm中的甲醇,0
‑
10
‑
20%,80g柱),获得目标化合物(3)(1.05g,30%,2
‑
步,来自硝基化合物1)。ms m/z 413(m h)
。
[0790]1‑
(2
‑
(2
‑
(叔
‑
丁氧基羰基氨基)乙氧基)乙基)
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉5
‑
氧化物(4):将化合物3(1.05g,2.54mmol)溶解在dcm(20ml)中,并用mcpba(750mg,2.83mmol)处理。将反应在室温下保持4h。混合物用nahco3饱和溶液(15mlx3)洗涤,干燥并浓缩,获得用于下一步(4)的粗浆(900mg,83%)。ms m/z 429(m h)
。
[0791]2‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基氨基甲酸叔丁酯(5):在压力管中,将化合物4(900mg,2.18mmol)溶解在二氯乙烷(25ml)中,用浓缩的氢氧化铵(28%,1ml)处理,并将温度升至80℃。在冷却之后,向此混合物中缓慢加入甲苯磺酰氯(470mg,2.46mmol)持续5min。加入浓缩的氢氧化铵(0.5ml),并将管密封。将管在80℃加热4h。在冷却之后,将混合物用dcm(60ml)稀释,用水(40ml)洗涤,干燥,通过硅胶柱色谱法纯化,获得目标化合物(5)(750mg,80%)。ms m/z 428(m h)
。
[0792]1‑
(2
‑
(2
‑
氨基乙氧基)乙基)
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
胺(a):将化合物5(750mg,1.75mmol)用在etoh(20ml)中的1.25m hcl在室温下处理17h。接着将反应物真空干燥,将残渣重悬在etoh/et2o(1/10;20ml)中,并过滤。收集固体,获得目标化合物(a)(600mg,85%)。hplc(方法1):5.8min,ms m/z 328(m h)
。
[0793][0794]4‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基氨基甲酰基)哌嗪
‑1‑
甲酸叔丁酯(6):将化合物a的hcl盐(100mg,0.25mmol)溶解在dcm(10ml)中,并用tea(68ul,0.511mmol)处理。向悬浮液中加入4
‑
(氯羰基)哌嗪
‑1‑
甲酸叔丁酯(75mg,0.286mmol)。将反应在室温下保持17h,用dcm/meoh(4ml/1ml)稀释,然后用盐水洗涤溶液。将有机相通过硅胶柱色谱法纯化,获得纯产物6(130mg,0.24mmol,96%)。ms m/z 540(m h)
。
[0795]
n
‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)哌嗪
‑1‑
甲酰胺(7):将化合物(6)(10mg,0.018mmol)用在etoh中的hcl(约1.5m,1ml)在室温下处理1小时,然后在60℃处理1h,并且真空干燥。将残渣用乙醚洗涤并干燥,获得目标化合物(7)(9mg,0.018mmol,定量)。ms m/z 440(m h)
。
[0796][0797]
n
‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)吗啉
‑4‑
甲酰胺(8):通过使化合物a与吗啉
‑4‑
碳酰氯反应,使用与6所述相似的程序,制备化合物7,获得目标化合物7(7mg,42%)。ms m/z 441(m h)。
[0798][0799]4‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基氨基甲酰基)哌嗪
‑1‑
甲酸4
‑
((r)
‑2‑
((r)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺基)苄基酯(9):将化合物7(22mg,0.02mmol)溶解在dcm(1ml)中,用dipea(3.5ul,0.02mmol)处理,然后用2,5
‑
二氧代吡咯烷
‑1‑
基2
‑
(叔丁氧羰基氨基氧基)乙酸酯(3.3mg,0.011mmol)处理。将反应在室温下保持17h。向混合物中加入tfa(0.3ml),并搅拌15min。在真空干燥之后,通过prep
‑
hplc将残渣纯化,获得化合物9(15mg,22%来自7)。ms m/z 918(m h)
。
[0800][0801]2‑
丁基
‑1‑
(2
‑
(2
‑
(哌嗪
‑1‑
甲酰胺基)乙氧基)乙基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
基氨基甲酸4
‑
((r)
‑2‑
((r)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺基)苄基酯(10)):将化合物6(65mg,0.097mmol)溶于dmf(2ml)中,用dipea(34ul,0.194mmol)然后用fmoc
‑
vc
‑
pab
‑
pnp(94mg,0.116mmol)处理。将反应物在室温下搅拌1h,加入水(10ml)。收集固体,用水(2ml)洗涤,并干燥。将黄色固体溶解在dmf(2ml)中,并用二乙胺(100ul,0.97mmol)在室温下处理30min。通过prep
‑
lc纯化反应混合物,得到中间体val
‑
cit
‑
pab
‑
oco
‑
(化合物6)。将该中间体(11mg,0.01mmol)溶解在dcm(1ml)中,用dipea(3.5ul,0.02mmol)处理,然后用2,5
‑
二氧代吡咯烷
‑1‑
基2
‑
(叔丁氧羰基氨基氧基)乙酸酯(3.3mg,0.011momol)处理。将反应在室温下保持17h。加入tfa(0.3ml)并且将混合物搅拌15min。在真空干燥之后,通过prep
‑
hplc将残渣纯化,获得化合物10(15mg,16%来自化合物
6)。ms m/z 918(m h)
。
[0802][0803]1‑
(2
‑
(2
‑
氨基乙氧基)乙基)
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
基氨基甲酸4
‑
((r)
‑2‑
((r)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺基)苄基酯(11):使用5作为起始原料,采用与10所述相似的程序,制备化合物11,获得目标化合物11(22mg,21%来自5)。ms m/z 806(m h)
。
[0804][0805]2‑
丁基
‑1‑
(2
‑
(2
‑
(哌嗪
‑1‑
甲酰胺基)乙氧基)乙基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
基氨基甲酸4
‑
((r)
‑2‑
((r)
‑2‑
((2,5
‑
二氧代
‑
2,5
‑
二氢
‑
1h
‑
吡咯
‑1‑
基)丙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺基)苄基酯(12):使用5,3
‑
(2,5
‑
二氧代
‑
2,5
‑
二氢
‑
1h
‑
吡咯
‑1‑
基)丙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯作为起始原料,用与10所述相似的程序制备化合物12,获得目标化合物12(无tfa处理)(15mg,17%来自5)。ms m/z 984(m h)
。
[0806][0807]
己二酸
‑
双
‑
(n
‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)哌嗪
‑1‑
甲酰胺(13):在23℃下,向化合物7(8mg,0.018mmol)和己二酸(2mg,0.07mmol)在dmf(1ml)中的溶液中加入edc(3mg,0.016mmol)、hobt(1mg,0.018mmol)和diea(4ul,0.23mmol)。在24h之后,将混合物通过prep
‑
lc纯化,干燥,获得化合物13(5mg,0.004mmol,23%)。ms m/z 1217(m h)
。
[0808][0809]4‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基甲酸叔丁酯(14):使用boc
‑
1,4
‑
丁二胺和原丙酸三乙酯作为起始原料,按照与5所述相似的程序,制备化合物14,获得目标化合物14(420mg,1.095mmol,14.5%来自起始原料)。ms m/z 384(m h)
。
[0810]1‑
(4
‑
氨基丁基)
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
胺,2hcl(b):在23℃下,将化合物14(400mg,1.043mmol)加入到dcm(0.5ml)和在二噁烷中的4m hcl(10ml,40mmol)中。在1h之后,lcms显示反应完成。在真空中除去溶剂,在高真空泵下干燥6h,获得为淡黄色固体的化合物b(400mg,1.129mmol,99%)。ms m/z 284(m h)
。
[0811][0812]2‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基)乙酸叔丁酯(15):向化合物b(31mg 0.109mmol)在dcm(5ml)中的溶液中加入溴乙酸叔丁酯(15ul,0.102mmol),然后在23℃下加入tea(88ul,0.681mmol)。在24h之后,将混合物通过prep
‑
lc纯化,获得为黄色固体的化合物15(4mg,0.006mmol,6%)。ms m/z 398(m h)
。
[0813][0814]5‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)吡啶酰胺(16):在23℃下,向化合物b(25mg,0.071mmol)和5
‑
氨基吡啶
‑2‑
甲酸(10mg,0.072mmol)在dmf(1ml)中的溶液中加入hatu(20mg,0.083mmol)和diea(50ul,0.287mmol)。在1h之后,将混合物通过prep
‑
lc纯化,并干燥,获得为白色固体的化合物16(21mg,0.033mmol,46%)。ms m/z 404(m h)
。
[0815][0816]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑
5,6,7
‑
三甲氧基
‑
1h
‑
吲哚
‑2‑
甲酰胺(17)a:使用化合物b和5,6,7
‑
三甲氧基
‑
1h
‑
吲哚
‑2‑
甲酸作为起始原料,采用与16所述相似的程序,制备化合物17,获得目标化合物17(13mg,0.017mmol,44%)。ms m/z 517(m h)
。
[0817][0818]5‑
氨基
‑
n
‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)吡啶酰胺(18):使用化合物a和5
‑
氨基吡啶
‑2‑
甲酸作为起始原料,和与16所述相似的程序,制备化合物18,获得目标化合物18(24mg,0.036mmol,82%)ms m/z 448(m h)
。
[0819][0820]3‑
(4
‑
(n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)氨磺酰基)苯基)丙酸甲酯(19):在23℃下,向化合物b(14mg,0.035mmol)和5
‑
氨基吡啶
‑2‑
甲酸(6mg,0.043mmol)在dmf(1ml)中的溶液中加入diea(40ul,0.230mmol)。在30min之后,将混合物通过prep
‑
lc纯化,并干燥,获得为淡黄色固体的化合物19(15mg,0.020mmol,58%)。ms m/z 510(m h)
。
[0821][0822]1‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
(3
‑
(吡咯烷
‑1‑
基甲基)苄基)脲(20):在23℃下,向(3
‑
(吡咯烷
‑1‑
基甲基)苯基)甲胺(19mg,0.100mmol)和硝基苯基氯甲酸酯(21mg,0.104mmol)在dmf(1ml)中的溶液中加入diea(34ul,0.195mmol)。在10min之后,lcms显示硝基苯酚活化完成。向该混合物中加入化合物b。在2h之后,将混合物通过prep
‑
lc纯化,并干燥,获得为白色固体的化合物20(3mg,0.006mmol,6%)。
[0823][0824]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)吡嗪
‑2‑
甲酰胺(21):使用化合物b和吡嗪甲酸作为起始原料,采用与16所述相似的程序,制备化合物28,获得目标化合物21(11mg,0.013mmol,23%)。ms m/z 390(m h)
。
[0825]
[0826]2‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基氨基甲酸叔丁酯(22):使用4
‑
氯
‑3‑
硝基喹啉、2
‑
氨基乙基氨基甲酸叔丁酯和原戊酸三乙酯作为起始原料,采用与化合物5所述相似的程序,制备化合物22,获得目标化合物22(140mg,0.365mmol,33%)ms m/z 384(m h)
。
[0827]1‑
(4
‑
氨基丁基)
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
胺,3hcl(c):使用化合物22作为起始原料,采用与a所述相似的程序,制备化合物c,获得目标化合物c(60mg,0.169mmol,定量)。ms m/z 284(m h)
。
[0828][0829]
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)吡嗪
‑2‑
甲酰胺(23):使用化合物c和吡嗪甲酸作为起始原料,采用与16所述相似的程序,制备化合物23,获得目标化合物23(8mg,0.09mmol,29%)。ms m/z 390(m h)
。
[0830][0831]
(s)
‑1‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基氨基)
‑5‑
胍基
‑1‑
氧代戊烷
‑2‑
基氨基甲酸叔丁酯(24):使用化合物c和boc
‑
arg
‑
oh作为起始原料,采用与16所述相似的程序,制备化合物24,获得目标化合物24(75mg,0.098mmol,79%)。ms m/z 540(m h)
。
[0832]
(s)
‑2‑
氨基
‑
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)
‑5‑
胍基戊酰胺,3hcl(25):在23℃下,将化合物24(60mg,0.111mmol)加入在二噁烷中的4m hcl(1ml,4mmol)。在2h之后,将反应物真空干燥。将残渣在高真空泵下干燥,获得为白色固体的化合物25(64mg,0.110mmol,定量)。
[0833][0834]
(s)
‑1‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基氨基)
‑5‑
胍基
‑1‑
氧代戊烷
‑2‑
基氨基甲酸叔丁酯(26):使用化合物a和boc
‑
arg
‑
oh作为起始原料,采用与16所述相似的程序,制备化合物26,获得目标化合物26(20mg,0.019mmol,
59%)。ms m/z 584(m h)
。
[0835]
(s)
‑2‑
氨基
‑
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)
‑5‑
胍基戊酰胺(27):使用化合物26作为起始原料,采用与25所述相似的程序,制备化合物27,获得目标化合物27(14mg,0.016mmol,定量)。ms m/z 484(m h)
。
[0836][0837]
n
‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)吡嗪
‑2‑
甲酰胺(28):使用化合物a和吡嗪甲酸作为起始原料,采用与16所述相似的程序,制备化合物28,获得目标化合物28(1.3mg,0.001mmol,4%)。ms m/z 434(m h)
。
[0838][0839]1‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)胍(29):在80℃下,向化合物a(15mg,0.038mmol)和亚氨基硫代氨基甲酸酯双(硫酸)甲酯(25mg,0.090mmol)在dmf(1ml)和水(1ml)中的溶液加入tea(50ul,0.358mmol)。在18h之后,将混合物通过prep
‑
lc纯化,获得化合物29(12mg,0.017mmol,45%)。ms m/z 370(m h)
。
[0840][0841]1‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)胍(30):使用化合物c作为起始原料,采用与29所述相似的程序,制备化合物30,获得目标化合物30(10mg,0.013mmol,29%)。ms m/z 434(m h)
。
[0842][0843]1‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)胍(31):使用化合物b作为起始原料,采用与29所述相似的程序,制备化合物31,获得目标化合物31(8mg,0.010mmol,29%)。ms m/z 326(m h)
。
[0844][0845]
(s)
‑2‑
乙酰胺基
‑
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)
‑5‑
胍基戊酰胺(32):使用化合物c和乙酰基
‑
精氨酸作为起始原料,采用与16所述相似的程序,制备化合物32,获得目标化合物32(18mg,0.025mmol,69%)。ms m/z 482(m h)
。
[0846][0847]
(s)
‑2‑
乙酰胺基
‑
n
‑
(2
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙氧基)乙基)
‑5‑
胍基戊酰胺(33):使用化合物a和乙酰基
‑
精氨酸作为起始原料,采用与16所述相似的程序,制备化合物33,获得目标化合物33(4mg,0.019mmol,67%)。ms m/z 526(m h)
。
[0848][0849]
(s)
‑2‑
乙酰胺基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑5‑
胍基戊酰胺(34):使用化合物b和乙酰基
‑
精氨酸作为起始原料,采用与16所述相似的程序,制备化合物34,获得目标化合物34(5mg,0.007mmol,30%)。ms m/z 482(m h)
。
[0850][0851]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(35):使用化合物b和苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物35,获得目标化合物35(8mg,0.013mmol,53%)。ms m/z 388(m h)
。
[0852][0853]4‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基甲酰基)苯乙基氨基甲酸叔丁酯(36):使用化合物b和4
‑
((2
‑
boc
‑
氨基)乙基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物36,获得目标化合物36(25mg,0.033mmol,38%)。ms m/z 531(m h)
。
[0854][0855]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
胍基丁酰胺(37):使用化合物b和4
‑
胍基丁酸作为起始原料,采用与16所述相似的程序,制备化合物37,获得目标化合物37(10mg,0.016mmol,45%)。ms m/z 411(m h)
。
[0856][0857]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑
2,3,5,6
‑
四氟苯甲酰胺(38):使用化合物b和2,3,5,6
‑
四氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物38,获得目标化合物38(7mg,0.010mmol,36%)。ms m/z 460(m h)
。
[0858][0859]4‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基甲酸叔丁酯(39):使用4
‑
氯
‑3‑
硝基喹啉、4
‑
氨基丁基氨基甲酸叔丁酯和原戊酸三乙酯作为起始原料,采用与化合物5所述相似的程序,制备化合物39,获得目标化合物39(177mg,0.430mmol,20%来自起始原料)。ms m/z 412(m h)
。
[0860]1‑
(4
‑
氨基丁基)
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
胺,3hcl(d):使用化合物39作为起始原料,采用与a所述相似的程序,制备化合物d,获得目标化合物d(180mg,0.431mmol,定量)。ms m/z 312(m h)
。
[0861][0862]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
碘苯甲酰胺(40):使用化合物b和4
‑
碘苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物40,获得目标化合物40(15mg,0.020mmol,72%)。ms m/z 514(m h)
。
[0863][0864]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(2
‑
胍基乙基)苯甲酰胺(41):使用化合物b和4
‑
(2
‑
胍基乙基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物41,获得目标化合物41(7mg,0.009mmol,29%)ms m/z 473(m h)
。
[0865][0866]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(42):使用化合物d和苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物42,获得目标化合物42(6mg,0.012mmol,38%)。ms m/z 416(m h)
。
[0867][0868]4‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基甲酰基)苯乙基氨基甲酸叔丁酯(43):使用化合物d和4
‑
((2
‑
boc
‑
氨基)乙基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物43,获得目标化合物43(9mg,0.010mmol,38%)。ms m/z 559(m h)
。
[0869][0870]4‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基甲酸叔丁酯(44):使
用4
‑
氯
‑3‑
硝基
‑
1,5
‑
萘啶、4
‑
氨基丁基氨基甲酸叔丁酯和原戊酸三乙酯作为起始原料,采用与5所述相似的程序,制备化合物44,获得目标化合物44(120mg,0.159mmol,5.3%来自起始原料)。ms m/z 413(m h)
。
[0871]1‑
(4
‑
氨基丁基)
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑4‑
胺,4hcl(e):使用化合物44作为起始原料,采用与a所述相似的程序,制备化合物e,获得目标化合物e(145mg,0.296mmol,定量)。ms m/z313(m h)
[0872][0873]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)吡嗪
‑2‑
甲酰胺(45):使用化合物d和吡嗪羧酸作为起始原料,采用与16所述相似的程序,制备化合物45,获得目标化合物45(4mg,0.004mmol,14%)ms m/z 418(m h)
。
[0874][0875]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
甲氧基苯甲酰胺(46):使用化合物b和4
‑
氨基
‑3‑
甲氧基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物46,获得目标化合物46(12.1mg,0.014mmol,58%)。ms m/z 433(m h)
。
[0876][0877]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(47):使用化合物b和4
‑
氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物47,获得目标化合物47(6mg,0.007mmol,30%)。ms m/z 403(m h)
。
[0878][0879]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(48):使用化合物d和4
‑
氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物
48,获得目标化合物48(0.4mg,0.0005mmol,2%)。ms m/z 431(m h)
。
[0880][0881]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
甲氧基苯甲酰胺(49):使用化合物d和4
‑
氨基
‑3‑
甲氧基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物49,获得目标化合物49(4.2mg,0.005mmol,21%)。ms m/z 461(m h)
。
[0882][0883]2‑
乙酰基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(50):使用化合物d和2
‑
乙酰基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物50,获得目标化合物50(7.6mg,0.01mmol,43%)。ms m/z 458(m h)
。
[0884][0885]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(2
‑
氨基乙基)苯甲酰胺,4hcl(51):使用化合物43作为起始原料,采用与a所述相似的程序,制备化合物51,获得目标化合物51(10mg,0.017mmol,定量)。ms m/z 459(m h)
[0886][0887]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)苯甲酰胺苯甲酰胺(52):使用化合物e和苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物52,获得目标化合物52(1.5mg,0.002mmol,8%)。ms m/z 417(m h)
。
[0888][0889]4‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基氨基甲酰基)苯乙基氨基甲酸叔丁酯(53):使用化合物e和4
‑
((2
‑
boc
‑
氨基)乙基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物53,获得目标化合物53(3.8mg,0.004mmol,16%)。ms m/z 560(m h)
。
[0890][0891]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)吡嗪
‑2‑
甲酰胺(54):使用化合物d和吡嗪羧酸作为起始原料,采用与16所述相似的程序,制备化合物54,获得目标化合物54(3mg,0.004mmol,20%)。ms m/z 419(m h)
。
[0892][0893]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)
‑4‑
胍基丁酰胺(55):使用化合物e和4
‑
胍基甲酸作为起始原料,采用与16所述相似的程序,制备化合物55,获得目标化合物55(2mg,0.003mmol,12%)。ms m/z 440(m h)
。
[0894][0895]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)
‑4‑
(2
‑
氨基乙基)苯甲酰胺,4tfa(56):在23℃下,向化合物53(3.6mg,0.006mmol)加入dcm(0.5ml)和tfa(1ml)。在20min之后,将反应物真空干燥,然后在高真空泵下干燥过夜,获得目标化合物56(7mg,0.008mmol,定量)。ms m/z 460(m h)
。
[0896][0897]2‑
乙酰基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)苯甲酰胺(57):使用化合物e和2
‑
乙酰苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物57,获得目标化合物57(5.2mg,0.006mmol,27%)。ms m/z 459(m h)
。
[0898][0899]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)
‑3‑
甲氧基苯甲酰胺(58):使用化合物e和4
‑
氨基
‑3‑
甲氧基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物58,获得目标化合物58(4.3mg,0.04mmol,20%)ms m/z 462(m h)
。
[0900][0901]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(59):使用化合物e和4
‑
氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物59,获得目标化合物59(1.6mg,0.002mmol,8%)ms m/z 432(m h)
。
[0902][0903]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)苯甲酰胺(60):使用化合物b和4
‑
二甲基氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物60,获得目标化合物60(1mg,0.001mmol,6%)。ms m/z 431(m h)
。
[0904][0905]
(e)
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
(4
‑
(二甲基氨基)苯基)丙烯酰胺(61):使用化合物b和4
‑
二甲基氨基肉桂酸作为起始原料,采用与16所述相似的程序,制备化合物61,获得目标化合物61(2mg,0.003mmol,11%)。ms m/z 457(m h)
。
[0906][0907]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)苯甲酰胺(62):使用化合物d和4
‑
二甲基氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物62,获得目标化合物62(3.5mg,0.004mmol,20%)。ms m/z 459(m h)
。
[0908][0909]
(e)
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
(4
‑
(二甲基氨基)苯基)丙烯酰胺(63):使用化合物d和4
‑
二甲基氨基肉桂酸作为起始原料,采用与16所述相似的程序,制备化合物63,获得目标化合物63(4.5mg,0.005mmol,25%)。ms m/z 485(m h)
[0910][0911]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)
‑4‑
(二甲基氨基)苯甲酰胺(64):使用化合物e和4
‑
二甲基氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物64,获得目标化合物64(0.5mg,0.001mmol,7%)。ms m/z 460(m h)
。
[0912][0913]
(e)
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c][1,5]萘啶
‑1‑
基)丁基)
‑3‑
(4
‑
(二甲基氨基)苯基)丙烯酰胺(65):使用化合物e和4
‑
二甲基氨基肉桂酸作为起始原料,采用与16中所述相似的程序,制备化合物65,获得目标化合物65(0.5mg,0.001mmol,7%)。ms m/z 486(m h)
。
[0914][0915]
(e)
‑
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)
‑3‑
(4
‑
(二甲基氨基)苯基)丙烯酰胺(66):使用化合物c和4
‑
二甲基氨基肉桂酸作为起始原料,采用与16中所述相似的程序,制备化合物66,获得目标化合物66(3mg,0.004mmol,16%)。ms m/z 457(m h)
。
[0916][0917]4‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基氨基甲酰基)苯乙基氨基甲酸叔丁酯(67):使用化合物c和4
‑
(2
‑
叔丁氧基羰基氨基)乙基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物67,获得目标化合物67(1.5mg,0.002mmol,11%)。ms m/z 457(m h)
。
[0918]
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)
‑4‑
(2
‑
氨基乙基)苯甲酰胺(68):使用化合物67作为起始原料,采用与56所述相似的程序,制备化合物68,获得目标化合物68(2.9mg,0.003mmol,定量)。ms m/z 431(m h)
。
[0919][0920]1‑
(4
‑
氨基丁基)
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
胺(f):使用4
‑
氯
‑3‑
硝基
‑
1,5
‑
萘啶、4
‑
氨基丁基氨基甲酸叔丁酯和原乙酸三乙酯作为起始原料,采用与a所述相似的程序,制备化合物f,获得目标化合物f(130mg,0.316mmol,9%来自起始原料)。ms m/z 270(m
h)
。
[0921][0922]
n
‑
(4
‑
(4
‑
氨基
‑2‑
甲基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(69):使用化合物f和苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物69,获得目标化合物69(6.5mg,0.008mmol,42%)。ms m/z 374(m h)
。
[0923][0924]
n
‑
(4
‑
(4
‑
氨基
‑2‑
甲基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)苯甲酰胺(70):使用化合物f和4
‑
二甲基氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物70,获得目标化合物70(6.5mg,0.009mmol,39%)。ms m/z 417(m h)
。
[0925][0926]
n
‑
(4
‑
(4
‑
氨基
‑2‑
甲基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(吡咯烷
‑1‑
基)苯甲酰胺(71):使用化合物f和4
‑
(1
‑
吡咯烷基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物71,获得目标化合物71(3.1mg,0.004mmol,18%)。ms m/z 443(m h)
。
[0927][0928]
n
‑
(4
‑
(4
‑
氨基
‑2‑
甲基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二乙基氨基)苯甲酰胺(72):使用化合物f和4
‑
(二乙基氨基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物72,获得目标化合物72(6.4mg,0.008mmol,41%)。ms m/z 445(m h)
。
[0929][0930]
3,4
‑
二氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰
胺(73):使用化合物b和3,4
‑
二氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物73,获得目标化合物73(1.1mg,0.001mmol,4%)。ms m/z 418(m h)
。
[0931][0932]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(吡咯烷
‑1‑
基)苯甲酰胺(74):使用化合物b和4
‑
(吡咯烷
‑1‑
基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物74,获得目标化合物74(4.5mg,0.005mmol,19%)。ms m/z 457(m h)
。
[0933][0934]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(甲基氨基)苯甲酰胺(75):使用化合物b和4
‑
甲基氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物75,获得目标化合物75(7.6mg,0.009mmol,33%)。ms m/z 417(m h)
。
[0935][0936]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
氟苯甲酰胺(76):使用化合物b和3
‑
氟
‑4‑
氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物76,获得目标化合物76(7mg,0.008mmol,31%)。ms m/z 421(m h)
。
[0937][0938]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)
‑
3,5
‑
二氟苯甲酰胺(77):使用化合物b和4
‑
(二甲基氨基)
‑
3,5
‑
二氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物77,获得目标化合物77(9.5mg,0.010mmol,41%)。ms m/z 467(m h)
。
[0939][0940]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)
‑3‑
氟苯甲酰胺(78):使用化合物b和4
‑
(二甲基氨基)
‑3‑
氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物78,获得目标化合物78(12mg,0.013mmol,49%)。ms m/z 449(m h)
。
[0941][0942]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)
‑3‑
硝基苯甲酰胺(79):使用化合物b和4
‑
(二甲基氨基)
‑3‑
硝基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物79,获得目标化合物79(11mg,0.012mmol,50%)。ms m/z 476(m h)
。
[0943][0944]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二乙基氨基)苯甲酰胺(80):使用化合物b和4
‑
(二乙基氨基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物80,获得目标化合物80(10mg,0.011mmol,42%)。ms m/z 459(m h)
。
[0945][0946]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑2‑
(二甲基氨基)苯甲酰胺(81):使用化合物b和2
‑
(二乙基氨基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物81,获得目标化合物81(12.2mg,0.014mmol,57%)。ms m/z 431(m h)
。
[0947][0948]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(82):使用化合物b和4
‑
氨基
‑
3,5
‑
二氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物82,获得目标化合物82(10.2mg,0.011mmol,49%)。ms m/z 439(m h)
。
[0949][0950]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)
‑3‑
氟苯甲酰胺(83):使用化合物d和4
‑
(二甲基氨基)
‑3‑
氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物83,获得目标化合物83(9mg,0.011mmol,50%)。ms m/z 477(m h)
。
[0951][0952]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)
‑
3,5
‑
二氟苯甲酰胺(84):使用化合物d和4
‑
(二甲基氨基)
‑
3,5
‑
二氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物84,获得目标化合物84(7mg,0.008mmol,38%)。ms m/z 495(m h)
。
[0953][0954]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)
‑3‑
硝基苯甲酰胺(85):使用化合物d和4
‑
(二甲基氨基)
‑3‑
硝基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物85,获得目标化合物85(10mg,0.012mmol,54%)。ms m/z 504(m h)
。
[0955][0956]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(吡咯烷
‑1‑
基)苯甲酰胺(86):使用化合物d和4
‑
(1
‑
吡咯烷基氨基)苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物86,获得目标化合物86(2mg,0.002mmol,11%)。ms m/z 485(m h)
。
[0957][0958]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
氟苯甲酰胺(87):使用化合物d和4
‑
氨基
‑3‑
氟
‑
苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物87,获得目标化合物87(6mg,0.008mmol,20%)。ms m/z 449(m h)
。
[0959][0960]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(88):使用化合物d和4
‑
氨基
‑
3,5
‑
二氟
‑
苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物88,获得目标化合物88(6mg,0.007mmol,34%)。ms m/z 467(m h)
。
[0961][0962]
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(二甲基氨基)苯甲酰胺(89):使用化合物b和4
‑
氨基
‑
3,5
‑
二氟苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物89,获得目标化合物89(10mg,0.011mmol,27%)。ms m/z 431(m h)
。
[0963][0964]5‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
乙基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)吡嗪
‑2‑
甲酰胺(90):使用化合物b和5
‑
氨基吡嗪
‑2‑
甲酸作为起始原料,采用与16所述相似的程序,制备化合物90,获得目标化合物90(8mg,0.009mmol,37%)。ms m/z 405(m h)
。
[0965]4‑
(3
‑
氨基喹啉
‑4‑
基氨基)丁基氨基甲酸叔丁酯(91):使用4
‑
氯
‑3‑
硝基喹啉和4
‑
氨基丁基氨基甲酸叔丁酯,采用与2所述相似的程序,制备化合物91,获得目标化合物91(5050mg,15.284mmol,97%来自起始原料)。ms m/z 331(m h)
。
[0966]4‑
(2
‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基氨基甲酸丁基氨基甲酸叔丁酯(92):在23℃下,向4
‑
(3
‑
氨基喹啉
‑4‑
基氨基)丁基氨基甲酸叔丁酯(1070mg,3.238mmol)在无水thf(12ml)中的溶液加入三乙胺(885ul,8.746mmol)和2
‑
乙氧基乙酰氯(500mg,4.078mmol)。在20h之后,在真空中除去溶剂。将残渣溶解在dcm(50ml)中,用饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,经mgso4干燥,获得为粗产物的中间体(4
‑
(3
‑
(2
‑
乙氧基乙酰胺基)喹啉
‑4‑
基氨基)丁基氨基甲酸叔丁酯)。将此粗产物溶解在meoh(5ml)中,然后在密封管中加入氧化钙(0.5g)。将反应混合物在120℃加热2.5h。在过滤除去cao之后,真空除去溶剂,将残渣通过prep
‑
lc纯化,获得化合物92(346mg,0.868mmol,27%)。ms m/z 399(m h)
。
[0967]1‑
(4
‑
氨基丁基)
‑2‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑4‑
胺,3hcl(g):使用化合物92,采用与a所述相似的程序,制备化合物g,获得目标化合物g(5270mg,0.595mmol,4%来自起始原料)。ms m/z 312(m h)
。
[0968][0969]3‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(93):使用化合物d和3
‑
氨基苯甲酸作为起始原料,采用与16所述相似的程序,制备化合物93,获得目标化合物93(5mg,0.006mmol,19%)ms m/z 431(m h)
。
[0970][0971]3‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
氟苯甲酰胺(94):在23℃下,向d(10mg,0.024mmol)和3
‑
氨基
‑4‑
氟
‑
苯甲酸(4mg,0.026mmol)在dmf(1ml)中的溶液加入dmtmmt(7mg,0.029mmol)和diea(30ul,0.172mmol)。在10min之后,将混合物通过prep
‑
lc纯化,获得目标化合物94(5mg,0.006mmol,21%)ms m/z 449(m h)
。
[0972][0973]3‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
氟苯甲酰胺(95)):使用化合物g和3
‑
氨基
‑4‑
氟
‑
苯甲酸作为起始原料,采用与94所述相似的程序,制备化合物95,获得目标化合物95(6mg,0.007mmol,26%)。ms m/z 451(m h)
。
[0974][0975]3‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
氟苯甲酰胺(96)):使用化合物g和3
‑
氨基苯甲酸作为起始原料,采用与94所述相似的程序,制备化合物96,获得目标化合物96(5mg,0.006mmol,19%)。ms m/z 433(m h)
。
[0976][0977]3‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)苯甲酰胺(97)):使用化合物g和4
‑
氨基
‑3‑
甲氧基苯甲酸作为起始原料,采用与94所述相似的程序,制备化合物97,获得目标化合物97(5mg,0.005mmol,23%)。ms m/z 463(m h)
。
[0978][0979]5‑
氨基
‑
n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)吡嗪
‑2‑
甲酰胺(98)):使用化合物c和5
‑
氨基
‑
吡嗪
‑2‑
甲酸作为起始原料,采用与16所述相似的程序,制备化合物98,获得目标化合物98(2.13mg,0.002mmol,11%)。ms m/z 405(m h)
。
[0980][0981]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑3‑
氟苯甲酰胺(99):使用化合物g和3
‑
氟
‑4‑
氨基苯甲酸作为起始原料,采用与94所述相似的程序,制备化合物99,获得目标化合物99(7.37mg,0.008mmol,37%)。ms m/z 451(m h)
。
[0982][0983]4‑
氨基
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
(乙氧基甲基)
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(100):使用化合物g和4
‑
氨基
‑
3,5
‑
二氟苯甲酸作为起始原料,采用与94所述相似的程序,制备化合物100,获得目标化合物100(9.7mg,0.011mmol,51%)。ms m/z 469(m h)
。
624(m h)
。
[0990]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
((s)
‑2‑
((s)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺基)
‑
3,5
‑
二氟苯甲酰胺(106):在23℃下,向fmoc
‑
aoa
‑
val
‑
oh(11mg,0.027mmol)和化合物105(24mg,0.022mmol)在dmf(1ml)中的溶液加入hatu(9mg,0.037mmol)和diea(40ul,0.023mmol)。在10min之后,向此混合物加入哌啶(50ul,5%)。在5min之后,将混合物通过prep
‑
lc纯化,获得化合物106(9mg,0.007mmol,27%)。ms m/z796(m h)
。
[0991][0992]
(s)
‑1‑
(1h
‑
咪唑
‑1‑
基)
‑1‑
氧代丙
‑2‑
基氨基甲酸叔丁酯(107):使用boc
‑
丙氨酸,采用与102所述相似的程序,制备化合物107,获得目标化合物107(730mg,3.051mmol,75%粗制物)。ms m/z 326(m h)
。
[0993]
(s)
‑4‑
(2
‑
(叔丁氧基羰基氨基)
‑5‑
脲基戊酰胺基)
‑
3,5
‑
二氟苯甲酸(108):使用107,采用与103所述相似的程序,制备化合物108,获得目标化合物108(17mg,0.049mmol,9%)。ms m/z 431(m h)
。
[0994]
(s)
‑
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
(2
‑
(2
‑
(氨基氧基)乙酰胺基)丙酰胺基)
‑
3,5
‑
二氟苯甲酰胺(109):使用108和化合物d,采用与106所述相似的程序,制备化合物109,获得目标化合物109(9mg,0.007mmol,31%来自108)。ms m/z 796(m h)
。
[0995][0996]
(s)
‑2‑
(3
‑
(2
‑
(1,3
‑
二氧代异吲哚啉
‑2‑
基氧基)乙氧基)丙酰胺基)
‑3‑
甲基丁酸叔丁酯(110):在23℃下,向3
‑
(2
‑
(1,3
‑
二氧代异吲哚啉
‑2‑
基氧基)乙氧基)丙酸(295mg,1.056mmol)和val
‑
otbu,hcl(225mg,1.078mmol)在dcm(5ml)中的溶液加入dmtmmt(304mg,1.260mmol)和diea(460ul,2.641mmol)。在1.5h之后,将溶液用etoac(100ml)稀释,并且用1n hcl(100ml)、饱和碳酸氢钠(100ml)和盐水(50ml)洗涤。将有机层用mgso4干燥,过滤,并在真空中除去溶剂。将残渣通过快速色谱法纯化,获得化合物110(379mg,0.872mmol,83%)。ms m/z 435(m h)
。
[0997]
(s)
‑2‑
(3
‑
(2
‑
(1,3
‑
二氧代异吲哚啉
‑2‑
基氧基)乙氧基)丙酰胺基)
‑3‑
甲基丁酸(111):在23℃下,向化合物110((379mg,0.872mmol)的溶液加入在二噁烷(5ml,20mmol)中的4m hcl。在20h之后,在真空中除去溶剂,使用高真空泵干燥,获得化合物111(320mg,
0.846mmol,97%)。ms m/z 379(m h)
。
[0998]
n
‑
(4
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)丁基)
‑4‑
((s)
‑2‑
((s)
‑2‑
(3
‑
(2
‑
(氨基氧基)乙氧基)丙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺基)
‑
3,5
‑
二氟苯甲酰胺(112):在23℃下,向化合物111(3mg,0.007mmol)和化合物105(5mg,0.005mmol)在dmf(1ml)中的溶液加入dmtmmt(3mg,0.012mmol)和diea(20ul,0.115mmol)。在1.5h之后,向此混合物加入肼h2o(2ul,0.506mmol)。在5min之后,将混合物通过prep
‑
lc纯化,获得化合物112(2mg,0.002mmol,21%)。ms m/z 855(m h)
。
[0999][1000]
(s)
‑
n
‑
(4
‑
((n
‑
(2
‑
(4
‑
氨基
‑2‑
丁基
‑
1h
‑
咪唑并[4,5
‑
c]喹啉
‑1‑
基)乙基)氨基甲酰亚胺基氨基甲酰氧基)甲基)苯基)
‑2‑
((s)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)
‑3‑
甲基丁酰胺基)
‑5‑
脲基戊酰胺(113):使用化合物30作为起始原料,采用与106所述相似的程序,制备化合物113,获得目标化合物113(2mg,0.001mmol,2%来自化合物30)。ms m/z 805(m h)
。
[1001]
表3
–
tlr激动剂
‑
核心1化合物
[1002]
[1003]
[1004]
[1005]
[1006]
[1007]
[1008]
[1009]
[1010]
[1011]
[1012]
[1013]
[1014]
[1015]
[1016][1017]
实施例3:包含以下代表性结构
–
核心5的tlr激动剂(图1)的合成:
[1018]
核心5
[1019][1020]
在一些实施方案中,x是o、s、nh或h;
[1021]
r1是c1至c
12
烷基、取代的c1至c
12
烷基、含氧的c1至c
12
烷基、杂环、取代的杂环、环烷基、取代的环烷基、
‑
n3、末端c1至c
12
烷基、末端取代的c1至c
12
烷基,或不存在;
[1022]
r2或r3各自连接形成c4至c8环烷基或独立地是
‑
h、c1至c
12
烷基、含硝基烷基、含氧烷基、芳香环;
[1023]
r4是
‑
onh2、末端c1至c
12
烷基、c1至c
12
烷基、c1至c
12
取代的烷基、c4至c8环烷基、芳香环、取代的芳香环、芳香杂环、取代的芳香杂环或不存在;
[1024]
z1是c1至c6烷基、c3至c
10
环烷基、c3至c
10
含硝基杂环;
[1025]
z2是芳香环、芳香杂环、c2至c8环烷基或不存在;
[1026]
z3是c1至c
12
烷基、取代的c1至c
12
烷基、c3至c8环烷基、取代的c3至c8环烷基、取代的c3至c8含硝基杂环、c3至c8含硝基杂环或不存在。
[1027]
如以下方案中所公开合成具有核心5结构的tlr激动剂。
[1028][1029]4‑
(2,6
‑
二氯
‑
9h
‑
嘌呤
‑9‑
基)甲基)苄基氨基甲酸叔丁酯、4
‑
(2,6
‑
二氯
‑
7h
‑
嘌呤
‑7‑
基)甲基)苄基氨基甲酸叔丁酯(114):在23℃下,向4
‑
(羟甲基)苄基氨基甲酸叔丁酯(1280mg,5.394mmol)和2,6
‑
二氯嘌呤(1050mg,5.556mmol)在thf(10ml)中的溶液加入pph3(1560mg,5.948mmol)。在30min之后,在0℃下经5min加入diad(1600ul,8.126mmol)。将混合物在50℃下搅拌。在2h之后,在真空中除去溶剂。将残渣混合物用etoac(100ml)稀释,并使用半饱和碳酸氢钠(100ml)和盐水(20ml)洗涤。将有机层用mgso4干燥,并过滤。在真空中除去溶剂。将残渣通过快速色谱法纯化,获得化合物114(2268mg,<5.555mmol,具有pph3的粗混合物)。ms m/z 409(m h)
。
[1030]4‑
((6
‑
氨基
‑2‑
氯
‑
9h
‑
嘌呤
‑9‑
基)甲基)苄基氨基甲酸叔丁酯(115):将化合物114(pph3的粗混合物,2268mg,<5.555mmol)放入配备有搅拌棒的耐压玻璃容器中。向该容器中加入meoh(12ml,84mmol)中的7n nh3。将管密封并在120℃下加热。在1h之后,在真空中除去溶剂,并将残渣溶解在dcm(100ml)中。通过过滤除去沉淀物。将液体通过快速色谱法纯化,获得化合物115(1043mg,2.682mmol,50%来自2,6
‑
二氯嘌呤)。ms m/z 400(m h)
。
[1031]4‑
((6
‑
氨基
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑9‑
基)甲基)苄基氨基甲酸叔丁酯(116):在23℃在干燥氮气下将化合物115(1043mg,2.682mmol)溶解在20%正丁醇钠(5ml,10.4mmol)中,将温度升至110℃。在1.5h之后,向混合物中加入1ml水,然后加入boc酸酐(170mg,0.779mmol)。在5min之后,在真空中除去溶剂。将残渣溶解在dcm(30ml)中,用半饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂。将残渣通过快速色谱法纯化,获得化合物116(560mg,1.313mmol,49%)。ms m/z 427(m h)
。
[1032]4‑
(6
‑
氨基
‑8‑
溴
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑9‑
基)甲基)苄基氨基甲酸叔丁酯(117):在23℃下,向化合物116(560mg,1.313mmol)在dcm(10ml)中的溶液加入溴(135ul,0.507mmol)。在10min之后,将反应物真空干燥。将残渣溶解在dcm(50ml)中,用半饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去溶剂。将残渣通过快速色谱法纯化,获得化合物117(440mg,0.871mmol,66%),其为hbr盐,ms m/z 506(m h)
。
[1033]6‑
氨基
‑9‑
(4
‑
(氨基甲基)苄基)
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(118):将化合物117(240mg,0.410mmol)溶解在37%(10ml)浓hcl溶液中,并且进行回流。在4.5h之后,在真空中除去溶剂。向残渣加入水(10ml)和meoh(4ml),通过加入28%nh3溶液(9ml)将其中和。在真空中除去溶剂。将残渣通过prep
‑
lc纯化,获得化合物118(11mg,0.019mmol,5%)。ms m/z 343(m h)
。
[1034]
n
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)
‑2‑
(氨基氧基)乙酰胺(119):在23℃下,向化合物118(5mg,0.007mmol)和2
‑
(叔丁氧基羰基氨基氧基)乙酸
‑
2,5
‑
二氧代吡咯烷
‑1‑
基酯(3mg,0.010mmol)在dmf(1ml)中的溶液加入diea(5ul,
0.057mmol)。在10min之后,在真空中除去溶剂。在23℃下,向残渣加入dcm(1ml)和tfa(1ml)。在10min之后,将混合物通过prep
‑
lc纯化,获得化合物119(3.6mg,0.005mmol,65%)。ms m/z 416(m h)
。
[1035][1036]
n
‑
(4
‑
(6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)
‑3‑
(2
‑
(氨基氧基)乙氧基)丙酰胺(120):在23℃下,向118(5mg,0.007mmol)和3
‑
(2
‑
(1,3
‑
二氧代异吲哚啉
‑2‑
基氧基)乙氧基)丙酸(3mg,0.011mmol)在dmf(1ml)中的溶液加入dmtmmt(3mg,0.012mmol)和diea(8ul,0.046mmol)。在15min之后,向该混合物加入肼h2o(3ul,0.06mmol)。在20min之后,将混合物通过prep
‑
lc纯化,获得化合物120(3.5mg,0.004mmol,59%)。ms m/z 474(m h)
。
[1037][1038]
2,6
‑
二氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
9h
‑
嘌呤(121):向磁力搅拌的2,6
‑
二氯嘌呤(2950mg,15.608mmol)在乙酸乙酯(100ml)中的溶液加入苯磺酸(30mg,0.19mmol),并在干燥氮气下将混合物加热至50℃。在50℃下,经1小时向搅拌的混合物加入3,4
‑
二氢
‑
2h
‑
吡喃(2200ul,26.153mmol)。将温度降至23℃。在1h之后,将混合物用半饱和nahco3(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂,将残渣在高真空泵中干燥,获得化合物121(4170mg,15.269mmol,98%)。ms m/z 274(m h)
。
[1039]2‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
9h
‑
嘌呤
‑6‑
胺(122):将化合物121(4170mg,15.269mmol)放入配备有搅拌棒的耐压玻璃容器中。向该容器中加入在meoh(12.84mmol)中的7n nh3。将管密封并在110℃下加热。在3.5h之后,将混合物冷却至室温并允许其静置过夜。将沉淀物过滤并用meoh(5ml)洗涤。将残渣在高真空泵上干燥,获得化合物122(3450mg,13.6mmol,89%)。ms m/z 254(m h)
。
[1040]2‑
氯
‑9‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
9h
‑
嘌呤
‑6‑
胺(123):将化合物122(1746mg,6.882mmol)放入配备有搅拌棒的耐压玻璃容器中。向该容器中加入正丁胺(7ml,70.86mmol)和diea(2.3ml,13.25mmol)。将管密封并在150℃下加热。在5h之后,将混合物冷却至室温,并在真空中除去溶剂。将残渣溶解在dcm(100ml)中,用水(30ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂。将残渣(中间体)溶解在meoh(10ml)和tfa(2ml)中,并在23℃搅拌过夜。在18h之后,在真空中除去溶剂。向残渣加入etoac(10ml),并且向沉淀物加入己烷(50ml)。通过过滤收集沉淀物,并在真空泵中干燥,获得为2tfa盐的化合物123(1640mg,3.777mmol,55%)。ms m/z 207(m h)
。
[1041]
n2
‑
丁基
‑9‑
((6
‑
氯吡啶
‑3‑
基)甲基)
‑
9h
‑
嘌呤
‑
2,6
‑
二胺(124):向化合物123(1640mg,3.777mmol)和2
‑
氯
‑5‑
(氯甲基)吡啶(900mg,5.556mmol)在dmf(5ml)中的溶液加入k2co3(2600mg,18.813mmol),并在50℃在氮气下搅拌混合物。在24h之后,向混合物加入冰水(100ml),并且分离沉淀物。将沉淀物溶解在dcm(100ml)中,用盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂。通过快速色谱仪(硅胶)以1%至10%meoh/dcm梯度将残渣纯化,获得化合物124(1020mg,3.074mmol,81%)。ms m/z 332(m h)
。
[1042]6‑
氨基
‑2‑
(丁基氨基)
‑9‑
((6
‑
氯吡啶
‑3‑
基)甲基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(125):在23℃下,向化合物124(1020mg,3.074mmol)在dcm(10ml)中的溶液加入溴(250ul,0.939mmol)。在1.5h之后,在真空中除去溶剂。将残渣在高真空泵上干燥。将8
‑
溴
‑
n2
‑
丁基
‑9‑
((6
‑
氯吡啶
‑3‑
基)甲基)
‑
9h
‑
嘌呤
‑
2,6
‑
二胺hbr(1500mg,<3.074mmol)的粗制中间体溶解在37%(15ml)的浓hcl溶液中,并将溶液回流。在8h之后,在真空中除去溶剂。向残渣加入水(10ml)和meoh(4ml),然后通过加入28%的nh3溶液(5ml)进行中和。通过离心机(5min,4000rpm)分离沉淀的固体,并用meoh(2ml)和水(10ml)洗涤。将沉淀物干燥,获得化合物125(1100mg,2.421mmol,79%)。ms m/z 348(m h)
。
[1043]6‑
氨基
‑9‑
((6
‑
(4
‑
(2
‑
氨基乙基)哌嗪
‑1‑
基)吡啶
‑3‑
基)甲基)
‑2‑
(丁基氨基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(126):将化合物125(30mg,0.086mmol)和2
‑
(哌嗪
‑1‑
基)乙基氨基甲酸叔丁酯(26mg,0.113mmol)的混合物在140℃加热。在20h之后,将混合物冷却至23℃。将dcm(0.5ml)和tfa(0.5ml)添加至残渣。30min后。在真空中除去溶剂,并将残渣通过prep
‑
lc纯化,获得为tfa盐的化合物126(9mg,0.010mmol,12%)。ms m/z 441(m h)
。
[1044]
n
‑
(2
‑
(4
‑
(5
‑
((6
‑
氨基
‑2‑
(丁基氨基)
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)吡啶
‑2‑
基)哌嗪
‑1‑
基)乙基)
‑2‑
(氨基氧基)乙酰胺(127):在23℃下,向化合物126(9mg,0.010mmol)和2
‑
(叔丁氧基羰基氨基氧基)乙酸
‑
2,5
‑
二氧代吡咯烷
‑1‑
基酯(2.5mg,0.011mmol)在dmf(1ml)中的溶液加入diea(10ul,0.060mmol)。在15min之后,在真空中除去溶剂。将dcm(1ml)和tfa(1ml)添加至残渣。在5min之后,在真空中除去溶剂。将残渣通过prep
‑
lc纯化,获得为tfa盐的化合物127(8mg,0.008mmol,82%)。ms m/z 514(m h)
。
[1045][1046]6‑
氨基
‑9‑
((6
‑
(2
‑
((2
‑
氨基乙基)(甲基)氨基)乙基氨基)吡啶
‑3‑
基)甲基)
‑2‑
(丁基氨基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(128):将化合物125(30mg,0.086mmol)和2,2
’‑
二氨基
‑
n
‑
甲基二乙胺(100ul,0.853mmol)的混合物在130℃加热。在20h之后,将混合物冷却至23℃,并通过prep
‑
lc纯化,获得化合物128(40mg,0.045mmol,52%)。ms m/z 423(m h)
。
[1047]
n
‑
(2
‑
((2
‑
(5
‑
((6
‑
氨基
‑2‑
(丁基氨基)
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)吡啶
‑2‑
基氨基(乙基)(甲基)氨基)乙基)
‑2‑
(氨基氧基)乙酰胺(129):使用化合物128作为起始原料,采用与127所述相似的程序,制备化合物129,获得目标化合物129(13mg,0.012mmol,54%)。ms m/z502(m h)
。
[1048][1049]
nh2o
‑
peg3
‑
pr
‑
(6
‑
氨基
‑9‑
((6
‑
(2
‑
((2
‑
氨基乙基)(甲基)氨基)乙基氨基)吡啶
‑3‑
基)甲基)
‑2‑
(丁基氨基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮)乙酰胺(130):在23℃下,向化合物128(20mg,0.023mmol)和phth
‑
peg4
‑
osu(10mg,0.022mmol)在dmf(1ml)中的溶液加入diea(50ul,0.287mmol)。在5min之后,在23℃将肼h2o(10ul)添加至混合物。在5min之后,将混合物通过prep
‑
lc纯化,获得化合物130(20mg,0.016mmol,70%)。ms m/z 692(m h)
。
[1050][1051]6‑
氨基
‑2‑
(丁基氨基)
‑9‑
((6
‑
(2
‑
((2
‑
羟乙基)(甲基)氨基)乙氧基)吡啶
‑3‑
基)甲基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(131):在23℃下,向化合物125(124mg,0.215mmol)在dmf(4ml)中的溶液加入n
‑
甲基二乙醇胺(200ul,1.007mmol)和60%(350mg,8.750mmol)的nah。将混合物在60℃在干燥氮气下搅拌。在3h之后,向混合物加入1n hcl(4ml),并通过prep lc纯化,获得化合物131(75mg,0.085mmol,39%)。ms m/z431(m h)
。
[1052][1053]2‑
丁氧基
‑
9h
‑
嘌呤
‑6‑
胺(132):在23℃在干燥氮气下将化合物122(690mg,2.720mmol)溶于20%正丁醇钠(8ml,16.7l mmol)中。加入之后,将温度升至100℃。在20h之后,在真空中除去溶剂。将残渣溶解在dcm(30ml)中,用半饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂。将meoh(5ml)和tfa(1ml)添加至残渣,并在23℃搅拌。在18h之后,在真空中除去溶剂。将残渣溶解在dcm(30ml)中,用碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂,获得为粗制物的2
‑
丁氧基
‑
9h
‑
嘌呤
‑6‑
胺(1300mg,2.987,定量)。ms m/z 208(m h)
。
[1054]
n2
‑
丁基
‑9‑
((6
‑
氯吡啶
‑3‑
基)甲基)
‑
9h
‑
嘌呤
‑
2,6
‑
二胺(133):使用化合物132作为起始原料,采用与124所述相似的程序,制备化合物133,获得目标化合物133(468mg,1.406mmol,47%)。ms m/z 333(m h)
。
[1055]
n
‑
(2
‑
(4
‑
(5
‑
((6
‑
氨基
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑9‑
基)甲基)吡啶
‑2‑
基)哌嗪
‑1‑
基)乙基)
‑2‑
(氨基氧基)乙酰胺(134):使用化合物133作为起始原料,采用与127所述相似的程序,制备化合物134,获得目标化合物134(12mg,0.012mmol,10%来自化合物133)。ms m/z 514(m h)
。
[1056][1057]6‑
氨基
‑2‑
丁氧基
‑9‑
((6
‑
氯吡啶
‑3‑
基)甲基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(135):在23℃下,向化合物133(124mg,0.373mmol)在dcm(10ml)中的溶液加入溴(30ul,0.113mmol)。在2h之后,将反应物真空干燥。将8
‑
溴
‑2‑
丁氧基
‑9‑
((6
‑
氯吡啶
‑3‑
基)甲基)
‑
9h
‑
嘌呤
‑6‑
胺hbr(150mg,<0.373mmol,粗制物)的粗残渣溶解在3n hcl溶液(15ml)中,并且进行回流。在20h之后,在真空中除去溶剂。将混合物通过prep
‑
lc纯化,获得化合物135(47mg,0.110mmol,29%)。ms m/z 349(m h)
。
[1058]6‑
氨基
‑9‑
((6
‑
(4
‑
(2
‑
氨基乙基)哌啶
‑1‑
基)吡啶
‑3‑
基)甲基)
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(136):将化合物135(46mg,0.132mmol)和4
‑
(2
‑
boc
‑
氨基乙基)
‑
哌啶(120mg,0.526mmol)混合,并且将混合物在140℃搅拌。在25h之后,在冷却至23℃之后,向混合物加入dcm(1ml)和tfa(1ml)。在10min之后,在真空中除去有机溶剂。将混合物通过prep
‑
lc纯化,获得化合物136(11mg,0.014mmol,11%)。ms m/z441(m h)
。
[1059]
n
‑
(2
‑
(1
‑
(5
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)吡啶
‑2‑
基)哌啶
‑4‑
基)乙基)
‑3‑
(2
‑
(氨基氧基)乙氧基)丙酰胺(137):使用化合物136和3
‑
(2
‑
(1,3
‑
二氧代吲哚啉
‑2‑
基氧基)乙氧基)丙酸作为起始原料,采用与130所述相似的程序,制备化合物137,获得目标化合物137(9mg,0.010mmol,70%)。ms m/z 572(m h)
。
[1060][1061]9‑
(4
‑
(2
‑
氨基乙基)苄基)
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑6‑
胺(138):在23℃在干燥氮气下将化合物122(3330mg,13.126mmol)溶解在20%正丁醇钠(25ml)中,并将温度升至100℃。在1.5h之后,在真空中除去溶剂。将残渣溶解在dcm(30ml)中,用半饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有机溶剂。通过快速色谱法以1%至4%的meoh/dcm梯度将残渣纯化,获得化合物138(2687mg,9.224mmol,70%)。ms m/z 292(m h)
。
[1062]8‑
溴
‑2‑
丁氧基
‑9‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
9h
‑
嘌呤
‑6‑
胺(139):在23℃下,向化合物138(2687mg,9224mmol)在dcm(50ml)中的溶液加入n
‑
溴代琥珀酰亚胺(2000mg,11069mmol)。在1h之后,向混合物加入饱和硫代硫酸钠(20ml)。将原料用dcm(20ml)萃取。将有机层用饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。在真空中除去有
m/z 585(m h)
。
[1071][1072]2‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基氨基甲酸叔丁酯(147):使用化合物142和6
‑
氨基己基氨基甲酸叔丁酯作为起始原料,采用与143所述相似的程序,制备化合物147,获得目标化合物147(23mg,0.026mmol,14%)。ms m/z542(m h)
。
[1073]6‑
氨基
‑9‑
(4
‑
((6
‑
氨基己基氨基)甲基)苄基)
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(148):使用化合物147作为起始原料,采用与144所述相似的程序,制备化合物148,获得目标化合物148(24mg,0.027mmol,定量)。ms m/z 442(m h)
。
[1074]
n
‑
(6
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基氨基)己基)
‑2‑
(氨基氧基)乙酰胺(149):使用化合物148作为起始原料,采用与127所述相似的程序,制备化合物149,获得目标化合物149(7mg,0.008mmol,31%)。ms m/z 515(m h)
。
[1075][1076]
n
‑
(2
‑
(1
‑
(4
‑
(6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑3‑
(2
‑
(氨基氧基)乙氧基)丙酰胺(150):在23℃下,向化合物144(14mg,0.018mmol)和n
‑
boc
‑
n,2
‑
二甲基
‑
丙氨酸(5.5mg,0.020mmol)在dmf(1ml)中的溶液加入dmtmmt(5mg,0.021mmol)和diea(20ul,0.115mmol)。在30min之后,将混合物通过prep
‑
lc纯化,获得化合物150(7mg,0.007mmol,37%)。ms m/z 653(m h)
。
[1077]
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑2‑
甲基
‑2‑
(甲基氨基)丙酰胺(151):使用化合物150作为起始原料,采用与144所述相似的程序,制备化合物151,获得目标化合物151(5.5mg,0.005mmol,定量)。ms m/z 553(m h)
。
[1078]
[1079]
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)新戊酰胺(152):在23℃下,向化合物144(14mg,0.018mmol)和三甲基乙酰氯(2.8ul,0.022mmol)在dmf(1ml)中的溶液加入diea(20ul,0.115mmol)。在1h之后,将混合物通过prep
‑
lc纯化,获得化合物152(7mg,0.008mmol,36%)。ms m/z 538(m h)
。
[1080][1081]
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)乙酰胺(153):在23℃下,向化合物144(20mg,0.022mmol)和乙酸酐(2.1ul,0.021mmol)在dmf(1ml)中的溶液加入diea(20ul,0.115mmol)。在1h之后,将混合物通过prep
‑
lc纯化,获得化合物153(8mg,0.010mmol,43%)。ms m/z 496(m h)
。
[1082][1083]4‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑
3,5
‑
二氟苯甲酰胺(154):使用化合物144和4
‑
氨基
‑
3,5
‑
二氟苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物154,获得目标化合物154(8mg,0.008mmol,38%)。ms m/z 609(m h)
。
[1084][1085]
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)异丁酰胺(155):使用化合物144和异丁酸作为起始原料,采用与150所述相似的程序,制备化合物155,获得化合物155(6mg,0.007mmol,32%)。ms m/z 524(m h)
。
[1086][1087]
1,7
‑
双(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基氨基)
‑
1,7
‑
二氧代庚烷
‑4‑
基氨基甲酸叔丁酯(156):使用化合物144和4
‑
(n
‑
boc
‑
氨基)
‑
1,6
‑
庚二酸作为起始原料,采用与150所述相似的程序,制备化合物156,获得目标化合物156(15mg,0.009mmol,34%)。ms m/z 1147(m h)
。
[1088]4‑
氨基
‑
n1,n7
‑
双(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)庚二酰胺(157):使用化合物156作为起始原料,采用与144所述相似的程序,制备化合物157,获得目标化合物157(15mg,0.01mmol,定量)。ms m/z 1047(m h)
。
[1089]
n1,n7
‑
双(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑4‑
(2
‑
(氨基氧基)乙酰胺基)庚二酰胺(158):使用化合物157和2
‑
(叔丁氧基羰基氨基氧基)乙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯作为起始原料,采用与145所述相似的程序,制备化合物158,获得目标化合物158(5mg,0.003mmol,34%)。ms m/z 1120(m h)
。
[1090][1091]
(s)
‑2‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)丙酰胺(173):在23℃下,向化合物144(20mg,0.022mmol)和n
‑
boc
‑
丙氨酸(5mg,0.026mmol)在dmf(1ml)中的溶液加入dmtmmt(6mg,0.025mmol)和diea(20ul,0.115mmol)。在10min之后,在真空中除去溶剂。将dcm(1ml)和tfa(1ml)添加至残渣。在10min之后,在真空中除去溶剂,并将残渣通过prep
‑
lc纯化,获得化合物173(8mg,0.009mmol,42%)。ms m/z 525(m h)
。
[1092][1093]
(s)
‑2‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑5‑
胍基戊酰胺(174):使用化合物144和n
‑
boc
‑
精氨酸作为起始原料,采用与173所述相似的程序,制备化合物174,获得目标化合物174(10mg,0.011mmol,48%)。ms m/z 610(m h)
。
[1094][1095]
在23℃下,将4
‑
(2
‑
(异丁基氨基)乙基)哌啶
‑1‑
甲酸叔丁酯(175):4
‑
(2
‑
氨基乙基)
‑1‑
boc
‑
哌啶(520mg,2.278mmol)和异丁醛(230ul,3.190mmol)溶解在甲醇(10ml)中。在
2h之后,向该混合物加入硼氢化钠(142mg,3.754mmol)。在10min之后,在真空中除去溶剂。将残渣溶解在dcm(100ml)中,用饱和nahco3(50ml)和盐水(50ml)洗涤,经mgso4干燥,并过滤。在真空中除去溶剂。将残渣通过prep
‑
lc纯化,获得为玻璃状无色固体的化合物175(499mg,1.755mmol,55%)。ms m/z 285(m h)
。
[1096]4‑
(2
‑
(3
‑
(2
‑
(1,3
‑
二氧代吲哚啉
‑2‑
基氧基)乙氧基)
‑
n
‑
异丁基丙酰胺基)乙基)哌啶
‑1‑
甲酸叔丁酯(176):在23℃下,向化合物175(80mg,0.201mmol)和phth
‑
peg1
‑
cooh(56mg,0.201mmol)在etoac(10ml)中的溶液加入cmpi(62mg,0.243mmol)和diea(70ul,0.402mmol)。在3h之后,通过过滤除去沉淀物,并将滤液用快速色谱法纯化,获得为白色固体的化合物176(65mg,0.119mmol,59%)。ms m/z 546(m h)
。
[1097]3‑
(2
‑
(1,3
‑
二氧代吲哚啉
‑2‑
基氧基)乙氧基)
‑
n
‑
异丁基
‑
n
‑
(2
‑
(哌啶
‑4‑
基)乙基)丙酰胺(177):使用化合物176作为起始原料,采用与144所述相似的程序,制备化合物177,获得目标化合物177(66mg,0.118mmol,定量)。ms m/z 446(m h)
。
[1098]
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑3‑
(2
‑
(氨基氧基)乙氧基)
‑
n
‑
异丁基丙酰胺(178):使用化合物177和化合物142作为起始原料,采用与143所述相似的程序,随后如130所述用肼h2o(10ul)处理,从而制备化合物178,获得化合物178(19mg,0.019mmol,7%)。ms m/z 641(m h)
。
[1099][1100]6‑
氨基
‑2‑
丁氧基
‑9‑
(4
‑
(哌啶
‑1‑
基甲基)苄基)
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(179):使用化合物142和哌啶作为起始原料,采用与143所述相似的程序,制备化合物179,获得化合物179(31mg,0.041mmol,54%)。ms m/z 411(m h)
。
[1101][1102]4‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
(6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑3‑
甲氧基苯甲酰胺(180):使用化合物144和4
‑
氨基
‑3‑
甲氧基苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物180,获得目标化合物180(8mg,0.008mmol,39%)。ms m/z 603(m h)
。
[1103][1104]
(s)
‑
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)丙酰胺(181):使用化合物173和2
‑
(叔丁氧基
羰基氨基氧基)乙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯作为起始原料,采用与127所述相似的程序,制备化合物181,获得目标化合物181(5mg,0.005mmol,58%)。ms m/z 598(m h)
。
[1105][1106]
(s)
‑
n
‑
(2
‑
(1
‑
(4
‑
((6
‑
氨基
‑2‑
丁氧基
‑8‑
氧代
‑
7h
‑
嘌呤
‑
9(8h)
‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑2‑
(2
‑
(氨基氧基)乙酰胺基)
‑5‑
胍基戊酰胺(182):使用化合物174和2
‑
(叔丁氧基羰基氨基氧基)乙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯作为起始原料,采用与127所述相似的程序,制备化合物182,获得目标化合物182(5mg,0.004mmol,42%)。ms m/z 683(m h)
。
[1107][1108]9‑
(4
‑
(4,4'
‑
二哌啶
‑1‑
基甲基)苄基)
‑6‑
氨基
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(183):使用化合物142和4,4'
‑
二哌啶作为起始原料,采用与143所述相似的程序,制备化合物183,获得化合物183(13mg,0.016mmol,7%)。ms m/z 494(m h)
。
[1109][1110]6‑
氨基
‑9‑
(4
‑
((1'
‑
(2
‑
(氨基氧基)乙酰基)
‑
4,4'
‑
二哌啶
‑1‑
基)甲基)苄基)
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(184):使用化合物183和2
‑
(叔丁氧基羰基氨基氧基)乙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯作为起始原料,采用与127所述相似的程序,制备化合物184,获得目标化合物184(5mg,0.005mmol,18%)。ms m/z 567(m h)
。
[1111][1112]3‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)苯甲酰胺(185):使用化合物144和3
‑
氨基苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物185,获得目标化合物185(8mg,0.009mmol,53%)。ms m/z 572(m h)
。
[1113]
[1114]
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑4‑
(2
‑
氨基乙基)苯甲酰胺(186):使用化合物144和4
‑
(2
‑
boc
‑
氨基)乙基苯甲酸作为起始原料,采用与173所述相似的程序,制备化合物186,获得目标化合物186(9mg,0.010mmol,58%)。ms m/z 600(m h)
。
[1115][1116]4‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)苯甲酰胺苯甲酰胺(187):使用化合物144和4
‑
氨基苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物187,获得目标化合物187(8mg,0.009mmol,53%)。ms m/z 572(m h)
。
[1117][1118]3‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑4‑
氟苯甲酰胺(188):使用化合物144和3
‑
氨基
‑4‑
氟苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物188,获得目标化合物188(11mg,0.011mmol,64%)。ms m/z 590(m h)
。
[1119][1120]
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑4‑
(2
‑
(2
‑
(氨基氧基)乙酰胺基)乙基)苯甲酰胺(189):使用化合物186和2
‑
(叔丁氧基羰基氨基氧基)乙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯作为起始原料,采用与127所述相似的程序,制备化合物189,获得目标化合物189(11mg,0.010mmol,94%)。ms m/z 673(m h)
。
[1121][1122]6‑
氨基
‑9‑
(4
‑
((4
‑
(4
‑
氨基苯基)哌啶
‑1‑
基)甲基)苄基)
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(190):使用化合物142和4
‑
(4
‑
氨基苯基)
‑
哌啶作为起始原料,采用与143所述相似的程序,制备化合物190,获得化合物190(3mg,0.004mmol,5%)。ms m/z 502(m h)
。
[1123][1124]6‑
氨基
‑9‑
(4
‑
((1'
‑
(3
‑
(2
‑
(氨基氧基)乙氧基)丙酰基)
‑
4,4'
‑
二哌啶
‑1‑
基)甲基)苄基)
‑2‑
丁氧基
‑
7h
‑
嘌呤
‑
8(9h)
‑
酮(191):使用化合物183作为起始原料,采用与137所述相似的程序,制备化合物191,获得目标化合物191(13mg,0.012mmol,48%)。ms m/z 625(m h)
。
[1125][1126]
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑4‑
羟基苯甲酰胺(213):使用化合物144和4
‑
羟基苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物213,获得目标化合物213(10mg,0.011mmol,66%)。ms m/z 573(m h)
。
[1127][1128]
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑3‑
(4
‑
羟基苯基)丙酰胺(214):使用化合物144和3
‑
(4
‑
羟基苯基)丙酸作为起始原料,采用与150所述相似的程序,制备化合物214,获得目标化合物214(9mg,0.010mmol,58%)。ms m/z 601(m h)
。
[1129][1130]
boc
‑
lys(boc
‑
氨基氧基乙酰基)
‑
oh(215):在23℃下,向在dmf(5ml)中的2
‑
(叔丁氧基羰基氨基氧基)乙酸2,5
‑
二氧代吡咯烷
‑1‑
基酯(399mg,1.384mmol)和boc
‑
lys
‑
oh(335mg,1.360mmol)加入diea(750ul,4.306mmol)。在2h之后,在真空中除去溶剂。将残渣溶解于etoac(50ml)中,并用1n hcl(50ml)和盐水(20ml)洗涤。有机层经mgso4干燥,过滤,并在真空中除去溶剂。将残渣通过快速色谱法纯化,获得为白色固体的化合物215(480mg,1.144mmol,83%)。ms m/z 420(m h)
。
[1131]
(s)
‑2‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)
‑6‑
(2
‑
(氨基氧基)乙酰胺基)己酰胺(216):使用化合物144和化合物215作为起始原料,采用与173所述相似的程序,制备化合物216,获得目标化合物216(6mg,0.006mmol,37%)。ms m/z 654(m h)
。
[1132][1133]
(s)
‑
n
‑
(5
‑
氨基
‑6‑
(1'
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)
‑
4,4'
‑
二哌啶
‑1‑
基)
‑6‑
氧代己基)
‑2‑
(氨基氧基)乙酰胺(217):使用化合物183和化合物215作为起始原料,采用与173所述相似的程序,制备化合物217,获得目标化合物217(6mg,0.006mmol,37%)。ms m/z 654(m h)
。
[1134][1135]5‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)烟酰胺(218):使用化合物144和5
‑
氨基烟酸作为起始原料,采用与150所述相似的程序,制备化合物218,获得目标化合物218(1mg,0.001mmol,7%)。ms m/z573(m h)
。
[1136][1137]5‑
氨基
‑
n
‑
(2
‑
(1
‑
(4
‑
((4
‑
氨基
‑6‑
丁氧基
‑2‑
氧代
‑
2,3
‑
二氢
‑
1h
‑
咪唑并[4,5
‑
c]吡啶
‑1‑
基)甲基)苄基)哌啶
‑4‑
基)乙基)吡嗪
‑2‑
甲酰胺(219):使用化合物144和5
‑
氨基
‑
吡嗪
‑
甲酸作为起始原料,采用与150所述相似的程序,制备化合物219,获得目标化合物219(10mg,0.11mmol,66%)。ms m/z 574(m h)
。
[1138]
表4
–
tlr激动剂
‑
核心5化合物
[1139]
[1140]
[1141]
[1142]
[1143]
[1144]
[1145][1146]
实施例4:包含以下结构
–
核心3的tlr激动剂(图1)的合成:
[1147]
核心3
[1148][1149]
在一些实施方案中,x是n或h;y是c或n;
[1150]
r1是c1至c
12
烷基、取代的c1至c
12
烷基、含氧的c1至c
12
烷基、杂环、取代的杂环或h
[1151]
r2是c1至c
12
烷基、c1至c
12
取代的烷基、c4至c8环烷基、芳香环、取代的芳香环、芳杂环、取代的芳杂环、
‑
onh2、末端c1至c
12
烷基或h
[1152]
如以下方案中所公开合成具有核心3结构的tlr激动剂。
[1153][1154]4‑
硝基
‑1‑
甲苯磺酰基
‑
1h
‑
吲哚(160):将化合物159(4
‑
硝基
‑
1h
‑
吲哚)(2.43g,15.0mmol)溶解在thf(15ml)中。在0℃下,将thf(30ml)中的氢化钠(900mg,22.5mmol)滴加到悬浮液中。将溶液加温至室温,另外搅拌1h。接着,缓慢加入在thf(15ml)中的甲苯磺酰氯(3.0g,15.75mmol),并将反应物搅拌过夜,使溶液在nahco3和et2o之间分配。将水层萃取(3x75ml),将合并的有机层用盐水洗涤,经mgso4干燥,并且真空浓缩。使所得固体吸收到accn中,超声处理,并过滤。未使用固体(起始原料)。将液体旋转蒸发,并将残渣(160)用于下一步(3.12g)。未观察ms m/z no2。
[1155]5‑
甲基
‑4‑
硝基
‑1‑
(苯基磺酰基)
‑
1h
‑
吲哚(161):在
‑
15℃下,将化合物160(3.11g,9.83mmol)溶解在thf(98.3ml)中。加入甲基氯化镁(4.9ml,14.75mmol),允许搅拌溶液持续1h 45min。接着加入ddq(3.79g,16.71mmol),同时保持温度低于
‑
10℃。将该反应加温至室温并且搅拌过夜。接下来,用dcm稀释反应物,使反应淬灭,然后旋转蒸发。使粗产物穿过用dcm洗脱的sio2塞。将洗脱液干燥并通过柱色谱法(在dcm中的己烷,0
‑
40%,40g柱)纯化,获得目标化合物161(2.06g,63%经过两个步骤)。未观察ms m/z no2。
[1156]
((e)n,n
‑
二甲基
‑2‑
(4
‑
硝基
‑1‑
(苯基磺酰基)
‑
1h
‑
吲哚
‑5‑
基)乙烯
‑1‑
胺(162):将5
‑
甲基
‑4‑
硝基
‑1‑
(苯基磺酰基)
‑
1h
‑
吲哚(161)(0.83g,2.63mmol)溶解在dmf(26.3ml)中。加入n,n
‑
二甲基甲酰胺二甲基缩醛(3.54ml,26.3mmol),并将反应物在115℃加热。将反应物通过旋转蒸发器蒸发。残渣(化合物162)用于下一步反应(0.98克)。未观察ms m/z no2。
[1157]4‑
硝基
‑1‑
(苯基磺酰基)
‑
1h
‑
吲哚
‑5‑
甲醛(163):向化合物162(0.98g,2.6mmol)在thf(13.2ml)和水(13.2ml)中的溶液加入偏高碘酸钠(1.7g mg,7.9mmol),并搅拌。将反应物过滤并用etoac(50ml)洗涤。将有机层用nahco3洗涤,经mgso4干燥,过滤,并且用旋转蒸发器蒸发。将残渣(化合物163,0.56g)在真空泵上干燥,并且用于下一步反应。未观察ms m/z no2。
[1158]4‑
氨基
‑1‑
(苯基磺酰基)
‑
1h
‑
吲哚
‑5‑
甲醛(164):向在meoh(47ml)中的化合物163(0.56g,1.7mmol)的溶液加入pd/c(0.03g)。将反应物在氢气(双气囊/1个大气压)气氛下搅
拌。将反应物用硅藻土过滤,并用meoh洗涤。在真空中使溶剂干燥,将残渣通过柱色谱法纯化(在etoac中的己烷,0
‑
50%,12g柱),获得目标化合物164(93mg,12%经过三个步骤),ms m/z 301(m h)
。
[1159]
7h
‑
吡咯并[2,3
‑
h]喹唑啉
‑2‑
胺(165):向化合物164(0.092g,0.31mmol)在dma(3.1ml)中的溶液加入碳酸胍(279mg,3.09mmol),并在150℃下搅拌。lcms显示反应完成,将混合物通过prep
‑
lc纯化,获得目标化合物165(6mg,11%),ms m/z 257(m h)
。
[1160]1‑
(2
‑
氨基
‑
7h
‑
吡咯并[2,3
‑
h]喹唑啉
‑7‑
基)
‑2‑
甲基丙
‑2‑
醇(166):在0℃下,向氢化钠溶液(矿物油中的60%分散液,2.2mg,0.054mmol)中加入5ml的己烷,并搅拌该溶液。除去己烷,以便洗去矿物油。接下来,将在dmf(1.1ml)中的化合物165(2mg,0.011mmol)滴加到所述溶液中,并搅拌1h。接着,滴加氧化异丁烯(1ul,0.011mmol),并搅拌反应物。将反应物过滤,并将混合物通过prep
‑
lc纯化,获得目标化合物166(1.5mg,23%),ms m/z 185(m h)
。
[1161]
表5
–
tlr激动剂
‑
核心3化合物
[1162][1163]
实施例5:包含以下代表性结构
–
核心2的tlr激动剂(图1)的合成:
[1164]
核心2
[1165][1166]
r1和r2各自连接形成c4至c8环烷基或独立地是
‑
h、c1至c
12
烷基、含硝基烷基、芳香环或
–
c(nh)nh2;
[1167]
r3是c1至c
12
烷基、取代的c1至c
12
烷基、含氧的c1至c
12
烷基、杂环取代的杂环或h。
[1168]
如以下方案中所公开合成具有核心2结构的tlr激动剂。
[1169][1170]2‑
(5,6
‑
二氨基嘧啶
‑4‑
基氨基)
‑2‑
氧代乙基氨基甲酸叔丁酯(167):在23℃下,向嘧啶
‑
4,5,6
‑
三胺(2050mg,16.383mmol)和boc
‑
gly
‑
oh(2880,16.440mmol)在dcm(50ml)中的溶液加入dcc(3800mg,18.417mmol)和dmap(80mg,0.655mmol)。在3h之后,经过滤除去沉淀物。将混合物通过prep
‑
lc纯化,获得目标化合物167(2458mg,8.707mmol,53%)ms m/z 283(m h)
。
[1171]
(6
‑
氨基
‑
9h
‑
嘌呤
‑8‑
基)甲基氨基甲酸叔丁酯(168):在23℃下,向化合物167(620mg,2.196mmol)在n
‑
buoh(20ml)中的溶液加入在meoh(2500ul,11.570mmol)中的25%naome。将温度升至70℃。在1h之后,将6n hcl(1.83ml,11mmol)加入到冰浴中,并将混合物用etoac(50ml)稀释。将混合物用饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,用mgso4干燥,并过滤。将混合物通过快速色谱法纯化,获得目标化合物168(250mg,0.946mmol,43%),ms m/z 265(m h)
。
[1172]
(6
‑
氨基
‑9‑
(2
‑
溴乙基)
‑
9h
‑
嘌呤
‑8‑
基)氨基甲酸甲叔丁酯(169):在23℃下,向化合物168(250mg,0.946mmol)在dmf(5ml)中的溶液加入二溴乙烷(2100mg,2.795mmol)和csco3(2400mg,1.842mmol)。在2.5h之后,将混合物用20ml dcm稀释,并用饱和碳酸氢钠(50ml)和盐水(50ml)洗涤。将有机层用mgso4干燥,并过滤。将混合物通过prep
‑
lc纯化,获得化合物169(144mg,0.545mmol,58%),ms m/z 372(m h)
。
[1173]4‑
氨基
‑
8,9
‑
二氢吡嗪并[1,2
‑
e]嘌呤
‑
7(6h)
‑
甲酸叔丁酯(170):向氢化钠溶液(在矿物油中的60%分散液,51.4mg,1.286mmol)中加入5ml的己烷。搅拌溶液,随后除去己烷,以便洗去矿物油。在23℃搅拌下,将在dmf(2.9ml)中的化合物169(159.2mg,0.429mmol)滴加到所述溶液中。在1h之后,lcms显示反应完成。通过使用gemini nx 150x30 c18柱,以5%至60%的水/90%acn 0.05%tfa梯度将混合物通过prep
‑
lc纯化,持续20min。合并含有产物的级分,并通过旋转蒸发器蒸发。将残渣在高真空泵上干燥,获得目标化合物170(36.7mg,0.05mmol,11%),ms m/z 291(m h)
。
[1174]
6,7,8,9
‑
四氢吡嗪并[1,2
‑
e]嘌呤
‑4‑
胺(171):向170(35.7mg,0.123mmol)的溶液加入1.2ml的dcm。在23℃搅拌下滴加三氟乙酸(45.7ul,0.615mmol)。在1h之后,lcms显示反应完成。将混合物通过旋转蒸发器蒸发,使用phme进行另外的共沸,获得化合物171(38.5mg,0.05mmol,41%),ms m/z 191(m h)
。
[1175]7‑
苄基
‑
6,7,8,9
‑
四氢吡嗪并[1,2
‑
e]嘌呤
‑4‑
胺(172):向171(10mg,0.053mmol)在dmf(1.1ml)中的溶液加入苯甲醛(6.7ul,0.066mmol)。加入diea(18.3ul,0.105mmol),并且将反应物搅拌15分钟。接着,加入硼
‑
吡啶络合物(6.7ul,0.067mmol),并且将反应物在23℃下搅拌过夜。通过使用gemini nx 150x30 c18柱,以5%至60%的水/90%acn 0.05%tfa
梯度将混合物通过prep
‑
lc纯化,持续20min。合并含有产物的级分,并通过旋转蒸发器蒸发。将残渣在高真空泵上干燥,获得化合物172(1.3mg,0.002mmol,3%),ms m/z 281(m h)
。
[1176]
表6
–
tlr激动剂
‑
核心2化合物
[1177][1178]
实施例6:包含以下代表性结构
–
核心4的tlr激动剂(图1)的合成:
[1179]
核心4
[1180][1181]
在一些实施方案中,r1是c1至c
12
烷基、取代的c1至c
12
烷基、含氧的c1至c
12
烷基、c3至c8环烷基、杂环、取代的杂环、卤素或h;
[1182]
r2是c1至c
12
烷基、c1至c
12
取代的烷基、c4至c8环烷基、芳香环、取代的芳香环、芳香杂环、取代的芳香杂环、
‑
onh2末端c1至c
12
烷基或h;
[1183]
r3是c1至c
12
烷基、取代的c1至c
12
烷基、含氧/氮/硫的c1至c
12
烷基、杂环、取代的杂环、环烷基、取代的环烷基、
‑
n3、末端c1至c
12
烷基、末端取代的c1至c
12
烷基,或不存在。
[1184]
如以下方案中所公开合成具有核心4结构的tlr激动剂。
[1185][1186]
n
‑
(4,6
‑
二氨基嘧啶
‑5‑
基)戊酰胺(193):在70℃下,将嘧啶
‑
4,5,6
‑
三胺(1015mg,8.112mmol)溶解在n
‑
甲基
‑2‑
吡咯烷酮(10ml)中。在溶液变清之后,冷却至23℃。向混合物加入戊酰氯(980ul,8.127mmol),并且将温度升至50℃。在20h之后,将温度降至23℃。向混合物加入etoac(50ml,沉淀物),并且通过过滤分离沉淀物。将固体用etoac(10ml))和丙酮(10ml)洗涤,并干燥,获得为浅棕色固体的化合物193(1780mg,7.237mmol,90%)。将产物用于下一个步骤而无需进一步纯化。ms m/z 210(m h)
。
[1187]8‑
丁基
‑
9h
‑
嘌呤
‑6‑
胺(194):在23℃下,向粗化合物193(1780mg,7.273mmol)在n
‑
buoh(30ml)中的溶液加入甲醇钠(1570mg,29.063mmol),并且加热至回流。在1h之后,将溶液冷却至室温,用6m hcl(3.2ml)中和,并加入盐水(20ml),获得二相混合物。将有机层分离,通过mgso
4干燥,
然后真空浓缩,获得为淡棕色固体的目标化合物194(1148mg 6.003mmol,83%)。ms m/z 192(m h)
。
[1188]4‑
(6
‑
氨基
‑8‑
丁基
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(195):在0℃下,向n
‑
boc
‑
氨基
‑
丁醇(1520mg,8.032mmol)和化合物1194(1420mg,7.426mmol)在thf(20ml)中的溶液加入pph3(2080mg,7.930mmol)。在30min之后,在0℃下,经5min向此混合物加入diad(2200ul,11.174mmol)。在3h之后,在真空中除去溶剂。将残渣用dcm(100ml)稀释,并且用半饱和碳酸氢钠(100ml)和盐水(20ml)洗涤。将有机层经mgso4干燥,并过滤。在真空中除去溶剂。将残渣通过快速色谱法纯化,获得为浅棕色固体的化合物195(1064mg,2.935mmol,37%)。ms m/z 363(m h)
。
[1189]4‑
(6
‑
(n
‑
苯甲酰基苯甲酰胺基)
‑8‑
丁基
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(196):在0℃下,向化合物195(1064mg,2.935mmol)在dcm(10ml)中的溶液加入苯甲酰氯(700ul,4.980mmol)和tea(900ul,17.788mmol),并将温度升至20℃。在2.5h之后,将混合物用饱和碳酸氢钠(50ml)和盐水(50ml)洗涤,经mgso4干燥,并过滤。在真空中除去溶剂,并将残渣通过快速色谱法纯化,获得为浅棕色油的化合物196(1650mg,2.891mmol,98%)。ms m/z 571(m h)
。
[1190]4‑
(6
‑
(n
‑
苯甲酰基苯甲酰胺基)
‑8‑
丁基
‑2‑
硝基
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(197):在23℃下,向四甲基硝酸铵(780mg,5.729mmol)在dcm(10ml)中的溶液加入三氟乙酸酐(1200ul,17.118mmol)。在1h之后,将混合物冷却至0℃,并加入化合物196(1650mg,2.891mmol)在dcm(20ml)中的溶液。将温度升至23℃。在2h之后,将混合物用dcm(20ml)稀释,用半饱和碳酸氢钠(20ml)和盐水(20ml)洗涤,经mgso4干燥,并过滤。在真空中除去溶剂,并将残渣通过快速色谱法纯化,获得为玻璃状淡黄色固体的化合物197(1067mg,
1.733mmol,60%)。ms m/z 616(m h)
。
[1191]9‑
(4
‑
氨基丁基)
‑8‑
丁基
‑
9h
‑
嘌呤
‑6‑
胺(198)和6
‑
氨基
‑9‑
(4
‑
氨基丁基)
‑8‑
丁基
‑
9h
‑
嘌呤
‑2‑
醇(199):在23℃下,向化合物197(220mg,0.357mmol)在etoh(20ml)中的溶液加入pd/c(10%,0.1g),并鼓入氢气。在18h之后,lcms显示产生了脱硝化合物。加入naome(30mg,0.6mmol),并将混合物搅拌4h。接下来,加入tfa(3ml)。在20min之后,在真空中除去溶剂,将混合物通过prep
‑
lc纯化,获得为浅棕色固体的化合物198(94mg,0.156mmo,44%),ms m/z 263(m h)
,以及为浅棕色固体的化合物199(0.024mmol,7%),ms m/z 279(m h)
。
[1192][1193]4‑
氨基
‑
n
‑
(4
‑
(6
‑
氨基
‑8‑
丁基
‑2‑
羟基
‑
9h
‑
嘌呤
‑9‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(200):使用化合物199和4
‑
氨基
‑
3,5
‑
二氟
‑
苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物200,获得为浅棕色固体的目标化合物200(14mg,0.018mmol,61%),ms m/z 434(m h)
。
[1194][1195]4‑
氨基
‑
n
‑
(4
‑
(6
‑
氨基
‑8‑
丁基
‑
9h
‑
嘌呤
‑9‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(201):使用化合物198和4
‑
氨基
‑
3,5
‑
二氟
‑
苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物201,获得为浅棕色固体的目标化合物201(13mg,0.017mmol,93%),ms m/z 418(m h)
。
[1196][1197]3‑
氨基
‑
n
‑
(4
‑
(6
‑
氨基
‑8‑
丁基
‑
9h
‑
嘌呤
‑9‑
基)丁基)苯甲酰胺(202):使用化合物198和3
‑
氨基苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物202,获得为浅棕色固体的目标化合物202(10mg,0.014mmol,88%),ms m/z 382(m h)
。
[1198][1199]5‑
氨基
‑
n
‑
(4
‑
(6
‑
氨基
‑8‑
丁基
‑
9h
‑
嘌呤
‑9‑
基)丁基)烟酰胺(203):使用化合物198和5
‑
氨基
‑
烟酸作为起始原料,采用与150所述相似的程序,制备化合物203,获得为浅棕色固体的目标化合物203(14mg,0.019mmol,定量),ms m/z 383(m h)
。
[1200][1201]4‑
(2,6
‑
二氯
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(204):在0℃下,向n
‑
boc
‑
氨基
‑
丁醇(1620mg,8.560mmol)和2,6
‑
二氯嘌呤(1495mg,7.910mmol)在thf(10ml)中的溶液加入pph3(2280mg,8.693mmol)。在30min之后,在0℃下经5min加入diad(2300ul,11.681mmol)。将混合物在50℃下搅拌。在6h之后,在真空中除去溶剂。将残渣用etoac(100ml)稀释,并且用半饱和碳酸氢钠(100ml)和盐水(20ml)洗涤。将有机层用mgso4干燥,并过滤。在真空中除去溶剂。将残渣通过快速色谱法纯化,获得为黄色油的化合物204(4500mg,<12.492mmol,<100%)。(约20%的杂质是pph3)。ms m/z 361(m h)
。
[1202]4‑
(6
‑
氨基
‑2‑
氯
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(205):将化合物204(pph3的粗混合物,4500mg,<12.492mmol)放入配备有搅拌棒的耐压玻璃容器中。向该容器中加入meoh(12ml,84mmol)中的7nnh3。将管密封,然后在120℃下加热。在30min之后,在真空中除去溶剂,并将残渣溶解在dcm(100ml)中。将溶液用饱和碳酸氢钠(100ml)和盐水(30ml)洗涤。将有机层经mgso4干燥,并过滤。在真空中除去溶剂。将残渣通过快速色谱法纯化,获得为淡黄色固体的化合物205(1310mg,3.844mmol,37%)。ms m/z 341(m h)
。
[1203]4‑
(6
‑
氨基
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(206):在23℃在干燥氮气下向化合物205(257mg,0.754mmol)在正丁醇(5ml)中的溶液加入金属钠(90mg,2.455mmol)。将温度升至100℃。在18h之后,在真空中除去溶剂。将残渣溶解在dcm(50ml)中,并且用半饱和碳酸氢钠(50ml)和盐水(50ml)洗涤。将有机层经mgso4干燥,并过滤。在真空中除去溶剂。获得为浅棕色粗固体的化合物206(310mg,<0.819mmol,定量)。ms m/z 379(m h)
。
[1204]4‑
(6
‑
氨基
‑8‑
溴
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(207):在23℃下,向化合物206(310mg,0.819mmol,粗制物)在dcm(10ml)中的溶液加入溴(150ul,0.563mmol)。在1h之后,在真空中除去溶剂。将混合物通过快速色谱法纯化,获得为玻璃状淡黄色固体的化合物207(250mg,0.547mmol,67%)。ms m/z 458(m h)
。
[1205]4‑
(6
‑
氨基
‑2‑
丁氧基
‑8‑
甲基
‑
9h
‑
嘌呤
‑9‑
基)丁基氨基甲酸叔丁酯(208):在23℃下,向化合物207(82mg,0.179mmol)在干燥thf(5ml)中的溶液加入在thf中的的三甲基铝1m(360ul,0.36mmol)和pdcl2(pph3)2(44mg,0.063mmol)。将混合物回流。在20h之后,将混合物用20ml的dcm稀释,并用半饱和碳酸氢钠(20ml)和盐水(20ml)洗涤。将有机层经mgso4干燥,
并过滤。在真空中除去溶剂。将残渣通过快速色谱法纯化,获得为浅棕色固体的化合物208(12mg,0.031mmol,17%)。ms m/z 393(m h)
。
[1206]9‑
(4
‑
氨基丁基)
‑2‑
丁氧基
‑8‑
甲基
‑
9h
‑
嘌呤
‑6‑
胺(209):在23℃下,向化合物208(12mg,0.031mmol)在dcm(0.5ml)中的溶液加入三氟乙酸(0.5ml)。在1h之后,在真空中除去溶剂。将残渣在高真空泵上干燥过夜,获得为浅棕色固体的化合物209(15mg,0.02mmol,定量)。ms m/z 293(m h)
。
[1207]4‑
氨基
‑
n
‑
(4
‑
(6
‑
氨基
‑2‑
丁氧基
‑8‑
甲基
‑
9h
‑
嘌呤
‑9‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(210):使用化合物209和4
‑
氨基
‑
3,5
‑
二氟
‑
苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物210,获得为浅棕色固体的目标化合物210(3.5mg,0.004mmol,22%),ms m/z 469(m h)
。
[1208][1209]9‑
(4
‑
氨基丁基)
‑8‑
溴
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑6‑
胺(211):使用化合物207,采用与209所述相似的程序,制备化合物211,获得为浅棕色固体的目标化合物211(8mg,0.011mmol,定量),ms m/z 358(m h)
。
[1210]4‑
氨基
‑
n
‑
(4
‑
(6
‑
氨基
‑8‑
溴
‑2‑
丁氧基
‑
9h
‑
嘌呤
‑9‑
基)丁基)
‑
3,5
‑
二氟苯甲酰胺(212):使用化合物211和4
‑
氨基
‑
3,5
‑
二氟
‑
苯甲酸作为起始原料,采用与150所述相似的程序,制备化合物212,获得为浅棕色固体的目标化合物212(8mg,0.009mmol,82%),ms m/z 513(m h)
。
[1211]
表7
–
tlr激动剂
‑
核心4化合物
[1212]
[1213][1214]
实施例7:本实施例公开了在本发明中使用的各种方法和技术。
[1215]
分子克隆
‑
从商业dna合成服务(idt,san diego,ca)获得cho细胞密码子优化的抗体重链和轻链cdna序列。将合成的dna片段用hind iii和ecor i(两者都来自new england biolabs(neb),ipswich,ma)消化,并通过pcr纯化试剂盒(qiagen,valencia,ca)纯化。将消化的抗体基因片段通过快速连接试剂盒(neb)连接至表达载体,产生用于表达野生型抗体重链和轻链的构建体。使所得质粒在大肠杆菌中增殖,并通过dna测序服务(eton)加以验证。
[1216]
含琥珀密码子突变体的生成
‑
基于抗her2 fab的晶体结构,选择了位于轻链恒定区的10个不同的表面可及位点,用于非天然氨基酸(例如,对乙酰
‑
苯丙氨酸(paf)或对叠氮基
‑
苯丙氨酸)的遗传掺入。这些位点对于抗原抗体结合并不重要。然后通过定点诱变将选择的位点的每个遗传密码子突变为琥珀密码子(tag),从而生成产生针对该抗体突变体的表达质粒。从idt购买引物。使用q5定点诱变试剂盒,按照说明书手册(neb)进行所有定点诱变实验。使针对突变体的表达质粒在大肠杆菌中增殖,并通过dna测序服务(eton)加以验证。表8列出了抗her2 fab的重链或轻链恒定区中的琥珀突变位点及其kabat编号和相应的
氨基酸序列seq id no.:2和4至11。seq id no.:1和3分别显示了抗her2 fab的野生型重链和轻链。抗her2fab包括以下重链和轻链序列:seq id no:2和seq id no:4;seq id no:2和seq id no:5;seq id no:2和seq id no:6;seq id no:2和seq id no:7;seq id no:2和seq id no:8;seq id no:2和seq id no:9;seq id no:2和seq id no:10;seq id no:2和seq id no:11;seq id no:2和seq id no:12;seq id no:2和seq id no:13。
[1217]
表8.具有用于非天然氨基酸掺入的琥珀位点的抗her2 fab重链(hc)和轻链(lc)氨基酸序列。下表中还公开了其中paf被任何其他非天然氨基酸替换的所有序列。
[1218]
[1219]
[1220]
[1221][1222]
x表示非天然氨基酸(nnaa);下划线表示表7中的fc突变
[1223]
除了在重链中的114位的琥珀突变以外,还产生了在抗her2抗体或抗体片段的不同位置的fc突变,用于改善药代动力学和/或增强抗体依赖性细胞吞噬作用(adcp)和/或抗体依赖性细胞毒性adcc活性(表9)。
[1224]
表9
–
抗her2 fc突变
[1225][1226]
瞬时表达
‑
将平台细胞系301
‑
14维持在补充有3mm l
‑
谷氨酰胺(gibco)和3mm glutamax(gibco)的ex
‑
cell 302(sigma)中。将细胞每3
‑
4天传代一次,其接种密度为每ml 40万个细胞。在转染前一天,将细胞以每ml 60万个细胞接种。在第0天,使用maxcyte电穿孔平台,按照说明书手册,用编码轻链和重链的抗体表达质粒转染细胞。在转染之后,将细胞放置在空的125ml摇瓶中,在静态培养箱中在37℃下孵育30min。然后将转染的细胞接种到摇瓶中的基础表达培养基(50%dynamis
–
50%excell 302,补充有50μm msx)中,密度为3x106/ml。将转染的细胞在37℃、5%co2下在设置为140rpm的轨道摇床上孵育。在第1天,将1mm paf与7g/l的cell boost5(ge healthcare)、120μg/l的long r3 igf
‑
1(sigma)和2mm glutamax一起加入培养物中。将培养箱内部的温度从37℃转变到32℃。在第3天加入另外的7g/l的cell boost 5和2mm glutamax,并在第5天收集上清液。使用葡萄糖计监测葡萄糖水平,并且当培养基中葡萄糖水平低于2g/l时,向培养物中加入另外的葡萄糖。通过vi
‑
cell仪测量活细胞计数和活力。使用蛋白g传感器通过octet测量生产率。
[1227]
稳定的大量池生成
‑
使用pvu i(neb)消化6小时将表达质粒线性化。在线性化之
后,使用苯酚提取来纯化dna,并将其溶解在浓度为2.5μg/μl的无内毒素水中。将平台细胞系bb
‑
117维持在补充有3mm l
‑
谷氨酰胺和3mm glutamax的ex
‑
cell 302中。将细胞每3
‑
4天传代,其接种密度为0.4x106/ml。在转染前一天,将细胞以0.6x106/ml个接种。在第0天,使用maxcyte电穿孔平台,按照说明书手册,用线性化抗体表达质粒转染细胞。在转染之后,使细胞静置在空的125ml摇瓶中,在静态培养箱中在37℃孵育30分钟。然后将30ml恢复培养基(50%ex
‑
302
‑
50%cd
‑
cho,补充有3mm谷氨酰胺和3mm glutamax)加入烧瓶中,并且振荡过夜。第一天,对转染的细胞进行计数、旋转下降、洗涤,并且再悬浮在选择培养基(50%ex
‑
302
–
50%cd
‑
cho,具有50
‑
100μm msx)中,用于稳定的大量集合生成。监测活细胞数和活力,每3
‑
4天更换一次培养基,直到稳定的大量集合的活力恢复到90%时为止。当选择结束时,制成冷冻细胞储备物,并且使用所得稳定的大量集合来生成用于补料分批表达的材料。
[1228]
分批补料表达
‑
在第0天,将先前生成的抗体稳定的大量集合以0.5x106/ml的密度接种到摇瓶中的基础表达培养基(50%dynamis
–
50%excell 302,补充有50μm msx)中。将转染的细胞在37℃、5%co2下在设置为140rpm的轨道摇床上孵育。在第3天,将0.5mm paf与10g/l的cell boost 4(ge health care)和0.52g/l cell boost7b(ge healthcare)一起加入培养物中。在第5天,将120μg/l的long r3 igf
‑
1加入培养物中。使用葡萄糖计监测葡萄糖水平,并且当培养基中葡萄糖水平低于2g/l时,向培养物中加入另外的葡萄糖。通过vi
‑
cell仪测量活细胞计数和活力。在第7天,收集上清液用于纯化。使用蛋白g传感器通过octet测量生产率。
[1229]
从eucode表达系统纯化含有nnaa的抗体
‑
将含有包含有nnaa的靶抗体的澄清细胞培养基装载到在20mm磷酸钠、100mm氯化钠(ph 7.5)中平衡的蛋白a prosep ultra柱(emd millipore)上。在装载之后,用缓冲液a(20mm磷酸钠、100mm氯化钠,ph 7.5)洗涤柱,然后用洗涤缓冲液b(5mm琥珀酸,ph 5.8)洗涤,以除去宿主细胞污染物。用洗脱缓冲液c(50mm甘氨酸,10mm琥珀酸,ph 3.2)从柱中洗脱靶抗体。合并靶抗体,用2.0m tris碱将ph调节至ph 5.0。通过将条件性蛋白a集合装载到在30mm乙酸钠(ph 5.0)中平衡的capto sp impres柱(ge healthcare)上,进一步纯化靶抗体。以线性梯度将靶抗体从柱中洗脱至100%缓冲液b(30mm乙酸钠,0.5m氯化钠,ph5.0),合并含有单体抗体的级分,经0.22μm过滤,在≤65℃储存,直到进一步使用时为止。
[1230]
tlr激动剂接头有效负载的位点特异性缀合
‑
将含有nnaa例如对乙酰基苯丙氨酸的抗体缓冲交换到缀合缓冲液(30mm乙酸钠,ph 4.0)中,并浓缩至10
‑
20mg/ml。向抗体加入最后100mm的乙酰肼,然后加入10摩尔当量的羟胺官能化的tlr激动剂药物接头。将缀合反应在25
‑
30℃孵育18
‑
20小时,随后通过经capto sp impres柱(ge healthcare)进行纯化,以除去多余的试剂。将纯化的adc缓冲交换到制剂缓冲液(50mm组氨酸,100mm nacl,5%海藻糖,ph 6.0)中,在≤65℃下储存,直到进一步使用时为止。图2和3说明了本发明的tc的位点特异性缀合。用不同的tlr激动剂来说明tlr激动剂药物
‑
接头抗体缀合。如图3所示,aa指示可裂解氨基酸接头;l指示不可裂解接头。
[1231]
实施例8:小分子tlr激动剂的体外功能测定法
[1232]
将hek
‑
blue
tm
htlr7细胞在hek
‑
blue
tm
检测培养基中孵育,并用渐增浓度的tlr7或tlr8或tlr7/8激动剂刺激。在24h孵育之后,通过quanti
‑
blue检测试剂(invivogen,san diego,ca)确定nf
‑
kb诱导的seap的水平,获得在655nm od时的读数。使用prism软件,从剂
量
‑
反应曲线确定ec
50
。以下表和图显示了在本文实施例1
‑
6中描述的本发明的示例性tlr激动剂的活性。与ec
50
值在50nm至1um或大于1um至3um的化合物相比,小于500nm的ec
50
值表明具有更高效力的化合物。
[1233]
图4显示了tlr7激动剂在hek
‑
blue htlr7报告细胞系中的刺激作用,其中使用了商业tlr7激动剂瑞喹莫德(r848),ec
50
为2.08um;化合物1,ec
50
为0.435um;以及化合物2,ec
50
为0.153um。
[1234]
测定了在以上实施例1
‑
6中公开的示例性tlr激动剂的活性。图5a、表10显示了与商业对照dsr
‑
6434、瑞喹莫德和莫托莫德相比的ec
50
值。
[1235]
表10
–
示例性tlr激动剂的活性
[1236][1237][1238]
图5b和表11显示了选择的tlr7激动剂的tlr7活性。axc
‑
887和axc
‑
877的ec
50
值低于10nm,表明这些化合物是非常有效的tlr7激动剂。在tlr8报告基因测定中,与ec
50
为1427nm的商业化合物莫托莫德相比,axc
‑
887显示出可测量的活性,ec
50
为3733nm。这表明acx
‑
887是tlr7/8双重激动剂。
[1239]
表11
–
示例性tlr激动剂的活性
[1240]
化合物ec
50
(nm)dsr
‑
643415.8axc
‑
88616.7axc
‑
8874.6
axc
‑
89015.3axc
‑
88511.3axc
‑
888467.9axc
‑
87850.4axc
‑
8775.5axc
‑
88187.3axc
‑
88319.3axc
‑
88471.3
[1241]
图6和表12显示了与接头附接的选择的tlr7激动剂的tlr7活性。在不同的tlr激动剂(有效负载)接头中,axc
‑
879展现出最高的效力。
[1242]
表12
–
示例性tlr激动剂和接头的活性
[1243]
化合物tlr激动剂有效负载 接头 ec
50
(nm)dsr
‑
643414.8axc
‑
876362.4axc
‑
879151.2axc
‑
880409.5axc
‑
882550.8axc
‑
874>10,000axc
‑
875>10,000axc
‑
862400.1axc
‑
863864.2axc
‑
867829.0axc
‑
868>5,000axc
‑
869>10,000axc
‑
889696.5
[1244]
图7和表13显示了另外的tlr7激动剂和与接头附接的tlr7激动剂的tlr7活性。axc
‑
895在测试的不同的有效负载中展现出最高的效力,并且axc
‑
901在不同的有效负载接头中展现出最高的效力。(dl)表示与接头附接的药物接头或tlr激动剂或与接头附接的tlr激动剂(有效负载)。
[1245]
表13
–
示例性tlr激动剂和接头的活性
[1246]
化合物ec
50
(nm)dsr
‑
64349axc
‑
901(dl)48axc
‑
900(dl)615axc
‑
8991302axc
‑
89511axc
‑
89147axc
‑
896(dl)325
axc
‑
897(dl)330axc
‑
89850axc
‑
89273axc
‑
893(dl)3120axc
‑
8941672
[1247]
图8和表14显示了另外的tlr7激动剂和与接头附接的tlr7激动剂的tlr7活性。所有测试化合物都具有tlr7激动剂活性。axc
‑
894、axc
‑
903、axc
‑
904、axc905和axc
‑
906展现了测试的不同有效负载的最高效力,其中ec
50
值低于10nm。
[1248]
表14
‑
示例性tlr激动剂和接头的活性
[1249]
化合物tlr7 ec
50
(nm)dsr
‑
643410.5axc
‑
9034.3axc
‑
907(dl)51.8axc
‑
8942.1axc
‑
909(dl)71.9axc
‑
9048.1axc
‑
9052.6axc
‑
9065.4axc
‑
90811.5axc
‑
860119.5axc
‑
9102591.1
[1250]
图9和表15比较了与接头(药物接头或dl)和含有最终代谢物(dl
‑
paf)的非天然氨基酸(例如paf)附接的不同tlr7激动剂的tlr7活性。在所有情况下,含有最终代谢物的paf比它们对应的带有药物接头的有效负载展现出更高的效力。在不同的tlr有效负载接头中,axc
‑
879展现出最高的效力。
[1251]
表15
‑
非天然氨基酸(nnaa)存在或不存在时的示例性tlr激动剂 接头的活性
[1252][1253][1254]
dl=药物接头;dl
‑
paf=含有最终代谢物的paf
[1255]
实施例9:tc的位点特异性缀合
[1256]
如实施例7所述,使用分析反相hplc进行tc的位点特异性缀合。图10a显示了具有在氨基酸位置ha114的非天然氨基酸(例如paf)的未缀合抗her2抗体在还原条件下的分析反相hplc色谱图。图10b和10c显示了在氨基酸位置ha114分别与tlr激动剂axc
‑
875和axc
‑
880缀合的抗her2抗体。表16显示了来自上述标准缀合条件的tlr缀合物的药物
‑
抗体比(dar)。可以看出在tlr缀合物之间主要由于相关的tlr接头
‑
有效负载的不同特性而不同的dar水平(0.3
‑
2.0)。
[1257]
表16.通过rp
‑
hplc确定的tlr缀合物的药物
‑
抗体比(dar)。
[1258]
缀合物药物抗体比her2
‑
ha114
‑
axc6381.7her2
‑
ha114
‑
axc8001.7her2
‑
ha114
‑
axc8011.9her2
‑
ha114
‑
axc8021.5her2
‑
ha114
‑
axc8742.0her2
‑
ha114
‑
axc8751.9her2
‑
ha114
‑
axc8620.8her2
‑
ha114
‑
axc8631.8her2
‑
ha114
‑
axc8671.5her2
‑
ha114
‑
axc8681.2her2
‑
ha114
‑
axc8690.3
[1259]
实施例10:利用各种肿瘤细胞的体外共培养测定法
[1260]
将raw
‑
blue
tm
细胞(invivogen,san diego,ca)与具有不同水平的her2表达水平的人肿瘤细胞以1:1的e:t比例共培养,在96孔板中每孔细胞总数为100万。向共培养细胞培养基中加入不同浓度的小分子tlr7激动剂和缀合的isac(免疫刺激抗体缀合物)。在24h孵育之后,通过quanti
‑
blue检测试剂(invivogen,san diego,ca),利用在od 655nm的读数,确定nf
‑
kb诱导的来自raw
‑
blue
tm
细胞的seap水平。使用prism软件生成剂量
‑
反应曲线。
[1261]
图11a
‑
11c比较了选定的有效负载接头在与抗her2抗体缀合时的肿瘤依赖性isac活性。skov3是her2高表达肿瘤细胞系(图11a);jimt
‑
1是her2中等/低表达肿瘤细胞系(图11b);并且a431是her2低表达肿瘤细胞系(图11c)。小分子tlr7激动剂在所有肿瘤细胞系的存在下都显示出强效的tlr7活性。在her2低表达或中等表达肿瘤细胞系的存在下,所有tlr7 isac显示无活性。具有有效负载药物接头axc
‑
863的isac仅在her2高表达肿瘤细胞系的存在下显示出强效的剂量依赖性活性。
[1262]
图12a和12b比较了另外的有效负载接头在与抗her2抗体缀合时的肿瘤依赖性isac活性。skbr3是her2高表达肿瘤细胞系(图12a),并且hcc1806是her2极低表达肿瘤细胞系(图12b)。小分子tlr7激动剂在所有肿瘤细胞系的存在下都显示出强效的tlr7活性。而所有tlr7 isac和未缀合的抗her2抗体在her2极低表达肿瘤细胞系中显示无活性。具有有效负载药物接头axc
‑
863的isac仅在her2高表达肿瘤细胞系的存在下显示出强效的剂量依赖性活性。
[1263]
图13a和13b比较了另外的有效负载接头在与抗her2抗体缀合时的肿瘤依赖性
isac活性。skbr3是her2高表达肿瘤细胞系(图13a),并且hcc1806是her2极低表达肿瘤细胞系(图13b)。小分子tlr7激动剂在所有肿瘤细胞系的存在下都显示出强效的tlr7活性。所有tlr7 isac和未缀合的抗
‑
her2抗体在her2极低表达肿瘤细胞系中显示无活性。所有isac只有在her2高表达肿瘤细胞系的存在下才显示出有力的剂量依赖性活性(图13a)。具有有效负载药物接头axc
‑
879的isac展现出最高的her2依赖性tlr7活性。
[1264]
图14a和14b比较了另外的有效负载接头在与抗
‑
her2抗体缀合时的肿瘤依赖性isac活性。skbr3是her2高表达肿瘤细胞系(图14a),并且hcc1806是her2极低表达肿瘤细胞系(图14b)。小分子tlr7激动剂在所有肿瘤细胞系的存在下都显示出强效的tlr7活性。tlr7 isac和未缀合的抗her2抗体在her2极低表达肿瘤细胞系的存在下显示出无活性。所有isac只有在her2高肿瘤细胞系的存在下才显示出有力的剂量依赖性活性。具有有效负载药物接头axc
‑
901的isac展现出最高的her2依赖性tlr7活性。
[1265]
图15a和15b比较了在skbr3 her2的高表达肿瘤细胞系(图15a)和hcc1806 her2极低表达的肿瘤细胞系(图15b)中与抗her2抗体缀合的三(3)个有效负载接头的肿瘤依赖性isac活性,其显示相比于代表已知的isac的her2
‑
axc
‑
860和her2
‑
axc
‑
910,her2
‑
axc
‑
879具有最佳的isac活性。表17描绘了用于生成比较的her2
‑
axc isac的tlr激动剂
‑
接头结构。
[1266]
表17
–
与图15a
‑
15b相关的tlr激动剂结构
[1267][1268]
实施例11:乳腺癌的治疗
[1269]
曲妥珠单抗连接的tlr激动剂衍生物用于乳腺癌治疗的安全性和/或有效性的人体临床试验
[1270]
目的:比较包含曲妥珠单抗连接的tlr激动剂衍生物的施用组合物的安全性和药代动力学。
[1271]
研究设计:本研究将是在乳腺癌患者中进行的i期单中心开放标签随机化剂量递增研究,随后为ii期研究。在进入研究之前,患者不应曾经暴露于曲妥珠单抗连接的tlr激
动剂衍生物。患者必须在试验开始前的2周内未曾接受过癌症治疗。治疗包括使用化学疗法、造血生长因子和生物疗法,比如单克隆抗体。患者必须已从与先前治疗相关的所有毒性恢复(达到0或1级)。所有受试者都关于安全性加以评估,并且如期收集用于药物代谢动力学分析的所有血液收集物。所有研究都在机构伦理委员会批准和患者知情同意的情况下进行。
[1272]
i期:在每个28天周期的第1天、第8天和第15天,患者接受静脉注射曲妥珠单抗连接的tlr激动剂衍生物。曲妥珠单抗连接的tlr激动剂衍生物的剂量可以根据如下概述的评估针对毒性加以保持或修改。在不存在不可接受的毒性的情况下,每28天重复治疗。3
‑
6名患者的群组接受递增剂量的曲妥珠单抗连接的tlr激动剂衍生物,直到确定曲妥珠单抗连接的tlr激动剂衍生物的最大耐受剂量(mtd)时为止。将mtd定义为在3名患者中的2名或6名患者中的2名经历剂量限制性毒性所处的剂量之前的剂量。根据由national cancer institute(nci)common terminology for adverse events(ctcae)版本3.0(2006年8月9日)设置的定义和标准来确定剂量限制性毒性。
[1273]
ii期:以在i期确定的mtd,患者接受与i期相同的曲妥珠单抗连接的tlr激动剂衍生物。在没有疾病进展或不可接受的毒性的情况下,每4周重复治疗,持续2
‑
6个疗程。在完成2个疗程的研究治疗之后,实现完全或部分应答的患者可以再接受4个疗程。在完成6个疗程的研究治疗之后,保持病情稳定超过2个月的患者,只要他们符合最初的资格标准,在疾病进展时可再接受6个疗程。
[1274]
血液采样在曲妥珠单抗连接的tlr激动剂衍生物施用之前和之后,通过直接静脉穿刺进行一系列的抽血。在给药前约10分钟和在给药后大约以下时间,获取用于测定血清浓度的静脉血样品(5ml):第1天、第8天和第15天。将每个血清样品分成两个等分试样。将所有血清样品都储存在
‑
20℃下。在干冰上运送血清样品。
[1275]
药代动力学:患者在开始治疗前以及在第1天、第8天和第15天接受血浆/血清样本采集,以进行药代动力学评价。在digital equipment corporation vax 8600计算机系统上,使用最新版本的bioavl软件,通过模型独立方法计算出药代动力学参数。确定以下药代动力学参数:峰值血清浓度(c
max
);达到峰值血清浓度的时间(t
max
);使用线性梯形法则计算的从时间零到最后一次采血时间(auc0‑
72
)的浓度
‑
时间曲线下面积(auc);和根据消除速率常数计算出的末端消除半衰期(t
1/2
)。通过对数
‑
线性浓度
‑
时间图的末端线性区中的连续数据点的线性回归来估计消除速率常数。计算每种治疗的药物代谢动力学参数的平均值、标准偏差(sd)和变异系数(cv)。计算参数平均值的比率(防腐制剂/非防腐制剂)。
[1276]
对联合治疗的患者响应:通过用x射线、ct扫描和mri成像来评估患者响应,并且在开始研究之前以及在第一周期结束时进行成像,其中每四周或在后续周期结束时另外进行成像。成像模态基于癌症类型和可行性/可用性加以选择,并且对于类似癌症类型以及在每个患者的整个研究过程中利用同一成像模态。使用recist标准确定响应率。(therasse等人,j.natl.cancer inst.2000年2月2日;92(3):205
‑
16;http://ctep.cancer.gov/forms/therasserecistjnci.pdf)。患者还经历癌症/肿瘤活检,以通过流式细胞术、western印迹和ihc评估祖癌细胞表型和克隆生长的变化,并通过fish评估细胞遗传学的变化。在完成研究治疗之后,定期随访患者持续4周。
[1277]
实施例12:乳腺癌的治疗
[1278]
曲妥珠单抗连接的tlr激动剂衍生物用于乳腺癌治疗的安全性和疗效的人体临床试验
[1279]
目的:比较在疾病进展时单独的曲妥珠单抗连接的tlr激动剂衍生物随后曲妥珠单抗和紫杉醇联合与曲妥珠单抗和紫杉醇一线联合在患有her2过表达转移性乳腺癌女性中的疗效和毒性。
[1280]
研究设计:本研究是一项随机、多中心研究。根据her2/neu过表达程度(2 与3 )、既往含蒽环类药物的辅助治疗(无既往治疗与没有左胸壁放射治疗的既往治疗与具有左胸壁放射治疗的既往治疗)、雌激素受体状态(阳性与阴性与未知)、既往治疗(一线与二线/三线)和中心,对患者进行分层。将患者随机分配到两个治疗组中的一个。组i:患者每周经30
‑
90分钟iv接受曲妥珠单抗连接的tlr激动剂衍生物。在疾病进展时,如同组ii,患者iv接受曲妥珠单抗连接的tlr激动剂衍生物和iv紫杉醇的联合。组ii:患者每周经30
‑
90分钟iv接受曲妥珠单抗连接的tlr激动剂衍生物。每周经1小时iv施用紫杉醇,持续3周,然后休息1周。
[1281]
在没有疾病进展或不可接受的毒性的情况下,继续在两个组中进行治疗。评估在基线和疗程2、3、4、5、6、8、10和12的第1天的生活质量。在第1、第3和第6个月进行患者随访,然后,此后每6个月随访。
[1282]
实施例13:膀胱癌的治疗
[1283]
目的:确定紫杉醇和放射治疗在有或没有本文所述的tlr激动剂衍生物的情况下在已经历针对肌肉浸润性膀胱移行细胞癌的既往的经尿道膀胱切除术的患者中的急性毒性。
[1284]
疾病特征:组织学或细胞学证实为膀胱原发性移行细胞癌(tcc);固有肌层入侵的组织学证据;满足以下分期标准的1个:t2
‑
4a期;nx、n0或n1;和m0疾病或临床t1期、3/3级疾病,并且,需要明确的局部治疗;前列腺尿道的肿瘤累及是允许的,前提是满足以下标准,在视觉上肿瘤被完全切除;无前列腺基质入侵的证据,通过胸部x射线或ct扫描和腹部/盆腔ct扫描,未见远处转移的证据;在过去3
‑
8周内经历了经尿道膀胱切除术(尽可能如判断为安全那样彻底),包括使用肿瘤映射的双手检查;可用于her2/neu分析的足够的肿瘤组织;不是根治性膀胱切除术的候选者。
[1285]
研究设计:本研究是一项非随机、多中心研究。根据her2/neu状态(her2/neu 2 或3 染色[第1组]与her2/neu 0或1 染色[第2组]),将患者分配到2个治疗组中的1个。
[1286]
组1:患者在第1天、第8天、第15天、第22天、第29天、第36天和第43天经1个小时iv接受紫杉醇,并且在第1天经90分钟然后在第8天、第15天、第22天、第29天、第36天和第43天经30分钟iv接受本文所述的曲妥珠单抗连接的tlr激动剂衍生物。患者还在第1
‑
5天、第8
‑
12天、第15
‑
19天、第22
‑
26天、第29
‑
33天、第36
‑
40天、第43
‑
47天和第50天经历放射治疗,每天一次。在没有疾病进展或不可接受的毒性的情况下,继续进行治疗。
[1287]
组2:与组1一样,患者接受紫杉醇并且经历放射治疗。在研究治疗完成之后,在第4
‑
5周进行患者随访,每3个月随访持续1年,每4个月随访持续1年,每6个月随访持续3年,然后,此后每年随访。
[1288]
实施例14:卵巢癌的治疗
[1289]
本文描述的曲妥珠单抗连接的tlr激动剂衍生物用于卵巢癌治疗的安全性和疗效
的人体临床试验
[1290]
目的:评价持续四周的每周一次iv剂量的包含本文所述的曲妥珠单抗连接的tlr激动剂衍生物的组合物在患有her2过表达卵巢癌的女性中的安全性和疗效。
[1291]
研究设计:本研究是一项非随机、开放标签、11周、多中心研究。本研究将评价持续四周的每周一次iv剂量的曲妥珠单抗连接的tlr激动剂衍生物的安全性概况、mtd、pk和免疫原性。将患者分配到单个组。患者每周一次接受一个剂量的曲妥珠单抗连接的tlr激动剂衍生物,持续4周。将在研究的第1天、第8天、第15天和第22天,通过iv输注施用曲妥珠单抗连接的tlr激动剂衍生物。将在第1天和第22天取尿样。
[1292]
血液采样在曲妥珠单抗连接的tlr激动剂衍生物施用之前和之后,通过直接静脉穿刺进行一系列的抽血。在给药前约10分钟和在给药后大约以下时间,获取用于测定血清浓度的静脉血样品(5ml):第1天、第2天、第4天、第5天、第8天、第15天、第22天、第36天、第43天和第50天。将每个血清样品分成两个等分试样。将所有血清样品都储存在
‑
20℃下。在干冰上运送血清样品。
[1293]
在没有疾病进展或不可接受的毒性的情况下,继续进行治疗。评估在基线和疗程2、3、4、5、6、8、10和12的第1天的生活质量。在第29天、第36天、第43天和第50天对患者进行随访。患者将被问及不良事件。患者将接受成像扫描和ecg,以评价肿瘤大小和心脏功能(第43天)。在研究结束时,患者将接受体格检查第50天)。具有疾病消退证据的患者可以接受继续治疗,直到记录到疾病进展的证据时为止。
[1294]
通过以下编号的实施方案进一步描述本发明。
[1295]
1.一种组合物,其包含具有掺入其中的一个或多个非天然编码氨基酸的一种或多种肿瘤靶向多肽,其中所述多肽通过共价键合到所述肿瘤靶向多肽的所述非天然氨基酸上的接头与tlr激动剂分子连接。
[1296]
2.根据实施方案1所述的组合物,其中所述肿瘤靶向多肽是与her2结合的抗体。
[1297]
3.根据实施方案1所述的组合物,其中所述肿瘤靶向多肽是曲妥珠单抗。
[1298]
4.根据实施方案1、2或3所述的组合物,其中所述tlr激动剂是tlr7或tlr8激动剂。
[1299]
5.根据实施方案1、2或3所述的组合物,其中所述tlr激动剂是图4的tlr7或tlr8激动剂。
[1300]
6.根据实施方案1所述的组合物,其中所述肿瘤靶向多肽与一种或多种水溶性聚合物缀合。
[1301]
7.根据实施方案6所述的组合物,其中所述至少一种水溶性聚合物与至少一个非天然编码氨基酸连接。
[1302]
8.根据实施方案7所述的组合物,其中所述水溶性聚合物是peg。
[1303]
9.根据实施方案1所述的组合物,其中所述接头是具有介于10至50之间的分子量的peg。
[1304]
10.根据实施方案1所述的组合物,其中所述组合物包含一个或多个增加所述组合物的稳定性或溶解度的氨基酸取代、添加或缺失。
[1305]
13.根据实施方案1所述的组合物,其中所述组合物包含一个或多个增加所述肿瘤靶向多肽在重组宿主细胞中的表达或体外合成的氨基酸取代、添加或缺失。
[1306]
14.根据实施方案1所述的组合物,其中所述非天然编码氨基酸对接头、聚合物或
生物活性分子具有反应性,而所述接头、聚合物或生物活性分子对多肽中20种常见氨基酸中的任何一种都不具有反应性。
[1307]
15.根据实施方案14所述的组合物,其中所述非天然编码氨基酸包含羰基、氨基氧基、肼基、酰肼基、氨基脲基、叠氮基或炔基。
[1308]
16.根据实施方案14所述的组合物,其中所述非天然编码氨基酸包含羰基。
[1309]
17.根据实施方案1所述的组合物,其中所述肿瘤靶向多肽与生物活性分子、细胞毒性剂、水溶性聚合物或免疫刺激剂连接。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。