1.本发明涉及生物酶仿生化学和新能源材料领域,具体是含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物的制备以及在催化制氢中的应用。
背景技术:
2.目前,氢气因其高燃烧热和环保型被认为是一种很有前途的替代能源载体。但当今工业化制氢主要是通过高成本、高能耗的贵金属铂催化剂电解水来实现的。基于此,人们致力于开发一种廉价耐用的质子还原催化剂,作为贵金属铂的替代品,可将水分解成h2。在自然界中铁铁氢化酶在各种微生物中催化可逆质子还原为h2,它具有独特的催化活性中心,是迄今为止将质子催化还原成氢气且催化效率最高的生物酶,故以期待其可替代贵金属铂的催化剂,可以缓解日益严峻的能源匮乏和环境污染等。因此科研工作者对天然铁铁氢化酶催化活性中心的基本结构和催化功能进行了广泛的生物酶仿生化学研究,并将不同配体引入到全羰基二铁二硫化合物中,合成了大量不同化合物。其中膦配体取代是一类重要的化合物,这是由于膦配体具有强的供电子能力,非常类似于天然铁铁氢化酶中氰根配体(cn
‑
)的电子结构和配位能力。
3.通过研究二铁二硫化合物中二铁核的不对称性结构有助于在铁铁氢化酶的二铁亚基中形成所需的“旋转”几何构型,以促进h2的快速生成。现已知有利于构造二铁核的非对称性结构主要通过用一些具有刚性主链或小咬合角的特殊双膦配体通过me3no诱导或紫外线照射取代二铁二硫六羰基化合物,来设计合成双膦配体取代的二铁二硫化合物。但到目前为止,还没有人注意到在膦配体取代的二铁二硫化合物中通过引入大位阻富电子性的双苯基取代二硫桥,作为一种方法来更有利于构造二铁核的非对称性结构,以期模拟高效催化制氢的天然酶中二铁亚基中所具有的“旋转”几何构型。
技术实现要素:
4.本发明针对现有的二铁二硫化合物催化制氢能力低下的问题,提供了含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物的制备以及在催化制氢中的应用。本发明首次在二硫桥引入大位阻富电子性的基团(苯基)得到新型二铁六羰基化合物,并进行双膦配体取代得到新型二铁二膦化合物。该类二铁二膦化合物结构中既包含良好供电子性的双膦配体又具有大位阻富电子性的双苯基取代二硫桥。利用电化学循环伏安法研究了它们电催化还原三氟乙酸质子生成氢气的能力,并用控制电解电位方法研究其催化产氢效率。
5.本发明是通过以下技术方案实现的:含双苯基取代二硫桥的二铁六羰基化合物,所述化合物的化学式为fe2{(μ
‑
schph)2o}(co)6,其结构式如下所示:
[0006][0007]
本发明还提供了含双苯基取代二硫桥的二铁二膦化合物,所述化合物的化学式为fe2{(μ
‑
schph)2o}(co)4{κ2‑
p^p},其中双膦配体p^p分别为:dppp[(ph2pch2)2ch2]、dppe[(ph2pch2)2]、pcncp[(ph2pch2)2n(ch2ph)];其结构式如下所示:
[0008][0009]
本发明进一步提供了所述含双苯基取代二硫桥的二铁六羰基化合物的合成方法,包括以下步骤:
[0010]
(1)在氮气气氛下,将起始原料fe2(μ
‑
sh)2(co)6溶解于四氢呋喃,得到混合液;
[0011]
(2)混合液置于
‑
78℃的低温浴中,加入苯甲醛,随后反应体系自然升温至室温,反应12h;
[0012]
(3)用冷阱抽除四氢呋喃溶剂,加入二氯甲烷,在冰水浴状态下,滴加浓h2so4,升温至室温反应9h;
[0013]
(4)减压除去反应二氯甲烷溶剂得到粗产物,用丙酮提取残余物,展开剂或洗脱剂进行制备型薄层层析或者柱层析色谱分离,得到含双苯基取代二硫桥的二铁六羰基化合物。
[0014]
作为含双苯基取代二硫桥的二铁六羰基化合物的合成方法技术方案的进一步改进,在步骤(1)、(2)、(3)中的fe2(μ
‑
sh)2(co)6、四氢呋喃、苯甲醛、二氯甲烷、浓硫酸的用量比为2mmol:40ml:1.62ml(16mmol):40ml:2.17ml(40mmol)。
[0015]
作为含双苯基取代二硫桥的二铁六羰基化合物的合成方法技术方案的进一步改进,所述展开剂或洗脱剂为体积比为10:1的石油醚和二氯甲烷的混合溶剂。
[0016]
本发明进一步提供了所述含双苯基取代二硫桥的二铁二膦化合物的合成方法,包括以下步骤:
[0017]
(5)所述含双苯基取代二硫桥的二铁六羰基化合物、me3no
·
2h2o和双膦配体p^p混合,然后注入乙腈溶液,磁力搅拌得到混合溶液,反应1h20min;
[0018]
(6)减压除去反应乙腈溶剂得到粗产物,用二氯甲烷提取残余物,展开剂或洗脱剂进行制备型薄层层析或者柱层析色谱分离,并用二氯甲烷和正己烷的混合溶剂进行重结晶,得到含双苯基取代二硫桥的二铁二膦化合物。
[0019]
本发明进一步提供含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物的合成路线:
[0020][0021]
作为含双苯基取代二硫桥的二铁二膦化合物的合成方法的进一步改进,在步骤(5)中的含双苯基取代二硫桥的二铁六羰基化合物、me3no
·
2h2o、双膦配体p^p、乙腈的用量比为0.20mmol:0.24mmol:0.24mmol:20ml。
[0022]
为含双苯基取代二硫桥的二铁二膦化合物的合成方法的进一步改进,所述展开剂或洗脱剂为体积比为5:1的石油醚和二氯甲烷的混合溶剂。
[0023]
为含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物的合成方法的进一步改进,所述制备型薄层层析为硅胶g薄层层析,柱层析为200
‑
300目硅胶柱层析。
[0024]
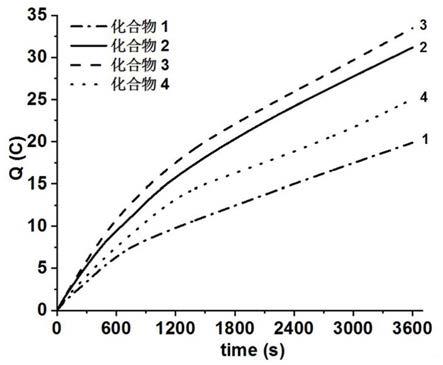
本发明进一步提供了含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物在电催化质子酸还原成氢气中的催化制氢应用。
[0025]
上述含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物的产氢能力及催化性能测试分别采用经典的电化学循环伏安法及控制电位电解库伦法,测试均为chi 660e电化学仪。循环伏安法:采用3mm直径的玻碳电极为工作电极、铂丝为对电极、非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的经典三电极体系在柱形槽中和氮气气氛下测定;在每次测试前都要用0.05μm的三氧化二铝粉末打磨玻碳电极,然后在水中用超声波清洗,最后丙酮冲洗,冷风吹干;测试体系的溶剂为色谱纯的乙腈、样品浓度为1mmol/l,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,质子酸为0、2、4、6、8、10mmol/l三氟乙酸;测试所得电位均为经二茂铁校正的还原电位。控制电位电解库伦法:采用15
×
20
×
1.0mm的玻碳片为工作电极、6mm直径的石墨棒为对电极、非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的三电极体系在柱形槽中和氮气气氛下测定:测试体系的溶剂为色谱纯的乙腈、样品浓度为0.3mmol/l并加入12mmol/l的三氟乙酸,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,测试电位均为
‑
2.3v versus fc/fc 。
[0026]
所述产氢能力表现为:在含有1mmol/l含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物的电化学测试体系中,随着质子酸浓度的不断增加,原始的还原峰逐渐消失,并出现两个新的还原峰,随着三氟乙酸逐渐加入其峰电流持续升高,相应的峰电位有轻微负移,这种现象正是均相催化析氢的显著特征,表明其具有电催化质子还原成氢气的能力。与此同时,利用控制电位电解库伦法的测试数据,通过计算公式ton=c/f
×
n1×
n2(c为通过电荷量,f为法拉第常数,n1为生成1mol h2所需的电子数,n2为催化剂物质的量)计算二铁金属化合物的ton,以此来表明它们的催化制氢效率。
[0027]
本发明所述含双苯基取代二硫桥的二铁六羰基化合物及含双苯基取代二硫桥的二铁二膦化合物,相对于现有技术,具有如下有益效果:
[0028]
(1)本发明所制备的含双苯基取代二硫桥的二铁六羰基化合物首次实现了在二硫桥引入大位阻、富电子性的双苯基基团,所制备的含双苯基取代二硫桥的二铁二膦化合物不仅在二铁核引入供电子性的螯合双膦配体,而且可通过取代二硫桥与螯合双膦配体二者间大的空间位阻和各自强的供电子性,构造二铁核的非对称性结构,以期模拟高效催化制
氢的天然酶中二铁亚基中所具有的“旋转”几何构型,进而更好地调节它们的产氢能力及催化活性。
[0029]
(2)本发明所述的制备方法简单简便、原料廉价易得、反应条件温和且易控、分离手段多样,产物收率适中,可适合于制备多种含其他有机基团取代二硫桥的二铁金属化合物。
附图说明
[0030]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0031]
图1为本发明实施例1所述化合物1傅里叶变换红外谱图。
[0032]
图2为本发明实施例2
‑
4所述化合物2
‑
4傅里叶变换红外谱图。
[0033]
图3为本发明实施例1所述化合物1的核磁共振氢谱图。
[0034]
图4为本发明实施例2
‑
4所述化合物2
‑
4的核磁共振氢谱图。
[0035]
图5为本发明实施例2
‑
4所述化合物2
‑
4的核磁共振膦谱图。
[0036]
图6为本发明实施例1
‑
4的化合物1
‑
4(1mm)在0.1m n
‑
bu4npf6/mecn溶液中cf3co2h(0,2,4,6,8,10mm)后的循环伏安曲线图,扫速50mvs
‑1。
[0037]
图7为本发明实施例1
‑
4的化合物1
‑
4(0.3mm)在0.1m n
‑
bu4npf6/mecn溶液中加入cf3co2h(12mm)电解所通过电量与时间关系图。
具体实施方式
[0038]
下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0039]
一般地,本发明的化合物可以通过本发明所描述的方法制备得到。下面的反应方案和实施例用于进一步举例说明本发明的内容。
[0040]
所属领域的专业人员将认识到:本发明所描述的化学反应可以用来合适地制备本发明的其他化合物,且用于制备本发明的化合物的其它方法都被认为是在本发明的范围之内。例如,根据本发明那些非例证的化合物的合成可以成功地被所属领域的技术人员通过修饰方法完成,如通过利用其他已知的试剂除了本发明所描述的,或将反应条件做一些常规的修改。另外,本发明所公开的反应或已知的反应条件也公认地适用于本发明其他同类型化合物的制备。
[0041]
所属领域的专业技术人员也应认识到:本发明实施例1
‑
4所证明的个别化合物的特点(化合物1
‑
4所采用的催化性能测试方法、测试结果),其他本发明那些非例证的化合物也同样具备,同样在催化质子酸还原成氢气方面有显著的催化作用。本发明的化合物(包含例证以及非例证的化合物)只是对实施例中化合物作变化或替换,不会对它们在催化质子酸还原成氢气方面的效果明显有不利的影响。
[0042]
在本发明所列实施例中,所使用的化学原料(即下表所列的化学原料)均以克为计
量单位。
[0043][0044][0045]
实施例1
[0046]
含双苯基取代二硫桥的二铁六羰基化合物(化合物1)的制备方法,其化学式为fe2{(μ
‑
schph)2o}(co)6,其制备过程如下所示:
[0047][0048]
其具体制备步骤如下:
[0049]
将2mmol fe2(μ
‑
sh)2(co)6加入到带有搅拌磁子的schlenk瓶中,抽换氮气3次后,注入40ml的四氢呋喃溶液并搅拌得到红色溶液。置于液氮
‑
丙酮浴中降温至
‑
78℃,缓慢加入1.62ml(16mmol)苯甲醛,混合液颜色加深,撤丙酮浴,自然升至室温反应12h。用冷阱抽除四氢呋喃溶剂,加入40ml二氯甲烷,在冰水浴状态下,缓慢滴加2.17ml(40mmol)浓h2so4,缓慢升温至室温反应9h,溶液变为黑红色;停止反应,旋蒸减压蒸馏脱去二氯甲烷溶剂,用丙酮提取残余物,用展开剂为石油醚:二氯甲烷(v:v=10:1)进行制备型薄层析色谱分离,收集橘红色主色带(r
f
=0.5),得到深红色泡沫状固体即为化合物1(0.234g,产率:21.7%)。
[0050]
化合物1的结构表征数据如下:ft
‑
ir(kbr disk)v
c≡o
/cm
‑1:2077(vs),2037(vs),2000(vs);1h
‑
nmr(600mhz,cdcl3,tms)δ
h
/ppm:7.34(s,10h,2xchph),5.11(s,1h,sch
e
),4.60(s,1h,sch
a
)。
[0051]
结合以上数据并由图1可知,化合物1的红外光谱在2077、2037、2000cm
‑1三处显示了二铁二硫骨架中铁羰基的特征伸缩振动吸收峰。尤其是,第一个红外吸收峰位于2077cm
‑1处,则表明了其是二铁二硫六羰基化合物。
[0052]
化合物1的电化学循环伏安法实验:
[0053]
该实验在chi 660e电化学仪上利用以3mm直径的玻碳电极为工作电极、以铂丝为对电极、以非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的经典三电极体系在柱形槽中和氮气气氛下测定;在每次测试前都要用0.05μm的三氧化二铝粉末打磨玻碳
电极,然后在水中用超声波清洗,最后丙酮冲洗,冷风吹干;测试体系的溶剂为色谱纯的乙腈、样品浓度为1mmol/l,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,质子酸为0、2、4、6、8、10mmol/l三氟乙酸;测试所得电位均为经二茂铁校正的还原电位。
[0054]
由图6可知,在含有1mmol/l化合物1的电化学测试体系中,随着浓度为0、2、4、6、8、10mmol/l三氟乙酸的逐渐加入,还原电位e
p
为
‑
1.65v的峰电流随着三氟乙酸逐渐加入其峰电流持续升高,相应的峰电位有轻微负移,这种现象正是均相催化析氢的显著特征。
[0055]
化合物1的电化学控制电位电解库伦法:
[0056]
采用15
×
20
×
1.0mm的玻碳片为工作电极、6mm直径的石墨棒为对电极、非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的三电极体系在柱形槽中和氮气气氛下测定:测试体系的溶剂为色谱纯的乙腈、样品浓度为0.3mmol/l并加入12mmol/l的三氟乙酸,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,电解电位为
‑
2.30v进行电解1h。
[0057]
由图7可知,在30ml的乙腈溶液中含有0.3mmol/l化合物1加入12mmol/l的三氟乙酸的电化学测试体系中q=19.88c,根据析氢反应ton的计算公式ton=c/f
×
n1×
n2可得其理论值为11.45。它可作为含双苯基取代二硫桥的二铁二膦化合物2
‑
4的参比值观察是否提高其催化产氢的效率。
[0058]
实施例2
[0059]
含双苯基取代二硫桥的二铁二膦化合物2的制备方法,其化学式为fe2{(μ
‑
schph)2o}(co)4{κ2‑
(pph2ch2)2ch2},其制备过程如下所示:
[0060][0061]
其具体制备步骤如下:
[0062]
在氮气气氛下,将0.108g(0.20mmol)化合物1、0.099g(0.24mmol)dppp和0.027g(0.24mmol)me3no
·
2h2o的混合物加入到带有搅拌磁子的schlenk瓶中,注入20ml乙腈并搅拌溶解得到红色溶液,室温反应1h20min,橘红色溶液变成黑红色溶液,停止反应,旋蒸减压脱去乙腈溶剂,用二氯甲烷提取残余物,用洗脱剂为石油醚:二氯甲烷(v:v=5:1)进行硅胶闪层析色谱分离,收集黄绿色带,得到黄绿色固体即为化合物2(0.015g,产率:8.4%)。
[0063]
化合物2的结构表征数据如下:ft
‑
ir(kbr disk)v
c≡o
/cm
‑1:2025(vs),1976(s),1954(vs),1884(vs);1h
‑
nmr(600mhz,cdcl3,tms)δ
h
/ppm:7.67(s,4h,2xschphh
‑
o),7.53(s,4h,2xschphh
‑
m),7.45(t,j=7.2hz,2h,2xschphh
‑
p),7.35
‑
7.25(m,8h,4xpphh
‑
o),7.22
‑
7.11(m,8h,4xpphh
‑
m),6.95(s,4h,4xpphh
‑
p),4.31(s,2h,2xsch),2.75
‑
2.62(m,4h,2xpch2),2.42(m,2h,
‑
ch2‑
);
31
p
‑
nmr(243mhz,cdcl3,85%h3po4)δ
p
/ppm:51.21(br s,apical
‑
basal isomer,89%),47.45(d,j=34.02hz,basal
‑
basal isomer,11%)。
[0064]
结合以上数据并由图2可知,化合物2的红外光谱在2025、1976、1954、1884cm
‑1四处显示了二铁二硫骨架中铁羰基的特征伸缩振动吸收峰,并且它的第一个红外吸收峰位于2025cm
‑1处表明了双膦螯合取代二铁二硫化合物2的成功生成。进一步地,由图5可知,化合
物2的核磁共振膦谱在51.21和47.45ppm两处分别给出了一个宽单峰和双峰膦信号,表明了化合物2有两种异构体存在,即该分子中双膦配体中两个膦原子以apical
‑
basal和basal
‑
basal两种几何构型螯合配位于同一铁原子。
[0065]
化合物2的电化学循环伏安法实验:
[0066]
该实验在chi 660e电化学仪上利用以3mm直径的玻碳电极为工作电极、以铂丝为对电极、以非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的经典三电极体系在柱形槽中和氮气气氛下测定;在每次测试前都要用0.05μm的三氧化二铝粉末打磨玻碳电极,然后在水中用超声波清洗,最后丙酮冲洗,冷风吹干;测试体系的溶剂为色谱纯的乙腈、样品浓度为1mmol/l,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,质子酸为0、2、4、6、8、10mmol/l三氟乙酸;测试所得电位均为经二茂铁校正的还原电位
[0067]
由图6可知,在含有1mmol/l化合物2的电化学测试体系中,随着浓度为0、2、4、6、8、10mmol/l三氟乙酸的逐渐加入,还原电位e
p
为
‑
2.09v的峰电流逐渐消失。加入2mmol/l的三氟乙酸时,在
‑
1.71v和
‑
1.86v处出现两个新的还原峰,并随着三氟乙酸逐渐加入其峰电流持续升高,相应的峰电位有轻微负移,这种现象正是均相催化析氢的显著特征,表明其具有电催化质子还原成氢气的能力。
[0068]
化合物2的电化学控制电位电解库伦法:
[0069]
采用15
×
20
×
1.0mm的玻碳片为工作电极、6mm直径的石墨棒为对电极、非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的三电极体系在柱形槽中和氮气气氛下测定:测试体系的溶剂为色谱纯的乙腈、样品浓度为0.3mmol/l并加入12mmol/l的三氟乙酸,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,电解电位为
‑
2.30v进行电解1h。
[0070]
由图7可知,在30ml的乙腈溶液中含有0.3mmol/l化合物2加入12mmol/l的三氟乙酸的电化学测试体系中q=31.18c,根据析氢反应ton的计算公式ton=c/f
×
n1×
n2可得其理论值为17.95。
[0071]
实施例3
[0072]
含双苯基取代二硫桥的二铁二膦化合物3的制备方法,其化学式为fe2{(μ
‑
schph)2o}(co)4{κ2‑
(pph2ch2)2},其制备过程如下所示:
[0073][0074]
其具体制备步骤如下:
[0075]
在氮气气氛下,将0.108g(0.20mmol)化合物1、0.096g(0.24mmol)dppe和0.027g(0.24mmol)me3no
·
2h2o的混合物加入到带有搅拌磁子的schlenk瓶中,注入20ml乙腈并搅拌溶解得到红色溶液,室温反应1h20min,橘红色溶液变成黑红色溶液,停止反应,旋蒸减压脱去乙腈溶剂,用二氯甲烷提取残余物,用洗脱剂为石油醚:二氯甲烷(v:v=5:1)进行硅胶闪层析色谱分离,收集黄绿色带,得到黄绿色固体即为化合物3(0.017g,产率:9.8%)。
[0076]
化合物3的结构表征数据如下:ft
‑
ir(kbr disk)v
c≡o
/cm
‑1:2020(vs),1948(vs),
1905(vs);1h
‑
nmr(600mhz,d6‑
acetone,tms)δ
h
/ppm:8.16
‑
7.92(m,4h,2xschphh
‑
o),7.70(t,1h,j=7.2hz,schphh
‑
p),7.59
‑
7.62(m,1h,schphh
‑
p),7.55(s,4h,2xschphh
‑
m),7.52
‑
7.51(m,1h,pphh
‑
p),7.44(t,4h,j=7.8hz,2xpphh
‑
o),7.40(t,4h,j=7.8hz,2xpphh
‑
o),7.33(t,2h,j=7.2hz,2xpphh
‑
p),7.28(t,4h,j=7.8hz,2xpphh
‑
m),7.23(d,4h,j=8.4hz,2xpphh
‑
m),7.20
‑
7.19(m,1h,pphh
‑
p),4.03(s,2h,2xsch),3.39(s,2h,pch2),2.94(s,2h,ppch2);
31
p
‑
nmr(243mhz,d6
‑
acetone,85%h3po4)δ
p
/ppm:91.00(s,apical
‑
basal isomer,95%),74.77(s,basal
‑
basal isomer,5%)。
[0077]
结合以上数据并由图2可知,化合物3的红外光谱在2020、1948、1905cm
‑1三处显示了二铁二硫骨架中铁羰基的特征伸缩振动吸收峰,并且它的第一个红外吸收峰位于2020cm
‑1处表明了双膦螯合取代二铁二硫化合物3的成功生成。进一步地,由图5可知,化合物3的核磁共振膦谱在91.00和74.77ppm两处分别给出了两个单峰膦信号,表明了化合物3有两种异构体存在,即该分子中双膦配体中两个膦原子以apical
‑
basal和basal
‑
basal两种几何构型螯合配位于同一铁原子。
[0078]
化合物3的电化学循环伏安法实验:
[0079]
该实验在chi 660e电化学仪上利用以3mm直径的玻碳电极为工作电极、以铂丝为对电极、以非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的经典三电极体系在柱形槽中和氮气气氛下测定;在每次测试前都要用0.05μm的三氧化二铝粉末打磨玻碳电极,然后在水中用超声波清洗,最后丙酮冲洗,冷风吹干;测试体系的溶剂为色谱纯的乙腈、样品浓度为1mmol/l,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,质子酸为0、2、4、6、8、10mmol/l三氟乙酸;测试所得电位均为经二茂铁校正的还原电位。
[0080]
由图6可知,在含有1mmol/l化合物3的电化学测试体系中,随着浓度为0、2、4、6、8、10mmol/l三氟乙酸的逐渐加入,还原电位e
p
为
‑
2.07v的峰电流逐渐消失。加入2mmol/l的三氟乙酸时,在
‑
1.75v和
‑
1.93v处出现两个新的还原峰,并随着三氟乙酸逐渐加入其峰电流持续升高,相应的峰电位有轻微负移,这种现象正是均相催化析氢的显著特征,表明其具有电催化质子还原成氢气的能力。
[0081]
化合物3的电化学控制电位电解库伦法:
[0082]
采用15
×
20
×
1.0mm的玻碳片为工作电极、6mm直径的石墨棒为对电极、非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的三电极体系在柱形槽中和氮气气氛下测定:测试体系的溶剂为色谱纯的乙腈、样品浓度为0.3mmol/l并加入12mmol/l的三氟乙酸,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,电解电位为
‑
2.30v进行电解1h。
[0083]
由图7可知,在38ml的乙腈溶液中含有0.3mmol/l化合物3加入12mmol/l的三氟乙酸的电化学测试体系中q=33.49c,根据析氢反应ton的计算公式ton=c/f
×
n1×
n2可得其理论值为15.22。
[0084]
实施例4
[0085]
含双苯基取代二硫桥的二铁二膦化合物4的制备方法,其化学式为fe2{(μ
‑
schph)2o}(co)4{κ2‑
(pph2ch2)2n(ch2ph)},其制备过程如下所示:
[0086][0087]
其具体制备步骤如下:
[0088]
在氮气气氛下,将0.108g(0.20mmol)化合物1、0.121g(0.24mmol)pcncp和0.027g(0.24mmol)me3no
·
2h2o的混合物加入到带有搅拌磁子的schlenk瓶中,注入20ml乙腈并搅拌溶解得到红色溶液,室温反应1h20min,橘红色溶液变成黑红色溶液,停止反应,旋蒸减压脱去乙腈溶剂,用二氯甲烷提取残余物,用洗脱剂为石油醚:二氯甲烷(v:v=5:1)进行硅胶闪层析色谱分离,收集黄绿色带,得到黄绿色固体即为化合物4(0.024g,产率:12.2%)。
[0089]
化合物4的结构表征数据如下:ft
‑
ir(kbr disk)v
c≡o
/cm
‑1:2022(vs),1963(vs),1942(vs),1894(vs);1h
‑
nmr(600mhz,cdcl3,tms)δ
h
/ppm:7.72(s,4h,2xschphh
‑
o),7.38(m,4h,2xschphh
‑
m),7.44
‑
7.37(m,8h,4xpphh
‑
o),7.22
‑
7.19(m,8h,4xpphh
‑
m),7.17
‑
7.14(m,5h,4xpphh
‑
p and ch2phh
‑
p),6.93(d,j=7.2hz,4h,ch2phh
‑
o,m),6.88(d,j=7.2hz,2h,2xschphh
‑
p),4.36(s,2h,2xsch),3.87
‑
3.65(m,4h,2xpch2),3.3(s,2h,nch2);
31
p
‑
nmr(243mhz,cdcl3,85%h3po4)δ
p
/ppm:52.04(s,apical
‑
basal isomer,95%),46.22(s,basal
‑
basal isomer,5%)。
[0090]
结合以上数据并由图2可知,化合物4的红外光谱在2022、1963、1942、1894cm
‑1四处显示了二铁二硫骨架中铁羰基的特征伸缩振动吸收峰,并且它的第一个红外吸收峰位于2022cm
‑1处表明了双膦螯合取代二铁二硫化合物4的成功生成。进一步地,由图5可知,化合物4的核磁共振膦谱在52.04和46.22ppm两处分别给出了两个单峰膦信号,表明了化合物4有两种异构体存在,即该分子中胺基双膦配体中两个膦原子以apical
‑
basal和basal
‑
basal两种几何构型螯合配位于同一铁原子。
[0091]
化合物4的电化学循环伏安法实验:
[0092]
该实验在chi 660e电化学仪上利用以3mm直径的玻碳电极为工作电极、以铂丝为对电极、以非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的经典三电极体系在柱形槽中和氮气气氛下测定;在每次测试前都要用0.05μm的三氧化二铝粉末打磨玻碳电极,然后在水中用超声波清洗,最后丙酮冲洗,冷风吹干;测试体系的溶剂为色谱纯的乙腈、样品浓度为1mmol/l,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,质子酸为0、2、4、6、8、10mmol/l三氟乙酸;测试所得电位均为经二茂铁校正的还原电位
[0093]
由图6可知,在含有1mmol/l化合物4的电化学测试体系中,随着浓度为0、2、4、6、8、10mmol/l三氟乙酸的逐渐加入,还原电位e
p
为
‑
2.08v和2.29v的峰电流逐渐消失。加入2mmol/l的三氟乙酸时,在
‑
1.98v和
‑
1.72v处出现两个新的还原峰,并随着三氟乙酸逐渐加入其峰电流持续升高,相应的峰电位有轻微负移,这种现象正是均相催化析氢的显著特征,表明其具有电催化质子还原成氢气的能力。
[0094]
化合物4的电化学控制电位电解库伦法:
[0095]
采用15
×
20
×
1.0mm的玻碳片为工作电极、6mm直径的石墨棒为对电极、非水ag/agno3(0.01m agno3/0.1m n
‑
bu4npf6/ch3cn)为参比电极的三电极体系在柱形槽中和氮气气
氛下测定:测试体系的溶剂为色谱纯的乙腈、样品浓度为0.3mmol/l并加入12mmol/l的三氟乙酸,支持电解质为用浓度为0.1mol/l的n
‑
bu4npf6,电解电位为
‑
2.30v进行电解1h。
[0096]
由图7可知,在30ml的乙腈溶液中含有0.3mmol/l化合物4加入12mmol/l的三氟乙酸的电化学测试体系中q=25.7c,根据析氢反应ton的计算公式ton=c/f
×
n1×
n2可得其理论值为14.44。
[0097]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。