1.本发明属于药物质量检测领域,具体是一种水柏枝蒙药材的质量检测方法。
背景技术:
2.水柏枝药材是藏药和蒙药的常用药材之一,具有较高药用价值,蒙药名为“水柏枝”,为柽柳科植物水柏枝的嫩枝,春、夏采收,剪取嫩枝晒干。被广泛用于藏药复方制剂中,如二十五味大汤丸、五味甘露药浴汤散、十九味草果散等。
3.目前水柏枝记载于蒙药典籍《认药白晶鉴》、《无误蒙药鉴》以及《中华本草》(蒙药卷),具清热、燥“协日乌素”、透疹、敛毒之功效,主治热毒、陈热、伏热、热症扩散、内毒症、“协日乌素”、血热、麻疹等症,味涩、甘,性凉,效钝、重固。
4.有关蒙药水柏枝化学成分的研究报道有:《中华本草》(蒙药卷)和《内蒙古植物药志》“polyphenols from myricaria alopecuroidess.iii.hydrolyzable tanning substances”(《khim.prir.soedin》1976年第1期)记载其枝中含鞣质,酸解产物中含去氢没食子酸及无水三没食子酸,说明其中含可水解鞣质;同属植物水柏枝中含有没食子酸及其衍生物。
5.《中华本草》(蒙药卷)、《中华人民共和国卫生部药品标准》(藏药分册)(1998年版)对药材仅有外观性状鉴别,无薄层鉴别,无检查项,更无含量测定。
6.现有技术“藏药水柏枝的质量标准研究”(《中国药房》杂志2017年第28卷第33期)鉴定10批水柏枝药材样品的原植物性状、显微特征,测定水分、总灰分和浸出物含量,采用薄层色谱法(tlc)和高效液相色谱法(hplc)进行定性、定量研究,但显微特征不全面并缺少茎横切面显微鉴别无法准确确定样本药材的基原,无酚酸类的理化鉴别确定药材中是否有酚酸类成分以及无重金属检查,高效液相色谱法采用等度洗脱,比梯度洗脱理论塔板数低。
7.现有技术“蒙药河柏中没食子酸的hplc含量测定”(《中药材》杂志2008年第31卷第2期)仅进行高效液相色谱法(hplc)测定没食子酸的含量,且药材的批次数量只有5批。
8.现代药理学研究表明,没食子酸及其衍生物具有多种药理活性,如抗炎、抗氧化、抗菌、抗病毒等多种生物学作用等,故没食子酸为水柏枝中的生物活性成分,其含量高低与蒙药水柏枝的功效具有一定的关系。因此对水柏枝进行全面的质量标准建立以监控水柏枝中的成分以及成分含量是有必要的。
9.近年来,我国中药材受到有害重金属元素污染时有报道,重金属超标一直是国际医药市场备受关注的热点。其中铅主要损害神经系统、造血系统、血管和消化系统;砷主要是扩张毛细血管,麻痹血管舒缩中枢,使腹腔脏器严重失血,引起肝、肾、心等实质器官的损害;镉对人有致畸、致癌、致突变作用;汞主要损害肾脏,造成肾功能衰竭;铜是人体必需的微量元素,然而过量的铜又会影响健康。然而,近年来对水柏枝重金属的检测几乎没有。准确限定重金属含量是保障人民用药安全有效和中药走向国际化的基础,因此本发明提供的一种水柏枝蒙药材的质量标准检测方法包括重金属检查并限定重金属含量。
10.综合来说,水柏枝多用在蒙药的复方制剂中,但成分复杂,目前还没有建立质量控
制的检测方法。所以,对水柏枝药材建立一个准确的、全面的、系统的标准对促进水柏枝的用药以及保障人体安全有重要的借鉴和指导意义。
技术实现要素:
11.本发明的目的是提供一种水柏枝蒙药材的质量标准检测方法,以解决现有技术存在的问题,提高水柏枝药品质量标准及安全有效性和质量可控性的水柏枝质量检测方法。
12.为了实现上述目的,本发明分别从性状、显微鉴别,理化鉴别,薄层色谱鉴别、含量测定、一般性检查(水分、总灰分、酸不溶灰分、浸出物、重金属)等多方面对水柏枝质量进行控制,为水柏枝的开发利用提供可靠的科学依据。
13.本发明的质量检测方法主要包括以下几部分内容,第一步性状鉴别、显微鉴别确定药材基原,第二步理化鉴别水柏枝药材中含酚酸类化合物成分,第三步薄层色谱定性鉴别没食子酸,第四步水柏枝中没食子酸含量测定,第五步水分、总灰分、酸不溶灰分、醇溶性浸出物测定以及重金属检查。
14.本发明的技术方案是一种水柏枝蒙药材的质量检测方法,由以下步骤组成:
15.(1)性状鉴别:对选取样本的外观形状、大小、颜色、表面特征、质地、断面特征、气味进行鉴定;性状特征符合以下特征的样本初步确认为是水柏枝蒙药材:
16.枝呈圆柱状,老枝灰褐色或红褐色,嫩枝红棕色或黄绿色,具有光泽和条纹;枝断面平坦,皮部所占比例较少,木部黄白色,髓部宽大为黄白色。叶窄条形互生小叶,密生于当年生绿色小枝上,多为卵形或狭长圆形,总状花序密集呈穗状,顶生于当年生枝条上。质脆,易折断。气微,味淡;见图1和图2。
17.(2)显微鉴别:包括粉末纤维鉴别和茎横切显微鉴别。
18.a.粉末显微鉴别:将步骤(1)所得样本制成粒径210~250μm的粉末,,挑取筛下粉末置于载玻片上,滴加甘油醋酸试液或水合氯醛试液,盖上盖玻片,加热透化,在显微镜下观察;符合以下特征的样本是水柏枝蒙药材:
19.表皮细胞呈不规则状,壁厚内陷,与细胞接触位置凸起;非腺毛,多为单细胞,内含不均匀分枝状腺,成丝状,壁稍厚,具有壁疣;导管多为具缘纹孔导管,宽铁链条状,螺纹状,少见网纹导管和螺纹导管;韧皮纤维,散在或成束,多碎断,不木化;薄壁细胞,不规则多边形,有时周围盾圆,壁稍厚鲜亮;木纤维成束存在;见图3。
20.b.茎横切显微鉴别:
21.a软化:选适当水柏枝药材材料放入水中浸泡4~5天左右,使材料变软,可用温水加快速度,需要每天换水。
22.b固定:将软化好的材料切成2~3mm的小段,浸泡于faa固定液(体积比为70%乙醇:冰醋酸:甲醛=18:1:1)12h以上。
23.c脱水:将药材从固定液中取出,用流动的自来水冲洗24h,从50%乙醇开始,经60%、70%、80%、95%直至纯酒精(无水乙醇),每次时间为1.5h,如不能及时进行各级脱水,材料可以放在70%乙醇中保存。
24.d透明:先经1/3二甲苯和2/3乙醇的混合液浸渍0.5h,再依次转入1/2二甲苯和1/2乙醇混合液、2/3二甲苯和1/3乙醇混合液中浸渍0.5h,最后转入纯二甲苯中浸渍0.5h。
25.e浸蜡:把组织材料块放在熔化的石蜡和二甲苯的等量混合液浸渍1.5小时,再先
后移入2个熔化的石蜡液中浸渍2h后过夜,浸蜡应在高于石蜡熔点3℃左右的温箱中进行。
26.f包埋、切片、贴片、烤片处理。
27.g染色:取已经干燥的切片,放于盛有二甲苯的染缸内脱蜡8min,脱完蜡后的切片可依次用纯乙醇和二甲苯的等量混合液、纯乙醇、95%乙醇、80%乙醇、70%乙醇洗5min,番红染液染色4h,水洗3s,依次用35%乙醇、50%乙醇、70%乙醇、80%乙醇、95%乙醇洗2min,固绿染色5s,染色后的切片需先后用2个纯乙醇洗30s,再用纯乙醇和二甲苯的等量混合液洗5min,纯二甲苯洗10min,洗去多余染料。
28.h封片:二甲苯透明后,迅速擦去材料周围多余液体,滴加适量(1~2滴)中性树胶,再将洁净盖玻片倾斜放下,以免出现气泡,封片后即制成永久性玻片标本,在显微镜下观察,符合以下显微特征确认为水柏枝蒙药材。
29.茎为类圆形,边缘呈波状凹凸。木栓层细胞数列,壁增厚,木化;皮层极狭小,为2~3列薄壁细胞。棱线处有下皮纤维束木质部,维管束14~16个,位于皮下纤维束下方,韧皮细胞细小排列整齐,木质部导管类圆,微木栓化;中央髓部宽广,薄壁细胞内含多数草酸钙柱晶;符合以上粉末显微和茎横切显微特征即确认样本为水柏枝蒙药材;见图4。
30.(3)理化检测:取步骤(1)所得样本制成粉末,加乙醇5~10滴与三氯化铁试液1滴,溶液颜色由黄绿色变为黑蓝色,证明样品中含有含酚酸类化合物成分,符合质量要求。
31.(4)没食子酸定性检测:
32.a.供试品溶液制备:取本品粉末过30目筛,准确称量0.5g药材,加入20ml水,加热回流提取4h,放冷至室温,过滤,即得。
33.b.对照品溶液制备:取没食子酸对照品适量,精密称定,加入甲醇制成质量浓度分别为2.230mg/ml的单一成分对照品贮备液,即得。
34.c.没食子酸薄层色谱定性鉴别:按照2020年版《中国药典》(四部)tlc法(通则0502)实验操作要求,分别吸取各单一成分对照品贮备液5μl和供试品溶液10μl,点于同一硅胶g薄层板上,在常温下饱和后展开,取出,晾干,检视,展开剂为氯仿:乙酸乙酯:甲酸(5:4:1,v/v/v),喷3%三氯化铁溶液显色后,105℃加热,日光灯下检视。供试品与各对照品在相应的位置上,显相同颜色的斑点,薄层展开斑点清晰,证明样品中含有没食子酸成分;见图5。
35.(5)水柏枝中没食子酸含量测定
36.a.色谱条件:采用十八烷基硅烷键合硅胶为填充剂,检测波长为210nm,柱温为30℃,进样体积5μl,流动相:以乙腈(a)-0.1%磷酸水溶液(b)为流动相,梯度洗脱(0~10min,10%
→
8%a),流速为1ml/min;在该色谱条件下,理论塔板数按没食子酸计算不低于6000,各峰与相邻峰的分离度均大于1.3,各成分测定不受其他组分的干扰。
37.b.对照品溶液制备:分别取没食子酸对照品各适量,精密称定,加入甲醇制成质量浓度分别为2.230mg/ml的单一成分对照品贮备液,取没食子酸对照品贮备液适量用甲醇逐步稀释至刻度,混匀,制备成6个系列对照品溶液,备用。
38.c.供试品溶液制备:取过30目筛水柏枝样品0.5g,精密称定,置于250ml圆底烧瓶中,加水20ml,加热回流提取4h,取出,放冷至室温,再次称质量,用水补足减失的质量,先后以滤纸、0.45μm微孔滤膜滤过,取续滤液,即得供试品溶液;结果见图6。
39.d.阴性对照溶液制备:取水20ml,置于250ml圆底烧瓶中,按制备供试品方法制备
阴性对照溶液。
40.e.测定:分别精密吸取对照品溶液、供试品溶液和阴性对照品溶液各5μl,注入液相色谱仪即得。
41.水柏枝中没食子酸定量分析方法学验证通过线性试验考察,y=17316x
‑
57127,相关系数r=0.9998,表明没食子酸在49.95~199.8μg
·
ml
‑
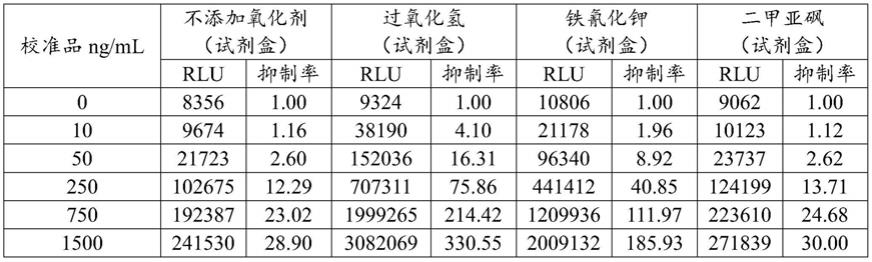
1范围内线性关系良好。通过仪器精密度试验,结果没食子酸峰面积的rsd(n=6)分别为1.55%,表明仪器精密度良好。通过重复性试验,结果样品中没食子酸的平均含量分别为4.37mg/g,rsd(n=6)分别为1.08%。表明本方法重复性良好。通过稳定性试验,计算没食子酸峰面积的rsd(n=6)分别为1.40%。表明供试品溶液室温下放置24h内稳定。通过回收率试验,平均回收率为96.72%。通过对10批样品进行测试,测得没食子酸c7h6o5平均含量为3.747mg/g,本品按干燥品计算,暂定本品中没食子酸c7h6o5不得少于2.23mg/g。
42.(6)水分、灰分检查:水分检查方法按照2020年版《中国药典》(四部)通则0832(烘干法)测定,不得超过8.49%,所述总灰分和酸不溶灰分检查方法按照2020年版《中国药典》(四部)通则2302测定,总灰分不得超过4.94%,酸不溶灰分含量不得超过0.50%。
43.(7)浸出物检查:按照2020年版《中国药典》(四部)通则2201,采用50%乙醇为溶剂以热浸法测定,不得低于14.11%。
44.(8)重金属检查:
45.按照2020年版《中国药典》(四部)通则2321第一法原子吸收分光光度法和第二法电感耦合等离子体质谱法测定重金属铅、镉、砷、汞、铜含量,铅不得过5mg/kg;镉不得过0.3mg/kg;砷不得过2mg/kg;汞不得过0.2mg/kg;铜不得过20mg/kg。
46.水柏枝药材的质量达到以上标准时,即水柏枝样本为合格的药材。
47.其中步骤(1)性状鉴别、步骤(2)显微鉴别中的a.粉末显微鉴别和b.花柄横切显微鉴别均属于药材的基原鉴别;步骤(3)理化检测和步骤(4)薄层色谱检识化学成分属于定性检测;步骤(3)~(8)属于化学成分定量检测。
48.本发明填补了水柏枝蒙药材质量检测方法的空白,具有全方面、系统化进行质量控制的优点,为水柏枝的开发利用提供了可靠的科学依据。
49.本发明的主要创新点在于:
50.1、现有技术“藏药水柏枝的质量标准研究”(《中国药房》杂志2017年第28卷第33期)鉴定10批水柏枝药材样品的原植物性状、显微特征,测定水分、总灰分和浸出物含量,采用薄层色谱法(tlc)和高效液相色谱法(hplc)进行定性、定量研究。但该方法显微特征不全面并缺少茎横切面显微鉴别无法准确确定样本药材的基原,无酚酸类的理化鉴别确定药材中是否有酚酸类成分以及无重金属检查,其高效液相色谱法采用等度洗脱,比梯度洗脱理论塔板数低。
51.2、现有技术“蒙药河柏中没食子酸的hplc含量测定”(《中药材》杂志2008年第31卷第2期)仅进行了高效液相色谱法(hplc)测定没食子酸的含量,且药材批次数量只有5批。
附图说明
52.图1为水柏枝原植物图。
53.图2为水柏枝性状图。
54.图3水柏枝粉末显微图。
55.图4水柏枝茎横切显微图。
56.图5水柏枝和对照品tlc图(1~10.供试品11.对照品)。
57.图6对照品(a)、水柏枝样品(b)、阴性溶液(c)的hplc图(1.没食子酸)。
具体实施方式
58.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,本实例是叙述性的,不是限定性的,不能以此限定本发明的保护范围。
59.实施例1
60.按照本发明提供的方法和标准对不同来源的某一批次水柏枝原料进行蒙药材的质量检测。
61.一、使用仪器:primostar型数码生物显微镜、eclipsee200型生物显微镜、yoko
‑
zs型薄层数码成像仪、g型硅胶板(规格50mm
×
100mm)、展开槽(规格110mm
×
120mm)、e2695型hplc仪(含2998型光电二极管矩阵检测器)、kh
‑
300de型台式数控超声波清洗器(、bt125d型十万分之一电子分析天平、uv
‑
2600型紫外分光光度计。
62.二、使用试药:没食子酸对照品(上海源叶生物科技有限公司,批号为y19m8c36143,纯度均大于98%),乙腈、甲醇(色谱纯),磷酸、甲醇(分析纯),超纯水。
63.表1水柏枝药材来源
[0064][0065]
三、检测步骤:
[0066]
(1)性状鉴别:对选取样本的外观形状、大小、颜色、表面特征、质地、断面特征、气味进行鉴定;性状特征符合以下特征的样本初步确认为是水柏枝蒙药材:
[0067]
枝呈圆柱状,老枝灰褐色或红褐色,嫩枝红棕色或黄绿色,具有光泽和条纹;枝断面平坦,皮部所占比例较少,木部黄白色,髓部宽大为黄白色。叶窄条形互生小叶,密生于当年生绿色小枝上,多为卵形或狭长圆形,总状花序密集呈穗状,顶生于当年生枝条上。质脆,易折断。气微,味淡,见图1和图2。
[0068]
(2)显微鉴别:
[0069]
a.粉末显微鉴别:将步骤(1)所得样本制成粉末,经筛分得到210~250μm的粉末样
品,挑取筛下粉末置于载玻片上,滴加甘油醋酸试液或水合氯醛试液,盖上盖玻片,加热透化,在显微镜下观察;符合以下特征的样本是水柏枝蒙药材:
[0070]
表皮细胞呈不规则状,壁厚内陷,与细胞接触位置凸起;非腺毛,多为单细胞,内含不均匀分枝状腺,成丝状,壁稍厚,具有壁疣;导管多为具缘纹孔导管,宽铁链条状,螺纹状,少见网纹导管和螺纹导管;韧皮纤维,散在或成束,多碎断,不木化;薄壁细胞,不规则多边形,有时周围盾圆,壁稍厚鲜亮;木纤维成束存在,见图3。
[0071]
b.茎横切显微鉴别:
[0072]
a软化:选适当步骤(1)所得样本放入水中浸泡4~5天左右,使材料变软,可用温水加快速度,需要每天换水。
[0073]
b固定:将软化好的材料切成2~3mm的小段,浸泡于faa固定液(体积比为70%乙醇:冰醋酸:甲醛=18:1:1)12h以上。
[0074]
c脱水:将药材从固定液中取出,用流动的自来水冲洗24h,从50%乙醇开始,经60%、70%、80%、95%直至纯酒精(无水乙醇),每次时间为1.5h,如不能及时进行各级脱水,材料可以放在70%乙醇中保存。
[0075]
d透明:先经1/3二甲苯和2/3乙醇的混合液浸渍0.5h,再依次转入1/2二甲苯和1/2乙醇混合液、2/3二甲苯和1/3乙醇混合液中浸渍0.5h,最后转入纯二甲苯中浸渍0.5h。
[0076]
e浸蜡:把组织材料块放在熔化的石蜡和二甲苯的等量混合液浸渍1.5小时,再先后移入2个熔化的石蜡液中浸渍2h后过夜,浸蜡应在高于石蜡熔点3℃左右的温箱中进行。
[0077]
f包埋、切片、贴片、烤片处理。
[0078]
g染色:取已经干燥的切片,放于盛有二甲苯的染缸内脱蜡8min,脱完蜡后的切片可依次用纯乙醇和二甲苯的等量混合液、纯乙醇、95%乙醇、80%乙醇、70%乙醇洗5min,番红染液染色4h,水洗3s,依次用35%乙醇、50%乙醇、70%乙醇、80%乙醇、95%乙醇洗2min,固绿染色5s,染色后的切片需先后用2个纯乙醇洗30s,再用纯乙醇和二甲苯的等量混合液洗5min,纯二甲苯洗10min,洗去多余染料。
[0079]
h封片:二甲苯透明后,迅速擦去材料周围多余液体,滴加适量(1~2滴)中性树胶,再将洁净盖玻片倾斜放下,以免出现气泡,封片后即制成永久性玻片标本,在显微镜下观察,符合以下显微特征确认为水柏枝蒙药材:
[0080]
茎为类圆形,边缘呈波状凹凸。木栓层细胞数列,壁增厚,木化;皮层极狭小,为2~3列薄壁细胞。棱线处有下皮纤维束木质部,维管束14~16个,位于皮下纤维束下方,韧皮细胞细小排列整齐,木质部导管类圆,微木栓化;中央髓部宽广,薄壁细胞内含多数草酸钙柱晶,见图4。符合以上粉末显微和茎横切显微特征即确认样本为水柏枝蒙药材。
[0081]
(3)理化检测:
[0082]
取步骤(1)所得样本的粉末少量溶于冰醋酸中,加入少量铜离子,立即加热,显蓝色为环烯醚萜类正反应;显蓝色说明含有环烯醚萜类,符合质量要求。
[0083]
表2水柏枝理化检识结果
[0084][0085]
(4)没食子酸定性检测:
[0086]
a.供试品溶液制备:取本品粉末过30目筛,准确称量0.5g药材,加入20ml水,加热回流提取4h,放冷至室温,过滤,即得。
[0087]
b.对照品溶液制备:取没食子酸对照品适量,精密称定,加入甲醇制成质量浓度分别为2.230mg/ml的单一成分对照品贮备液,即得。
[0088]
c.没食子酸薄层色谱定性鉴别:按照2020年版《中国药典》(四部)tlc法(通则0502)实验操作要求,分别吸取各单一成分对照品贮备液5μl和供试品溶液10μl,点于同一硅胶g薄层板上,在常温下饱和后展开,取出,晾干,检视,展开剂为氯仿:乙酸乙酯:甲酸(5:4:1,v/v/v),喷3%三氯化铁溶液显色后,105℃加热,日光灯下检视。供试品与各对照品在相应的位置上,显相同颜色的斑点,薄层展开斑点清晰,证明含有含没食子酸成分,结果见图5。
[0089]
(5)高效液相色谱法测定没食子酸含量:
[0090]
a.色谱条件:采用十八烷基硅烷键合硅胶为填充剂,检测波长为210nm,柱温为30℃,进样体积5μl,流动相:以乙腈(a)-0.1%磷酸水溶液(b)为流动相,梯度洗脱(0~10min,10%
→
8%a),流速为1ml/min;在该色谱条件下,理论塔板数按没食子酸计算不低于6000,各峰与相邻峰的分离度均大于1.3,各成分测定不受其他组分的干扰。
[0091]
b.溶液制备:
[0092]
a.对照品溶液制备:分别取没食子酸对照品各适量,精密称定,加入甲醇制成质量浓度分别为2.230mg/ml的单一成分对照品贮备液,取没食子酸对照品贮备液适量用甲醇逐步稀释至刻度,混匀,制备成6个系列对照品溶液,备用。
[0093]
b.供试品溶液制备:取过30目筛水柏枝样品0.5g,精密称定,置于250ml圆底烧瓶中,加水20ml,加热回流提取4h,取出,放冷至室温,再次称质量,用水补足减失的质量,先后以滤纸、0.45μm微孔滤膜滤过,取续滤液,即得供试品溶液。
[0094]
c.阴性对照溶液制备:取水20ml,置于250ml圆底烧瓶中,按制备供试品方法制备阴性对照溶液。
[0095]
c.系统适用性试验:
[0096]
取没食子酸对照品溶液、供试品溶液各5μl,按拟定的色谱条件进样测定,记录色谱图。结果表明,在该色谱条件下,理论塔板数按没食子酸计算不低于6000,各峰与相邻峰的分离度均大于1.3,各成分测定不受其他组分的干扰,结果见图6。
[0097]
d.线性关系考察:
[0098]
取没食子酸对照品储备液于6个5ml容量瓶中用甲醇稀释至刻度线,混匀,配制成系列浓度的混合对照品溶液,依照拟定的色谱条件分别进样5μl,测定峰面积。浓度(x)为横坐标,峰面积(y)为纵坐标,绘制标准曲线。计算没食子酸的线性回归方程、线性范围、相关系数。
[0099]
表3水柏枝中没食子酸线性关系考察结果
[0100][0101]
e.精密度试验:
[0102]
精密吸取混合对照品溶液5μl,按拟定的色谱条件进样5μl进行测定,连续进样6
次,测定没食子酸的峰面积。结果没食子酸峰面积的rsd(n=6)分别为1.55%,表明仪器精密度良好。
[0103]
f.重复性试验:
[0104]
分别称取同一批样品(编号s10)6份,每份0.5g,精密称定,置250ml圆底烧瓶中,按照拟定的方法制备6份供试品溶液,按拟定的色谱条件进样5μl进行测定,记录没食子酸的峰面积,计算平均含量及rsd。结果样品中没食子酸的平均含量分别为4.37mg/g,rsd(n=6)分别为1.08%。表明本方法重复性良好。
[0105]
g.稳定性试验:
[0106]
取同一份供试品溶液(编号s1),分别在0、2、4、8、12、24h按拟定的色谱条件进样5μl进行测定,记录没食子酸的峰面积,计算没食子酸峰面积的rsd(n=6)分别为1.40%。表明供试品溶液室温下放置24h内稳定。
[0107]
h.加样回收率试验:
[0108]
称取已知含量的样品(编号s2)6份,精密称定,按拟定的方法,精密加入没食子酸对照品溶液适量,制备6份供试品溶液,再按拟定的色谱条件进样5μl进行测定,计算加样回收率和rsd,结果见表4,表明本方法准确度良好。
[0109]
表4水柏枝中没食子酸加样回收率试验结果,n=6
[0110][0111]
i.样品含量测定:
[0112]
取10批药材样品各适量,分别按拟定的方法制备供试品溶液,再按拟定的色谱条件进样测定,记录峰面积,以峰面积代入标准曲线计算样品含量。每批样品平行测定3次,计算没食子酸的平均含量,结果见表5。
[0113]
表5水柏枝样品含量测定结果(n=3,mg/g)
[0114]
[0115]
计算没食子酸的平均含量为3.747mg/g,本品按干燥品计算,暂定本品中没食子酸c7h6o5不得少于2.23mg/g。
[0116]
(6)水分、灰分检查:
[0117]
依照2020年版《中国药典》(四部)通则0832(烘干法)进行水分测定,每批样品平行测定3次,取平均值,结果,本实施例中各批样品水分含量为6.16%~8.68%,平均值为7.07%,以平均值上浮约20%,药材水分含量不得超过8.49%,结果见表6;所述总灰分和酸不溶灰分检查方法为2020年版《中国药典》(四部)通则2302测定,每批样品平行测定3次,取平均值,结果,本实施例中各批样品总灰分含量为2.40%~5.97%,平均值为4.12%,以平均值上浮约20%,总灰分含量不得超过4.94%;本实施例中各批样品酸不溶灰分含量为0.03%~1.02%,平均值为0.42%,以平均值上浮约20%,酸不溶灰分含量不得超过0.50%,结果见表6。
[0118]
(7)浸出物检查:
[0119]
依照2020年版《中国药典》(四部)通则2201,采用50%乙醇为溶剂以热浸法进行提取,测定醇溶性浸出物的含量。每批样品平行测定3次,取平均值。结果,本实施例中各批样品醇溶性浸出物含量为11.45%~25.03%,平均值为17.64%,以平均值下浮约20%,暂定药材醇溶性浸出物含量不得低于14.11%,结果见表6。
[0120]
表6水柏枝中水分、总灰分、酸不溶灰分和浸出物含量测定结果(n=3,%)
[0121][0122]
(8)重金属检查:
[0123]
a.原子荧光分光光度计工作条件工作参数为:负高压:300v;总电流:30ma;载气流量:400ml/min;屏蔽气流量:1000ml/min;原子化气高度:10mm;读数时间:10s;延迟时间:1s。
[0124]
b.icp
‑
ms工作条件用调谐液调整仪器灵敏度、氧化物、双电荷、分辨率等各项指标。优化后的icp
‑
ms工作参数为:射频功率:1300w;等离子体流速:15l/min;辅助气流速:0.70l/min;载气流速:0.86l/min;雾室温度:2℃;采样深度:10mm;采样锥孔径:1.0mm;截取锥孔径:0.45mm。
[0125]
c.微波消解条件经优化调整的微波消解程序见表7。
[0126]
表7微波消解程序
[0127][0128]
d.标准溶液的配制分别精密吸取铅、砷、镉、汞、铜单元素标准溶液,用10%硝酸溶液逐级稀释制成混合标准溶液。同时,加入金标准溶液(1μg/ml),使金终浓度为4ng/ml。该标液临用时配制。
[0129]
e.供试品溶液的制备取供试品,粉碎成粗粉,取约0.25g,精密称定,置微波消解罐中,加20ml纯化硝酸,电热板200℃,消煮1h,不够继续加酸。待样品完全溶解后,加入2ml过氧化氢预消解,置微波消解仪内消解,消解程序见表7。消解结束后,用水稀释定容至25ml,摇匀,即得。同法制备试剂空白溶液。过滤时得过滤到玻璃管中,先用原子荧光光度计测定汞,随后用icp
‑
ms测定其他元素。
[0130]
f.内标溶液的制备取锗、铑混合标准溶液用水稀释,制成0.5μg/ml的混合内标溶液,即得。
[0131]
g.测定法用调谐液调整仪器,各项指标达到测定要求后,引入在线内标、标准系列及供试品溶液。测定时选取的同位素为cu、as以ge作为内标,pb以rh作为内标,仪器的内标进样管在仪器分析过程中始终插入内标溶液,依次将仪器的样品管插入各个浓度的标准品溶液和样品溶液进行测定,测量值取3次读数的平均值,并扣除试剂空白。
[0132]
h.线性关系考察取标准系列混合溶液,依法测定,以各元素与内标计数值的比值(y)为纵坐标,各元素质量浓度(x)为横坐标,绘制标准曲线,结果见表8,各元素的线性关系良好。
[0133]
表8线性关系考察结果
[0134][0135]
i.结果分析
[0136]
重金属检查方法为按照2020年版《中国药典》(四部)通则2321第一法原子吸收分光光度法和第二法电感耦合等离子体质谱法测定重金属铅、镉、砷、汞、铜含量,铅不得过5mg/kg;镉不得过0.4mg/kg;砷不得过2mg/kg;汞不得过0.2mg/kg;铜不得过20mg/kg;结果见表9。
[0137]
表9水柏枝中铅、砷、镉、汞、铜的含量(mg/kg,n=3)
[0138]
[0139][0140]
以上所述仅为本发明优选实施例,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。