1.本发明涉及一种高效液相色谱分析方法,尤其是一种夏桑菊颗粒中迷迭香酸的含量测定方法。
背景技术:
2.夏桑菊颗粒,由夏枯草、野菊花、桑叶组成,经现代工艺精制加工而成。夏桑菊颗粒是《中华人民共和国卫生部药品标准》中药成方制剂(第十五册)收载的中成药,具有清肝明目、疏风散热,除湿痹,解疮毒之功效,适用于治疗风热感冒引起的目赤头痛,高血压,头晕耳鸣,咽喉肿痛,疔疮肿毒等症状,并可作清凉饮料。现代研究表明,夏桑菊颗粒可抑制多种细菌的生长繁殖,对金黄色葡萄球菌和溶血性链球菌的抑菌作用较强,说明本品清热解毒作用及抗感染作用与其抗菌作用密切相关。
3.夏桑菊颗粒的君药是夏枯草,2010年版《中国药典》修订了夏枯草的含量测定项,其含量测定指标修订为迷迭香酸,研究检测夏枯草主要药效成分迷迭香酸来建立夏桑菊颗粒的质控指标。夏桑菊颗粒的服用方法为用水冲泡饮用,目前,关于夏桑菊中迷迭香酸含量的检测集中于颗粒制剂的检测,对于冲泡后的夏桑菊颗粒水液中迷迭香酸含量还未见报,如专利cn101862373a公开了《一种夏桑菊颗粒的检测方法》,采用高效液相色谱法测定夏桑菊颗粒中夏枯草的特征成分迷迭香酸的含量。因此,迷迭香酸的含量测定方法仍有待于进步一优化。
技术实现要素:
4.鉴于此,本发明的目的在于提供一种夏桑菊颗粒中迷迭香酸的含量测定方法,用于夏桑菊颗粒的质量监控。该方法具有操作简单,分析时间短,检出限低及耐用性、准确性良好的优点。
5.本发明的技术方案是这样实现的:
6.一种夏桑菊颗粒中迷迭香酸的含量测定方法,采用高效液相色谱法进行测定,采用zorbax bonus
‑
rp c18(4.6
×
250mm,5μm)色谱柱,用十八烷基硅烷键合硅胶作填充剂,流动相以乙酸铵
‑
磷酸盐缓冲溶液(磷酸调节ph=3.0~3.5)为a相,以甲醇为b相,检测波长为315~335nm,柱温为25~30℃,流速为1.1~1.3ml/min。
7.优选的,所述乙酸铵
‑
磷酸盐缓冲溶液的乙酸铵和磷酸盐溶液的体积比为2:(3~6)。
8.优选的,所述乙酸铵为浓度0.01~0.05mol/l的乙酸铵水溶液。
9.优选的,所述的磷酸盐溶液为0.01
‑
0.1mol/l磷酸二氢钾水溶液。
10.优选的,所述流动相中甲醇和乙酸铵
‑
磷酸盐缓冲溶液的体积比为25~30:75~70。
11.本发明所述的含量测定方法,通过以下步骤实现:
12.(1)对照品溶液的制备:精密称取迷迭香酸对照品,用溶剂配制成对照品溶液;
13.(2)供试品溶液的制备:取夏桑菊颗粒样品研细成粉,精密称取样品粉末,加入溶剂,称重,超声处理20~40min,取出,冷却至室温,用溶剂补重,摇匀,滤过,取续滤液;
14.(3)取对照品溶液和供试品溶液注入高效液相色谱仪,检测迷迭香酸的含量;
15.高效液相色谱仪的条件如下所示:
16.色谱柱:zorbax bonus
‑
rp c18(4.6
×
250mm,5μm),填充剂为十八烷基硅烷键合硅胶;
17.流动相a:体积浓度为0.1~0.2%的乙酸铵
‑
磷酸盐缓冲溶液(ph为3.0~3.5);
18.流动相b:体积浓度为60~75%的甲醇;
19.柱温:25~30℃;
20.流速:1.1~1.3ml/min;
21.检测波长为325~345nm;
22.理论板数按迷迭香酸峰计算应不低于5000;
23.洗脱程序:
24.在0~15min,流动相a的比例维持在85%,流动相b的比例维持在15%;
25.在15~25min,流动相a的比例由85%转变为50%,流动相b的比例由15%转变为50%;
26.在25~35min,流动相a的比例由50%转变为85%,流动相b的比例由50%转变为15%;
27.在35~45min,流动相a的比例维持在85%,流动相b的比例维持在15%。
28.优选的,每1ml所述对照品溶液含100~125μg迷迭香酸对照品。
29.优选的,步骤(2)中,样品粉末和溶剂的质量体积比为1g:40~100ml。
30.优选的,所述溶剂为水或甲醇。
31.优选的,所述甲醇的体积浓度为60~75%。
32.优选的,步骤(2)中,样品粉末和水的质量体积比为1g:50~100ml。
33.优选的,步骤(2)中,样品粉末和甲醇的质量体积比为1g:40~80ml
34.优选的,所述超声的功率为180~200w,频率为28~33khz,温度为40~60℃。
35.优选的,步骤(4)中,对照品溶液和供试品溶液的吸取量为1~20μl。
36.本发明的含量测定方法测定夏桑菊颗粒中迷迭香酸含量的检出限为1.2μg/ml。
37.与现有技术相比,本发明的有益效果是:
38.本发明采用高效液相色谱法对夏桑菊颗粒中迷迭香酸进行定量测定,能够准确测定夏桑菊颗粒剂和冲泡后汤剂中迷迭香酸的含量,可检测低浓度样品,分析时间短、检测灵敏度高,而且检测过程简单、快捷。
39.本发明的测定方法的专属性、精密度和耐用性等方面均经详细验证,且各项验证结果均符合相关法规和指导原则的要求,实际检测效果良好。
附图说明
40.图1为本发明实施例4中4.1专属性的对照品溶液的hplc图谱;
41.图2为本发明实施例4中4.1专属性的空白溶液的hplc图谱;
42.图3为本发明实施例4中4.1专属性的夏桑菊颗粒阴性对照溶液的hplc图谱
43.图4为本发明实施例4中4.1专属性的夏桑菊颗粒样品溶液的hplc图谱;
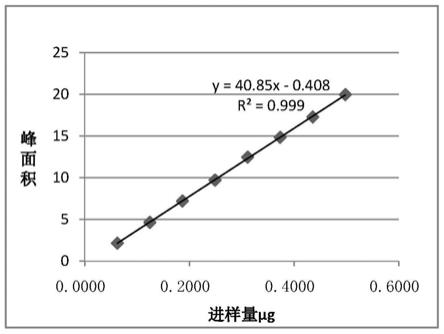
44.图5为本发明实施例4中4.2线性范围和检出限的线性图;
具体实施方式
45.为了更好理解本发明技术内容,下面提供具体实施例,对本发明做进一步的说明。
46.本发明实施例所用的实验方法如无特殊说明,均为常规方法。
47.本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
48.以下4个实施例均选取规格为3g/袋的同批次(170701)样品夏桑菊颗粒(自制)。
49.实施例1
‑
夏桑菊颗粒剂中迷迭香酸的含量测定
50.实验步骤:
51.(1)对照品溶液的制备:精密称取迷迭香酸对照品适量,用体积浓度75%的甲醇配制成1ml含100μg迷迭香酸对照品的对照品溶液;
52.(2)供试品溶液的制备:取夏桑菊颗粒样品研细成粉,精密称取0.375g样品粉末,加入15ml体积浓度75%的甲醇,在超声的功率为180w,频率为33khz,温度为40℃的条件下,超声处理30min,取出,冷却至室温,补重,摇匀,滤过,取续滤液;
53.(3)各取20μl对照品溶液和供试品溶液注入高效液相色谱仪,检测迷迭香酸的含量;
54.色谱条件如下所示:
55.仪器:高效液相色谱仪;
56.色谱柱:zorbax bonus
‑
rp c18(4.6
×
250mm,5μm),填充剂为十八烷基硅烷键合硅胶;
57.流动相:流动相a为体积浓度0.1%的乙酸铵
‑
磷酸盐缓冲溶液(乙酸铵:磷酸盐=2:3,ph3.5),比例为30%;流动相b为甲醇体积浓度75%甲醇水溶液,比例为70%;
58.柱温:28℃;
59.流速:1.2ml/min;
60.检测波长为325nm;
61.理论板数按迷迭香酸峰计算应不低于5000;
62.洗脱程序:
63.时间(min)流动相a(%)流动相b(%)08515158515255050358515458515
64.实施例2
‑
夏桑菊颗粒汤剂中迷迭香酸的含量测定
65.实验步骤:
66.(1)对照品溶液的制备:精密称取迷迭香酸对照品适量,用水配制成1ml含125μg迷迭香酸对照品的对照品溶液;
67.(2)供试品溶液的制备:取夏桑菊颗粒样品研细成粉,精密称取0.375g样品粉末,
加入18.8ml水,在超声的功率为200w,频率为33khz,温度为60℃的条件下,超声处理30min,取出,冷却至室温,补重,摇匀,滤过,取续滤液;
68.(3)各取1μl对照品溶液和供试品溶液注入高效液相色谱仪,检测迷迭香酸的含量;
69.色谱条件如下所示:
70.仪器:高效液相色谱仪;
71.色谱柱:zorbax bonus
‑
rp c18(4.6
×
250mm,5μm),填充剂为十八烷基硅烷键合硅胶;
72.流动相:流动相a为体积浓度0.2%的乙酸铵
‑
磷酸盐缓冲溶液(乙酸铵:磷酸盐=2:6,ph3.0),比例为25%;流动相b为甲醇体积浓度65%甲醇水溶液,比例为75%;
73.柱温:25℃;
74.流速:1.1ml/min;
75.检测波长为325nm;
76.理论板数按迷迭香酸峰计算应不低于5000;
77.洗脱程序:
78.时间(min)流动相a(%)流动相b(%)08515158515255050358515458515
79.对比例1
‑‑
夏桑菊颗粒剂中迷迭香酸的含量测定
80.参照《中国药典》2020年版一部中夏桑菊颗粒质量标准含量测定项进行测定;
81.实验步骤:
82.(1)对照品溶液的制备:精密称取迷迭香酸对照品适量,用体积浓度75%的甲醇配制成1ml含25μg迷迭香酸对照品的对照品溶液;
83.(2)供试品溶液的制备:取夏桑菊颗粒样品研细成粉,精密称取1.25g样品粉末,加入25ml体积浓度75%的甲醇,在超声的功率为250w,频率为33khz的条件下,超声处理30min,取出,冷却至室温,补重,摇匀,滤过,取续滤液;
84.(3)各取10μl对照品溶液和供试品溶液注入高效液相色谱仪,检测迷迭香酸的含量;
85.色谱条件如下所示:
86.仪器:高效液相色谱仪;
87.色谱柱:zorbax bonus
‑
rp c18(4.6
×
250mm,5μm),填充剂为十八烷基硅烷键合硅胶;
88.流动相:乙腈
‑
1%醋酸溶液(19:81);
89.柱温:35℃;
90.流速:0.9ml/min;
91.检测波长为329nm;
92.理论板数按迷迭香酸峰计算应不低于5000;
93.洗脱程序:
94.时间(min)流动相a(%)流动相b(%)08515158515255050358515458515
95.实施例1
‑
2和对比例1迷迭香酸的含量测定结果显示,本发明实施例1
‑
2的迷迭香酸峰和相邻峰达到基线分离,而且迷迭香酸峰的保留时间、理论塔板数、拖尾因子均符合要求,对比例1测定方法的理论塔板数不符合要求,拖尾因子较大,测定结果如下;
96.表1系统适用性结果
97.名称保留时间(min)峰面积理论塔板数拖尾因子实施例129.54812.17349301.10.954实施例228.18710.52759149.40.937对比例123.7468.47834545.81.745
98.实施例3
‑
样品处理过程中超声参数的考察
99.评价供试品超声处理参数对含量测定结果的影响,其中一个超声参数变化时,其他超声参数不变。
100.实验步骤:
101.(1)对照品溶液的制备:取迷迭香酸对照品约10mg,精密称定,置20ml容量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密量取1ml,置20ml容量瓶中,加甲醇稀释至刻度,摇匀,即得;
102.(2)供试品溶液制备的制备:取夏枯草颗粒样品,混匀,研细,精密称取0.375g,精密称定,置具塞锥形瓶中,精密加入75%甲醇30ml,密塞,称定重量,超声处理,取出,放冷至室温,再称定重量,用75%甲醇补足减失的重量,摇匀,0.22μm滤过器过滤,取续滤液,备用;
103.(3)各取10μl对照品溶液和供试品溶液注入高效液相色谱仪,检测迷迭香酸的含量;
104.色谱条件如下所示:
105.仪器:高效液相色谱仪
106.色谱柱:zorbax bonus
‑
rp c18(4.6
×
250mm,5μm),填充剂为十八烷基硅烷键合硅胶;
107.流动相:流动相a为体积浓度0.1%的乙酸铵
‑
磷酸盐缓冲溶液(乙酸铵:磷酸盐=2:3,ph3.5),比例为30%;流动相b为甲醇体积浓度75%甲醇水溶液,比例为70%;
108.柱温:28℃;
109.流速:1.2ml/min;
110.检测波长为325nm;
111.理论板数按迷迭香酸峰计算应不低于5000;
112.洗脱程序:
113.时间(min)流动相a(%)流动相b(%)08515158515255050358515458515
114.表2样品不同超声处理参数的测定结果
[0115][0116][0117]
由上表数据可知,本发明的超声处理参数rsd值在3.44%,测定的迷迭香酸的含量差异不大,说明本发明的超声处理参数较为合理。
[0118]
实施例4方法学考察
[0119]
4.1专属性试验
[0120]
实验步骤:
[0121]
(1)空白溶剂的制备:取75%甲醇,用0.22μm过滤器过滤。
[0122]
(2)阴性对照溶液的制备:取夏枯草阴性对照样品,取0.375g(即1/8袋),精密称定,置具塞锥形瓶中,精密加入75%甲醇25ml,密塞,称定重量,超声处理(功率180w、频率33khz、温度40℃)30min,取出,放冷至室温,再称定重量,用75%甲醇补足减失的重量,摇匀,0.22μm滤过,取续滤液,备用。
[0123]
(3)供试品溶液制备:取夏枯草颗粒样品,混匀,研细,精密称取0.375g,精密称定,置具塞锥形瓶中,精密加入75%甲醇30ml,密塞,称定重量,超声处理,取出,放冷至室温,再称定重量,用75%甲醇补足减失的重量,摇匀,0.22μm滤过器过滤,取续滤液,备用。
[0124]
(4)对照品溶液:取迷迭香酸对照品约10mg,精密称定,置20ml容量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密量取1ml,置20ml容量瓶中,加甲醇稀释至刻度,摇匀,0.22μm滤过器过滤,取续滤液,备用;
[0125]
(5)各取10μl空白溶剂、阴性对照溶液、对照品溶液和供试品溶液注入高效液相色
谱仪,检测迷迭香酸的含量;
[0126]
色谱条件如下所示:
[0127]
色谱柱:zorbax bonus
‑
rp c18(4.6
×
250mm,5μm),填充剂为十八烷基硅烷键合硅胶;
[0128]
流动相a为体积浓度0.1%的乙酸铵
‑
磷酸盐缓冲溶液(乙酸铵:磷酸盐=2:3,ph3.5),比例为30%;流动相b为甲醇体积浓度75%甲醇水溶液,比例为70%;
[0129]
柱温:28℃;
[0130]
流速:1.2ml/min;
[0131]
检测波长为325nm;
[0132]
理论板数按迷迭香酸峰计算应不低于5000;
[0133]
洗脱程序:
[0134]
时间(min)流动相a(%)流动相b(%)08515158515255050358515458515
[0135]
结果显示,以上处理方法获得的供试品溶液的色谱图中有与迷迭香酸对照品保留时间一致的色谱峰出现,而阴性对照溶液的色谱图中无与此对照品保留时间一致的峰出现,说明此方法对本品迷迭香酸的含量测定无干扰,图谱见图1
‑
图4。
[0136]
4.2线性范围和检出限
[0137]
称取迷迭香酸对照品12.63mg,用甲醇定容至20ml,再取1ml用甲醇定容至20ml,取续滤液2μl、4μl、6μl、8μl、10μl、12μl、14μl、16μl、18μl注入液相色谱仪,色谱条件同专属性试验,以对照品进样量为横坐标(x),峰面积为纵坐标(y),进行线性回归分析。再将迷迭香酸对照品溶液逐级稀释,进样10μl,分别注入液相色谱仪分析,以3倍信噪比计算检出限,计算得检出限为1.2μg/ml。
[0138]
表3含量测定线性试验结果(n=8)
[0139][0140][0141]
结果表明,迷迭香酸对照品在0.0623μg~0.4981μg进样量范围内,与峰面积呈良好的线性关系(r2=0.9998),线性图见图2。
[0142]
4.3精密度试验
[0143]
对照品溶液、供试品溶液的制备及色谱条件同专属性试验;
[0144]
仪器精密度:由不同分析人员,间隔一天,在同一实验室分别采用两台不同的高效液相色谱仪测定迷迭香酸对照品溶液,计算其峰面积的平均值,考察分析人员采用不同设备测定的精密度。称取迷迭香酸对照品溶液过滤后,取续滤液10μl,连续进样6次,结果见表4;峰面积的rsd应不大于2%,结果显示符合要求;
[0145]
重复性:取6份样品,分别制备成供试品溶液,其迷迭香酸含量按外标法以峰面积计算(mg/袋),测定结果见表4,所得6份供试液各成分含量的rsd应不超过4%,结果显示此测定方法重复性较好;
[0146]
中间精密度:由不同分析人员,间隔一天,在同一实验室分别采用两台不同的液相色谱仪测定样品的含量,每次取6份样品,计算平均值,考察其中间精密度,按外标法以峰面积计算其含量mg/袋,测定结果见表4,所得12个含量数据的rsd应不超过4%,结果表明中间精密度良好;
[0147]
表4精密度测定结果
[0148][0149][0150]
4.4稳定性试验
[0151]
供试品溶液及对照品溶液室温下放置,分别于放置的第0、2、4、6、8、10、12、14、16、18、30、91小时后进样,测定其峰面积,考察溶液的稳定性。
[0152]
对照品溶液:配制同专属性试验;
[0153]
供试品溶液:配制同专属性试验;
[0154]
色谱条件:同专属性试验。
[0155]
表5稳定性试验结果
[0156][0157]
结果显示,对照品溶液和供试品溶液在室温下放置的1
‑
91h,其溶液峰面积变化较小,说明在这段时间内,溶液较稳定。
[0158]
4.5加样回收试验
[0159]
取本品,按80%、100%和l20%分别制备三个浓度,每个浓度各制备三份样品,在9份样品中分别加入等比例的本品对照品,照专属性试验分别制备9份供试品溶液,进行加样回收试验。
[0160]
取制备好的供试品溶液,精密量取10μl注入液相色谱仪,照专属性试验的色谱条件进行测定,另取对照品溶液,同法测定,按外标法以峰面积计算,即得。
[0161]
表6加样回收试验
[0162][0163]
结果显示,在80%~120%的浓度范围内,回收率为94.12%~102.78%,rsd为1.88%~2.51%,实验结果表明,本品回收率试验良好。
[0164]
4.6耐用性试验
[0165]
对照品溶液的制备:同专属性试验;
[0166]
供试品溶液的制备:同专属性试验;
[0167]
测定方法:分别取10μl对照品溶液和供试品溶液注入高效液相色谱仪,按照下述条件测定
[0168]
分别考察检测波长变化
±
5nm、流速相对值变化
±
10%、流动相比例改变
±
1%、柱温变化对仪器色谱行为的影响,每个条件下各测试一次。
[0169][0170]
表7耐用性测定结果
[0171][0172]
结果显示,测定本品迷迭香酸含量时,流速、波长、流动相比例、柱温的变化的含量rsd为2.74%,说明本发明含量测定方法的耐用性良好。
[0173]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。