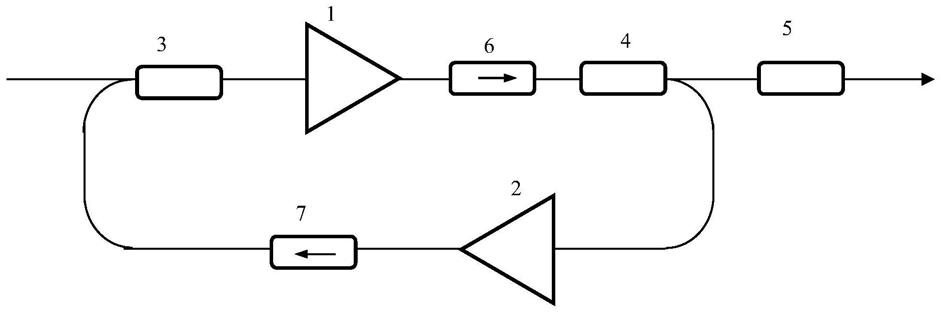
基于钴的锂金属氧化物阴极材料
1.本发明专利申请是国际申请号为pct/ib2016/053920,国际申请日为2016年6月30日,进入中国国家阶段的申请号为201680037898.7,发明名称为“基于钴的锂金属氧化物阴极材料”的发明专利申请的分案申请。
技术领域
2.本发明涉及基于钴的锂金属氧化物阴极材料。具体地,本发明涉及一种具有成层结构的基于锂钴的氧化物,该氧化物用作可再充电的锂离子电池组中的阴极材料。该氧化物具有核心
‑
壳构型,并且具有掺杂元素、氧化物和专属涂布层。
背景技术:
3.自从mizushima等人于1980年发现了锂钴氧化物的电化学性质后,锂钴氧化物就已用作二次li电池组的原型阴极材料。基于锂钴氧化物的材料具有成层结构,该成层结构沿六方单位晶胞(空间群r
‑
3m)的001方向有交替的共边(edge
‑
shared)coo6八面体和lio6八面体的coo2平板(slab)和lio2平板。此类成层结构理想地适于通过分别在电池组充电和放电期间脱嵌和嵌入来可逆地容纳锂。由于可再充电的锂电池组和锂离子电池组的高能量密度,其可用于多种携带式电子应用中,诸如移动电话、笔记本电脑、数字相机和摄影机。可商购获得的锂离子电池组通常由基于石墨的阳极和基于licoo2的阴极材料组成。由于当今的消费性电子产品需要具有较高能量密度的可再充电的电池组,为了需求增加的终端应用,具有增加的能量密度的基于licoo2的材料骤增。
4.基于licoo2的阴极材料的能量密度(单位为wh/l)被定义为循环期间的平均电压(单位为v)、比容量(单位为mah/g)和重力密度(单位为g/cm3)的乘积。提高能量密度的有效途径包括:(a)增加装填密度,这通常需要增加粉末粒子的粒径,以及(b)通过增加充电电压而增加比容量。在商业电池中,通常利用相对于石墨阳极约4.35v的上限截止电压(upper cutoff voltage)对licoo2进行循环,并且给出164mah/g的比容量。为了从licoo2获得更高的容量,人们必须将licoo2充电至高于4.35v的电位;通常是4.40v,其中比容量为172mah/g,并且甚至高达4.45v,其中比容量为182mah/g。然而,使用较高上限截止电压的反复充电
‑
放电循环导致快速的容量损失,被认为这是由脱嵌的licoo2的结构不稳定性所造成,并且是由于与电解质的副反应增加所造成。
5.因此这两种途径的工业适用性因为枝节问题而受到限制。后一种途径则由于在较高电压下,与电解质接触的带电的电极材料的不稳定行为而受到限制。在锂从li
x
coo2(其中x<1)移除时,li
x
coo2的电子结构发生改变,特征在于对于钴和氧的强烈电子电荷转移,导致了强烈的电子非定域化。带电的li
x
coo2为一种非常强烈的氧化剂,且具有高反应性表面。电解质在接触此类氧化表面时是热力学上不稳定的。与作为还原剂的电解质的反应,在能量上是特别优选的。即使在低温下,在高电压下的licoo2阴极正常循环期间,此寄生反应(parasite reaction)仍缓慢地但持续地进行。反应产物覆盖表面并且电解质被分解,并且这两种影响持续地导致电池组的电化学性能劣化,由此观察到容量损失以及电阻的大幅增
加(还称为极化)。
6.此外,在“solid state ionic,83,167(1996)”中,报告了带电li
x
coo2的严重的钴洗提现象(elution)。相对于锂阳极,当钮扣电池中的充电电位从4.1v增加至多达4.5v时,钴溶解以指数方式增加,且钴以钴金属的形式沉积到负电极上。推论出在此电压范围内,钮扣电池容量衰退与钴溶解之间的直接相关。在“journal of materials chemistry,21(2011)17754
‑
17759”中,amine等人强调在li离子电池组中过渡金属离子从阴极溶解是不利的现象,因为这些金属离子从阴极迁移到阳极,并在阳极表面电解质界面(sei)上被还原成金属态。沉积在石墨阳极表面上的金属或金属合金对sei的稳定性具有负面影响。因此,此金属从阴极溶解并沉积到阳极上导致不良的安全性、在较高电压下不良的循环稳定性、以及在高温下带电阴极的不良的储存性质。“j.electrochem.soc.,155,711,(2008)”描述了一种机制,其中电解质中所含有的杂散hf侵蚀licoo2并造成co溶解。
7.此背景技术研究的第一结论是,获得具有防止副反应和金属溶解的功能性表面的锂钴氧化物是所期望的。另一方面,增加粒径以增加装填密度削弱了可再充电的电池组的功率容量。为了满足功率需要,电池组整体,尤其是活性阴极材料本身必须具有足够高的倍率性能。增加平均粒径减小了有效的固态锂扩散动力学,这最终导致倍率性能降低。
8.仔细地研究已公开的关于阴极材料的结果,允使更好地理解基于licoo2的可再充电的电池组的限制。基于licoo2的材料的目前最佳技术的基本限制在于li过量和粒径的两难困境。此两难困境于yoshio,m等人(2009).lithium
‑
ion batteries.new york:springer science buisness media llc.中被详细回顾:用于合成的对应锂过量越高,(表示为摩尔比li:co>>1.00,而通常li:co为约1.05);则装填密度越高,且粒径越高;比表面积(或bet)越低,且碱含量越高,且电化学功率性质越低。支持在递增的li:co的情况下licoo2粒子生长并致密化的机制基于所谓的“锂
‑
助熔效应(lithium
‑
flux effect)”,其中li过量用作增强licoo2粒子的生长的助熔剂,而这最终增加了装填密度。所期望的是,具有致密和整体(monolithic)粒子形态的此类licoo2材料的比表面积减小,以及电解质与阴极材料的副反应面积减小。
9.在wo2010
‑
139404中,作者说明了装填密度、平均粒径、与目前最佳技术的用于制备mg和ti掺杂的licoo2的锂过量之间的关系。获得约3.70g/cm3的18μm粒子的典型装填密度。作者强调,大的压制密度是优选的,并且是在整体、马铃薯形、且非聚结的一次licoo2粒子的情况下获得的。但是,使用大的li:co过量以获得较大的整体粒子导致了不良的电化学性能、以及较低的功率(较低的c
‑
倍率)和较低的放电容量,这继而消除了通过增加粒径所获得的能量密度增益。这些功率和放电容量的限制起因于两个主要因素:(i)当粒径增加时,根据准菲克定律(quasi
‑
fick
‑
law)(d约l2/t)的基本固态扩散限制、以及(ii)在递增的li过量的情况下结构缺陷的引入;例如,如levasseur在chem.mater.,2002,14,3584
‑
3590中所讨论,其中li过量与coo2层中li的co取代以及与氧缺乏的li
1 x
co1‑
x
o2‑
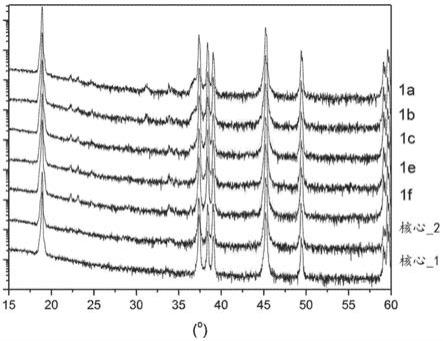
x’相关联;进一步限制粒子内li离子扩散平均自由程。最后,此类大的li:co值还增加了ph、自由(或可溶性)碱含量和碳含量,而这损害带电的阴极的安全性、储存和鼓胀性质。
10.在背景技术中,已提出若干途径来应对这些问题。其通常与基于licoo2的材料的掺杂、涂布、或表面改质相关联。例如,ep1137598b1中描述了诸如mg、ti和al的元素对于co的取代和co的取代对高电压性质的改善。在高电压下,此类掺杂防止结构坍塌,并改善基于
licoo2的材料的循环稳定性和可逆容量。但是工业适用性却受到限制,因为ti掺杂妨碍了licoo2的烧结,因此需要较高的烧结温度或较大的li过量。为了获得高电压稳定性,licoo2材料通常经(例如用al2o3)涂布、或换句话讲经化学改性(例如通过提供氟化表面)。问题在于,经涂布的致密licoo2粒子常常具有较大的极化和较低的li离子扩散,这导致较低的可逆容量和不良的倍率性能,使得通过充电至较高电压所获得的能量密度的增益的一部分被较低的本质容量消除。
11.结果就是,当年状态的现有技术合成的合成无法允许获得具有优异的能量密度、电化学功率和循环寿命性质的致密整体的基于licoo2的粒子。已获得部分的改善和优化,但是上文所提及的基本问题仍未完全解决。因此,明显存在对高能量密度的基于licoo2的阴极的需要,所述基于licoo2的阴极可以稳定的方式循环,即通过减少实际电池在较高电压下的副反应和金属溶解的稳定的方式循环。本发明的目标是限定一种用于高端二次电池组应用的阴极材料,该阴极材料具有高装填密度、在高充电电压下延长的循环的期间的高倍率性能、改善的放电容量、且展现高稳定性。
技术实现要素:
12.从第一方面观看,本发明可提供以下产物实施方案:
13.实施方案1:一种用于可再充电的电池组中的阴极材料的成层锂金属氧化物粉末,该成层锂金属氧化物粉末具有通式(1
‑
x)[li
a
‑
b
a
b
]
3a
[co1‑
c
m
c
]
3b
[o2‑
d
‑
e
n’e
]
6c
.xli3po4,其中0.0001≤x≤0.05,0.95≤a≤1.10,0<b c≤0.1,
‑
0.1≤d≤0.1,且e≤0.05,其中li对co a m 3p的摩尔比介于0.970与1.005之间,其中a和m是包括由mg、ti和al所组成的组中的至少一者的一种或多种元素;其中n’是由f、s、n和p所组成的组中的一种或多种掺杂物;该粉末包括核心和表面层,该核心具有包含元素li、co和氧的成层晶体结构并具有介于0与小于0.05之间的p对co的摩尔比,并且该表面层由元素li、co和氧的混合物所组成,该混合物还包括电子绝缘粒子,该电子绝缘粒子由包含由mg、ti和al所组成的组中的一种或多种元素的氧化物所组成,其中该氧化物还可包括li和co中的一者或两者,并且该混合物还包括以离散粒子的形式致密地附接至该核心的多个离子传导性li3po4粒子。li对co a m 3p的摩尔比对应于[((1
‑
x).(a
‑
b)) 3x]/[((1
‑
x).(1 b)) 3x]。在另外的实施方案中,b>0或c>0,或b>0且c>0。“致密地附接(densely attached)”应理解为,离散粒子完全无法或至少在不运用相当大的力的情况下无法从核心分开,与例如通过固态混合仅“松散地附接(loosely attached)”并可容易地分开的粒子是相对的。
[0014]
实施方案2:在该通式中:
[0015]
0.970≤[((1
‑
x).(a
‑
b)) 3x]/[((1
‑
x).(1 b)) 3x]≤1.000。
[0016]
实施方案3:在该通式中:
[0017]
0.970≤[((1
‑
x).(a
‑
b)) 3x]/[((1
‑
x).(1 b)) 3x]≤0.990。
[0018]
在这些实施方案中,通过将li对co a m 3p的摩尔比的范围限制为1.000或甚至0.990,在电化学测试中获得了更好的结果,如下文所示。
[0019]
实施方案4:在该粉末中,li3po4以膜层和离散粒子的组合附接至核心的表面,从而在核心上形成离子传导性电子绝缘层。
[0020]
实施方案5:在该粉末中,li3po4粒子可具有小于5μm,且优选小于1μm的尺寸。
[0021]
实施方案6:实施方案4的锂金属氧化物粉末,其中以膜存在的li3po4可覆盖核心的表面的至少50%。
[0022]
实施方案7:在实施方案4中,该膜可具有小于10nm但是>0nm的厚度。
[0023]
实施方案8:在该粉末中,该li3po4可为结晶化合物。
[0024]
实施方案9:在该粉末中,该li3po4可在氧位点上具有多达25at%的氮掺杂。
[0025]
实施方案10:在实施方案4中,该离子传导性电子绝缘表面层的厚度如通过xps所测定为1μm或更小,但是>0μm,该厚度是被限定为其中(a m)/co>(2.(b c))/(1
‑
c)mol/mol的摩尔比的深度。在该粉末的总式中,(a m)/co的摩尔比由(b c)/(1
‑
c)表示。因为a和m偏析至表面层,所以此层的深度可被定义为(a m)/co的摩尔比大于该通式中的值两倍的区域。在该表面层中,a、m和co的摩尔含量通过使用xps(x射线光电子光谱法)而测量。
[0026]
实施方案11:实施方案10的锂金属氧化物粉末,厚度为0.5μm或更小,但是>0μm。
[0027]
实施方案12:在该粉末的通式中,除了由mg、ti和al所组成的组中的至少一者以外,a可包括选自由下列所组成的组的至少一种元素:na、si、s、k、ca、v、cr、mn、fe、ni、cu、zn、sr、nb、zr、w、f和稀土金属。
[0028]
实施方案13:在该通式中,除了由mg、ti和al所组成的组中的至少一者以外,m还可包括选自由以下所组成的组的至少一种元素:li、na、si、s、k、ca、v、cr、mn、fe、ni、cu、zn、sr、nb、zr、w、f和稀土金属。
[0029]
实施方案14:粉末具有在室温下低于10
‑3s/cm、或低于10
‑4s/cm、或甚至是低于10
‑5s/cm的电子传导性。
[0030]
实施方案15:在该通式中,该p/co的摩尔比介于0.01mol%与5mol%之间。
[0031]
实施方案16:在该通式中,该li/(co 3p)的摩尔比介于0.980与1.020之间。
[0032]
实施方案17:该粉末可具有<20μmol/g、优选<15μmol/g、且更优选<12μmol/g的可溶性碱含量。
[0033]
可清楚得知,根据本发明的另外的产品实施方案可通过组合先前所述的不同产品实施方案所涵盖的个别特征来提供。
[0034]
从第二方面观看,本发明可提供以下方法实施方案:
[0035]
实施方案18:一种用于制造先前所述的锂金属氧化物粉末的方法,该方法包括以下步骤:
[0036]
‑
提供第一含co或含co、a和m的前体粉末与第一含li的前体粉末的第一混合物,该第一混合物具有>1.02、或甚至>1.04的li:co的摩尔比,
[0037]
‑
在含氧的气氛中在至少350℃、或至少600℃、或甚至至少900℃的温度t1下烧结该第一混合物,从而获得富含li的锂金属氧化物化合物,
[0038]
‑
提供第二含co或含co、a和m的前体粉末和含有磷酸盐的试剂,
[0039]
‑
将富含li的锂金属氧化物化合物、第二含co或含co、a和m的前体粉末、以及含有磷酸盐的试剂混合,从而获得第二混合物,其中li对co a m 3p的摩尔比介于0.970与1.005之间,
[0040]
‑
在含氧的气氛中在至少600℃的温度t2下烧结该第二混合物。也可将此温度设定介于700℃与1000℃之间、或介于850℃与950℃之间。在t2下的烧结步骤之前,可将第二含li的前体粉末添加至该第二混合物中。此方法中,含li的前体粉末可为li2co3。在此过程实
施方案中,该方法也可为将富含li的锂金属氧化物化合物、第二含co或含co、a和m的前体粉末、以及含有磷酸盐的试剂混合,从而获得第二混合物,其中li对co a m 3p的摩尔比介于0.970与1.005之间的步骤如下修改:不将含a和m的前体粉末添加至第二混合物,且该第二混合物是干燥的或喷雾干燥的,由此li3po4粒子沉淀在混合物中的固体上。其后,添加含a和m的前体以获得第三混合物,其中该li对co a m 3p的摩尔比介于0.970与1.005之间,接着进行最终烧结步骤。
[0041]
实施方案19:一种用于制造先前所述的锂金属氧化物粉末的方法,该方法包括以下步骤:
[0042]
‑
提供第一含co或含co、a和m的前体与第一含li的前体粉末的第一混合物,该第一混合物具有>1.01、或甚至>1.04的li:co的摩尔比,
[0043]
‑
在含氧的气氛中在至少350℃、或至少600℃、或甚至至少900℃的温度t1下烧结该第一混合物,从而获得富含li的锂金属氧化物化合物,
[0044]
‑
将富含li的锂金属氧化物化合物与含有磷酸盐和锂的试剂混合,从而将li3po4粒子(薄片)沉淀在富含li的锂金属氧化物化合物的表面上,并获得第二混合物,
[0045]
‑
提供第二含co或含co、a和m的前体粉末;以及
[0046]
‑
将第二混合物和第二含co或含co、a和m的前体粉末混合,从而获得第三混合物,由此该第三混合物具有介于0.970与1.005之间的li对co a m 3p的摩尔比,
[0047]
‑
在含氧的气氛中在至少600℃、或甚至至少700℃的温度t2下烧结该第三混合物。也可将此温度设定介于700℃与1000℃之间、或介于850℃与950℃之间。在t2下的烧结步骤之前,可将第二含li的前体粉末添加至该第三混合物中。此方法中,含li的前体粉末可为li2co3。因为此类含有磷酸盐和锂的试剂通常以液体形式被包含,所以在添加干燥的第二含co或含co、a和m的前体粉末以获得该第三混合物之前,可首先将该第二混合物干燥。在过程实施方案19中,该方法也可为,将co前体粉末与富含li的锂金属氧化物化合物、含有磷酸盐和锂的试剂混合,从而获得中间体混合物,该中间体混合物随后经喷雾干燥,从而将li3po4粒子(薄片)于沉淀在co前体粉末与富含li的锂金属氧化物化合物的混合物上,得到第二混合物,在此之后仅添加含a和m的前体粉末以获得第三混合物,接着进行烧结。在不同的过程实施方案中,该含a和m的前体可包括al氧化物,即al2o3,且也可包括mgo和ti2o3中的任一者。co前体可为co3o4。
[0048]
从第三方面观看,在实施方案20中,本发明可提供先前所述的锂金属氧化物粉末在li离子二次电池组、li聚合物二次电池组或固态二次电池组(还称为ssb)中的任一者中的用途。
附图说明
[0049]
图1:核心_1以及实施例1a、实施例1c和实施例1f的粒径分布;
[0050]
图2:核心_1(图1a)、核心_2(图1b)、样本1c(图1c)和样本1e(图1d)的fesem;
[0051]
图3:核心_1、核心_2、以及样本1a、样本1b、样本1c、样本1e和样本1f的xrd。显示在15
°
至60
°
的2
‑
θ范围内的反射强度,以log10为标度(以a.u.为单位);
[0052]
图4_1:核心_1的钮扣电池评估;
[0053]
图4_2:核心_2的钮扣电池评估;
[0054]
图4_3:样本1c的钮扣电池评估;
[0055]
图4_4:样本1g的钮扣电池评估;
[0056]
图5:样本1c至样本1g的1c能量衰退(下方图,%)和4.6v放电容量(上方图,mah/g)的演变;
[0057]
图6:核心_3(图6a)、核心_4(图6b)和样本2a(图6c)的fesem;
[0058]
图7:样本2a(下线)和样本2b(上线)的xrd图谱;
[0059]
图8:样本2a(下线)和样本2b(上线)的浮动电流(ma/g)随时间而变动的演变;
[0060]
图9a:样本2a的eds表面映射;
[0061]
图9b和图9c:样本2a的co(9b)和p(9c)的eds表面映射和空间分布;
[0062]
图10:样本2a的eds映像和谱图(map sum spectrum);
[0063]
图11:样本3a、样本3ac、样本3b、样本3c、样本3d和样本3e在5000倍(5000
×
)放大倍率下的sem图像;
[0064]
图12:样本3a、样本3b、样本3c、样本3d和样本3e的xrd图谱。以log
10
为标度,以随以2θ(度)的衍射角而变动的方式显示强度;
[0065]
图13:样本1c的xps深度曲线。绘制随深度而变动的元素含量xx=[ca,mg,na,p,s,ti]对co的摩尔比(溅射速率相对于sio2,且等于8.8nm/min);
[0066]
图14:样本2a的xps深度曲线。绘制随深度而变动的元素含量xx=[ca,mg,na,p,s,ti]对co的摩尔比(溅射速率相对于sio2,且等于8.8nm/min)。
[0067]
在图2、图6和图11中,放大倍率为5000倍(
×
5000),从而白条代表1μm。
具体实施方式
[0068]
本发明公开一种用于锂二次电池组的基于licoo2的正活性材料,该材料具有高能量密度,且在高电压下保持优异的循环稳定性。从第一方面观看,本发明可提供一种用于可再充电的电池组中的阴极材料的锂金属氧化物粉末,该锂金属氧化物粉末具有通式(1
‑
x)[li
a
‑
b
a
b
]
3a
[co1‑
c
m
c
]
3b
[o2‑
d
‑
e
n’e
]
6c
.xli3po4,其中添加在[]之后的字母代表位点,其中0.0001≤x≤0.05,0.90≤a≤1.10,0<b c≤0.1,且
‑
0.1≤d e≤0.1,其中a和m是包括由mg、ti和al所组成的组中的至少一者的一种或多种元素。基于锂钴氧化物的材料为具有菱形对称(空间群r
‑
3m,其中a约且c约)的成层材料,并且与α
‑
nafeo2是等结构的。在此类结构中,li和co离子占据来自氧紧密装填的八面体位点,且顺序是:li离子优先占据(x,y,z)=(0,0,0)结晶位点(3a wyckoff位置),co离子优先占据(0,0,1/2)(3b wyckoff位置),而o离子占据(0,0,z)(6c wyckoff位置)。a为3a位点上的掺杂物,选自na、mg、al、si、s、k、ca、ti、v、cr、mn、fe、ni、cu、zn、sr、nb、zr、w、f和稀土金属,且是由mg、ti和al所组成的组中的至少一者。m为3b位点上的掺杂物,选自li、na、mg、al、si、s、k、ca、ti、v、cr、mn、fe、ni、cu、zn、sr、nb、zr、w、f和稀土金属,且是由mg、ti和al所组成的组中的至少一者。n’为6c位点上的掺杂物,并且选自f、s、n和p。具体地,金属位点(3b位点)上的li掺杂已报告用于此类成层的基于钴的结构,并且已知导致氧缺乏;一般而言可以有多达0.1的li离子占据3b位点,导致约0.1的氧缺乏。吾等已发现,当(li):(co m a 3p)的摩尔比介于0.970与1.005之间、优选介于0.970与1.000之间时,本发明的材料的电化学性能大幅改善。当摩尔比大于1.005时,电化学性质被降级,该电化学性质主要是在高电压下的循环稳定性。另一
方面,对于低于0.970的值,材料的比容量显著减小。这些考虑指示,(1
‑
x)[li
a
‑
b
a
b
]
3a
[co1‑
c
m
c
]
3b
[o2‑
d
‑
e
n’e
]
6c
.xli3po4材料必须具有0.970≤[((1
‑
x).(a
‑
b)) 3x]/[((1
‑
x).(1 b)) 3x]≤1.005、优选0.97≤[((1
‑
x).(a
‑
b)) 3x]/[((1
‑
x).(1 b)) 3x]≤1.000的化学计量比以最佳地运作。
[0069]
本发明的材料包括第二相li3po4;该第二相li3po4以结晶态或非晶态、或结晶态和非晶态两者的组合的形式存在。存在两种经良好表征的li3po4结晶多形体,此类结晶多形体被识别为β(空间群pmn21)和γ(空间群pnma、pmnb、或pcmn,取决于xyz轴参考选择)。文献中[等人,j.raman spectrosc.,34,77(2003)]提及了α形式,但其晶体结构还未被充分阐述。在超过400℃至600℃的范围的温度下,β形式可以不可逆地转变成γ形式。在γ
‑
li3po4中,当使用wyckoff位置时,锂离子占据两个不同的结晶位点,此类位点识别为8d和4c,磷为在4c中而氧占据标记为8d和两个4c的三个不同的位点。通过a元素掺杂li3po4是可能的,且得到与li3po4等结构的li3p1‑
x
a
x
o4化合物。另外,其中n(氮元素)部分地取代o(至少多达25at%)的li3po4化合物也是可能的,且得到与li3po4等结构的化合物li3‑
α
po4‑
β
n
γ
,其中
‑
1≤α≤1,0<β≤1,且0<γ≤1。li3po4的特点在于相对高的li离子传导性和在高达4.7v相对于li/li
的良好的电化学稳定性,例如kuwata等人,ecs trans.,16,53(2009)报告了在25℃下4.5
×
10
‑7s/cm的电子传导性,适于用作所有固态li离子二次电池组(ssb)的潜在电解质。
[0070]
由p/co的摩尔比表示的li3po4的量可介于0.01mol与5mol%之间;当该量低于0.01mol%时,未观察到对电化学性质的影响,并且当该量超过5mol%时,阴极材料的比容量大幅减小。本发明的材料也可包括呈包括一种或多种a、m、li和co元素的氧化物的形式的次要化合物。典型的示例为但不限于li2o、li2so4、li2tio3、mgo、coo、mg1‑
xco
x
o、co3o4、co3‑
x
mg
x
o4、li2zro3、li
1 x
ni1‑
x
o2…
也可能存在lif。在一个实施方案中,次要相的量不超过5at%;如果该量过高,则阴极材料的比容量大幅减小。
[0071]
本发明的阴极材料由核心和离子传导性电子绝缘表面层所组成,该核心具有成层晶体结构且该表面层包含下列的混合物:核心的元素;包含由mg、ti和al所组成的组中的一种或多种元素的氧化物;以及li3po4。材料的粒子表面上的局部副反应(电解质的还原、钴洗提
…
)需要同时存在有锂阳离子和电子。锂阳离子存在于电解质中,并且电子存在于阴极主体中。但是如果电子绝缘层将阴极主体中的电子与电解质中的锂阳离子物理性地分开,则进一步的电解质还原是不可能的。如果电子绝缘阴极材料可成功地循环,则可预期高电压稳定性,因为电解质的氧化需要将电子供应至阴极。但是传统认知是,具有相对较低电子传导性的阴极无法具有良好的电化学性能。显然,具有电子绝缘和离子传导表面的阴极材料将解决此问题。此问提由本发明的材料通过提供此类官能化粒子表面和电子绝缘氧化物而解决,所述官能化粒子表面由核心的元素组成,尤其是li和co,所述电子绝缘氧化物也可为锂化的且在其式中具有co。
[0072]
licoo2的电子传导性在室温下在10
‑2s/cm至1s/cm的范围内(如us2012/0107691a1中所讨论)。本发明的材料具有非常低的电子传导性,其中传导性值低于目前最佳技术的基于锂钴氧化物的产品至少1(约10
‑3s/cm)至3(<10
‑5s/cm)个数量级,同时仍保持优异的锂离子传导性。作者相信,低电子传导性和高离子传导性的共存是本发明的重要特点,且所得的协同作用允许获得阴极材料的优异的高电压稳定性。此类低传导性的基于licoo2的阴极可
得到优异的电化学性能令人感到意外,因为普遍所接受的是,相对较高的电子传导性是阴极粒子内和跨介于电解质与阴极粒子之间的界面的li阳离子扩散所需的。
[0073]
li离子传导li3po4化合物以膜层、离散粒子、或膜层与离散粒子的组合的形式附接至核心的表面。在一个实施方案中,li3po4化合物致密地附接至粒子且均匀地覆盖表面。在至少两个实施方案中,作者猜想与膜组合的多个li3po4粒子的共存,所述粒子一般具有低于5μm的直径,致密地附接至基于锂钴氧化物的粒子,所述膜均匀地覆盖所述粒子,一般具有在0.01nm至20nm的范围内的厚度(根据xps且相对于sio2所测定)。li3po4化合物的此类分布和形态是尤其所期望的,因为作者已观察到,在高电压下金属(钴)溶解因此被大幅抑制。作者猜想,li3po4涂布层除了提供良好的离子传导性之外还实现若干功能,诸如吸收电解质中的水而降低hf含量、hf清除和保护活性材料免于恶化与电解质的副反应。在此阶段回想起以下内容是重要的:只有电子绝缘表面和离子传导性表面的组合允许获得在高电压下优异的循环稳定性和改善的(经抑制的)金属溶解。
[0074]
a、m和n’元素似乎是(自核心)偏析的,如通过xps所证明,其具有自粒子表面至核心的径向梯度掺杂分布。在至少两个实施方案中,作者观察到,在约500nm的深度(自表面,通过xps相对于sio2所测定)内,a和m元素对co的原子比超过标称含量(b c)/(1
‑
c)。也就是此类a、m和n’元素是偏析的,且发现在粒子表面处有较大浓度特别期望的。作者猜想,例如,就mg、ti和al而言,由于mg
‑
o、ti
‑
o和al
‑
o的金属
‑
氧键的稳定性质,与电解质接触的表面层的结构稳定性是改善的,且允许在高电压下长时间循环期间有改善的容量保持。除对成层相掺杂以外,所列举的a和m、以及来自核心的元素(li和co)中的一种或多种元素也可以次要相存在;这些次要相可能致密地附接至粒子的表面。来自a和m的列表的元素的次要相的实施例为但不限于li2o、lif、li2so4、li2tio3、mgo、coo、mg1‑
x
co
x
o(其中0≤x≤1)、co3o4、co3‑
x
mg
x
o4(其中0≤x≤1)、li2zro3、li2mgzro4、li
1 x
ni1‑
x
o2(其中0≤x≤1)、co2mgo4、li2tio3、zro2、sio2、al2o3、lialo2、li2sicoo4…
因为这些元素中大部分是电化学非活性的,而为了保持活性材料的高比容量;所以保持次要相的量低于5重量%、优选小于2.5重量%是重要的。据信,此类次要相致密地附接至粒子表面且聚积在1nm至100nm深度内。
[0075]
n’元素选自f、s、n和p的清单。例如所预期的是,当f部分取代o时,由于co
‑
f和li
‑
f键结的强且稳定的性质;在高电位下关于阴极材料的寄生反应(电解质分解)的电化学稳定性是改善的。
[0076]
在一些实施方案中,核心材料粒子具有整体形态,并且具有d50>10μm、或d50>15μm、或甚至>20μm的平均粒径。尤其所期望的是,随着压制密度得以改善,且brunauer
‑
emmett
‑
teller(bet)表面积得以控制且低于0.5m2/g,核心材料以此致密且整体的形态为特点。在其他实施方案中,在细粉和大粒子的混合物的情况下,粒径分布具有双峰轮廓,粒子的量通常介于1重量%与25重量%之间、或甚至是介于10重量%至20重量%之间,且大粒子与精细粒子之间的众数比(mode ratio)至少高于3μm/μm、或甚至是高于4μm/μm、和/或甚至是高于5μm/μm。此类策略使一些实施方案达到至少3.5g/cm3、或甚至是至少3.7g/cm3的压制密度。
[0077]
从第二方面观看,本发明可提供先前所述的过程方法。在一个具体实施方案中,所述过程包括下列步骤:
[0078]
‑
提供第一co氧化物、ti氧化物和mg氧化物与第一含li的前体粉末的第一混合物,
该第一混合物具有>1.05的li:co的摩尔比,
[0079]
‑
在含氧的气氛中在至少350℃、或至少600℃、或甚至至少900℃的温度下烧结该第一混合物,从而获得富含li的锂金属氧化物化合物,
[0080]
‑
将li3po4薄片沉淀在该富含li的锂金属氧化物化合物的表面上,
[0081]
‑
将携带li3po4薄片的富含li的锂金属氧化物化合物与下列混合:第二co氧化物、ti氧化物和mg氧化物与第二含li的前体粉末的第二混合物,从而获得第三混合物,该第三混合物具有介于0.970与1.005之间、优选介于0.985与1.000之间的li对co ti mg 3p的摩尔比,以及
[0082]
‑
在含氧的气氛中在至少600℃、或甚至至少900℃的温度下烧结该第三混合物。沉淀可通过以下获得:将富含li的锂金属氧化物化合物浸没于水中,添加锂化磷酸盐化合物诸如lih2po4和lioh的水溶液,接着在干燥器中蒸发水或进行喷雾干燥。
[0083]
本发明的方法(其中在第一烧结步骤期间,形成了锂超化学计量混合物,且在第二步骤中,过量的锂通过与第二含co的前体反应以形成锂化钴氧化物,且通过与含有磷酸盐的试剂反应以形成li3po4而消耗)得到由元素li、co和氧的非均匀混合物所组成的复杂表面结构,该混合物还包括由氧化物所组成的电子绝缘粒子,此类氧化物包括由mg、ti和al所组成的组群中的一种或多种元素,其中此类氧化物还可包括li和co中的一者或两者,且该混合物还包括以离散粒子的形式致密地附接至核心的多个离子传导性li3po4粒子。第一烧结步骤之后的li对金属的比率在第二烧结步骤期间未降低时,则不形成此类具有经偏析的a和m氧化物的表面层。如将在实施例中所示,具有此复杂核心
‑
表面层结构的粉末展现优于背景技术的粉末的电化学结果。还显示,li对co a m 3p的摩尔比对这些结果具有直接影响。
[0084]
a和m前体优选是具有微米或亚微米尺寸的粉末,诸如但不限于tio2、mgo、al2o3、zno、zro2、li2zro3、li2tio3…
含有磷酸盐的试剂的示例包括但不限于li3po4、lipo3、h3po4、li3‑
x
h
x
po4(其中0≤x≤3)、(nh4)3po4、h(nh4)2po4、h
x
(nh4)3‑
x
po4(其中0≤x≤3)
…
此类含有磷酸盐的试剂尤其适于通过湿式浸渍、沉淀、或诸如喷涂(在分散于含水介质中之后,进行喷嘴喷雾)的方法施加。其他含有a和m以及磷酸盐的试剂是可能的,诸如alpo4,其是具有通式m’po4的lisicon化合物(其中m’含有选自m的一种或多种金属),诸如但不限于li
1 x
ti2‑
x
al
x
(po4)3(其中0≤x≤1)和liti2‑
x zr
x
(po4)3(其中0≤x≤2)。在这些情况下,具有微米尺寸、或甚至是亚微米尺寸的此类试剂的粉末可用于上文过程中。
[0085]
该过程的关键方面在于控制含有磷酸盐的试剂与锂金属氧化物化合物的混合物的退火温度。退火可在350℃至高达1100℃的范围内的温度下进行。优选地,退火在600℃至高达1100℃的范围内进行,以便将li3po4化合物致密地附接至基于licoo2的材料粒子的表面。
[0086]
总而言之,本发明公开一种设计正活性材料的独特策略,此类正活性材料具有防止电解质和高反应性核心表面在高电压(通常,4.6v相对于li金属)下直接接触的离子传导性电子绝缘表面,而这抑制了寄生反应(电解质氧化、金属洗提
…
),且所述正活性材料的特点在于在高电压(通常,4.6v相对于li金属)下优异的容量、容量衰退和能量衰退。此外,高离子传导性电子绝缘正活性材料的表面允许快速的li离子转移反应,从而大幅减少不可逆容量,且大幅改善倍率性能。当上文所述的材料配合至li离子二次电池组、li聚合物二次电
池组、或所有固体li离子二次电池组(ssb)中时,此类增强的电化学性质是高度期望的。
[0087]
下文实施例中进一步说明本发明,其中使用于下实验方法:
[0088]
在agilent icp
‑
720es上进行感应耦合电浆光学发射光谱法(icp
‑
oes)以测定阴极材料的元素组成。
[0089]
使用jeol jsm 7100f扫描电子显微镜来进行扫描电子显微法(sem)。电子显微镜配备有来自oxford instruments的50mm
2 x
‑
max
n eds(能量色散x射线光谱仪)传感器。
[0090]
使用配备有cu(k
‑
α)靶x射线管和衍射光束单光仪的rigaku d/max 2200pc衍射计在室温下在15至85的2
‑
θ度范围内进行x射线衍射。不同相的晶格参数从x射线衍射图谱使用全图谱匹配和里特沃尔德精算法(rietveld refinement method)而计算。
[0091]
在配备有loresta gp mcp
‑
t610万用电表的mitsubishi mcp
‑
pd51粉末电阻率测量系统上以4探针构型测量导电性。直接对粉末测量的导电性对应于表面层的导电性。在63.7mpa的施加压力下对粉状阴极材料进行测量。
[0092]
残余的li2co3和lioh碱含量是可通过介于表面与水之间的反应产物的分析而定量测量的材料表面性质。如果将粉末浸没至水中,则发生表面反应。在反应期间,水的ph增加(因为碱性化合物溶解),通过ph滴定来定量碱。滴定结果是“可溶性碱含量(soluble base content)”(sbc)。可如下测量可溶性碱含量:将100ml去离子水添加至20g阴极粉末中,接着搅拌10分钟。通过在搅拌周期期间封闭烧瓶来谨慎地防止空气暴露,因为可能会发生从空气吸收co2的状况而使结果出错。随后通过使用布赫纳(buchner)抽滤移除水溶液,从而获得>90g的澄清溶液,该溶液含有可溶性碱。可溶性碱的含量通过记录在搅拌下以0.5ml/min的速率添加0.1m hcl期间的ph曲线来滴定直至ph达到3。参考电压曲线通过滴定以低浓度溶解于di水中的lioh和li2co3的合适混合物而获得。在几乎所有情况下,都观察到两个不同的平线区。介于ph 8至9之间的具有端点γ1(以ml为单位)的上平线区为oh
‑
/h2o,接着为co
32
‑
/hco3‑
,介于ph 4至6之间的具有端点γ2(以ml为单位)的下平线区为hco3‑
/h2co3。介于第一平线区与第二平线区之间的拐点γ1以及在第二平线区之后的拐点γ2获自ph曲线的微分dph/dvol的对应最小量。第二拐点大致上接近于ph 4.7。随后以lioh和li2co3的重量百分比,且针对sbc以μmol/g为单位表示结果。
[0093]
如下测量压制密度:将3克粉末装填至1.300cm的直径“d”的压片模(pellet die)中。施加2.8吨的单轴负荷(对应于207mpa的压力)30秒。在将负荷释放降至7.4mpa(100kg单轴负荷)之后,测量压制粉末的厚度“t”。随后如下计算压片密度:3/(n
×
(d/2)2×
t),以g/cm3为单位。
[0094]
在将粉末分散于含水介质中之后使用具有hydro 2000mu湿式分散配件的malvern mastersizer 2000测量粒径分布。在psd中,d50值是体积分布的中位数,即以微米为单位的d50是将分布的分开成两部分的大小,其中一半的分布高于此直径,而一半的分布低于此直径。类似地定义d10值和d90值。
[0095]
如下测量钮扣电池中的电化学性质。钮扣电池电极如下制备:将约27.27重量%的活性阴极材料、1.52重量%聚二氟亚乙烯聚合物(kf聚合物l#9305,kureha america inc.)、1.52重量%导电碳黑(super p,erachem comilog inc.)和69.70重量%n
‑
甲基
‑2‑
吡咯啶酮(nmp)(来自sigma
‑
aldrich)以高速均质机密切混合。接着将浆料在铝箔上以带铸法展布成薄层(一般100微米厚)。在蒸发掉nmp溶剂之后,使铸膜通过辊压(使用40微米间
隙)处理。使用圆形冲模切刀(测得直径为14mm)从膜上冲压出电极。接着将电极在90℃下干燥过夜。之后将电极称重以决定活性材料含量。通常,电极含有90重量%的活性材料,活性材料装载重量为约17mg(约11mg/cm2)。随后将电极放在充氩手套箱中,并组装至2325型钮扣电池体内。阳极为具有500微米厚度的锂箔(来源:hosen);分隔件为tonen 20mms微孔聚乙烯膜。钮扣电池填充有溶于碳酸伸乙酯和碳酸二甲酯(1:2体积比)的混合物中的lipf6的1m溶液(来源:techno semichem co.)。各电池在25℃下使用toscat
‑
3100计算机控制恒电流循环站(来自toyo)来进行循环。用于评估核心_1、核心_2和样本1a至样本1g的钮扣电池测试表详述于表4中。如下,此表使用160ma/g的1c电流限定(definition)且包括2个部分:
[0096]
(i)部分i在4.3v至3.0v/li金属窗口范围内在0.1c(在10hr内充分放电)、0.2c、0.5c、1c(1c对应于在1hr内的充分放电)、2c和3c下评估倍率性能。除了第1循环(其中初始充电容量cq1和放电容量dq1以恒定电流模式(cc)测量)之外,所有后续循环的特点在于在0.05c的端电流标准(end current criterion)的充电期间为恒定电流
‑
恒定电压。在各充电与放电之间允许第一循环有30分钟的静止时间,而所有后续循环有10分钟的静止时间。不可逆容量qirr.以%为单位表示为:
[0097][0098]
在0.2c、0.5c、1c、2c和3c下的倍率性能如下表示为分别nc=0.2c、0.5c、1c、2c和3c的保持的放电容量dqn(其中n=2、3、4、5和6)之间的比率:
[0099]
例如
[0100]
(ii)部分ii是在0.5c下的循环寿命评估。在4.6v/li金属下的放电容量在0.1c下在循环7和在1c下在循环8测量。在0.1c和1c下的容量衰退如下计算,且以每100次循环的%来表示:
[0101]
以%/100次循环为单位,
[0102]
以%/100次循环为单位。
[0103]
在0.1c和1c下的能量衰退如下计算,以%/100次循环为单位来表示。是在第n次循环的平均电压。
[0104]
以%/100次循环为单位,
[0105]
以%/100次循环为单位。
[0106]
浮动充电方法:
[0107]
在可商购获得的“3m电池组电解质hq
‑
115”的最新技术报告中,浮动充电方法被用来测试新型电解质在高电压下的稳定性。该方法是通过使lco/石墨袋式电池(pouch cell)或18650电池在4.2v和60℃下连续充电900小时而进行。比较充电的下所记录的电流。较高的电流反映出发生了更多副反应,因此这种方法能够识别出在高电压下电池组中发生的寄生反应。在“energy environ.sci.,6,1806(2013)”中,类似的浮动充电方法用于评估在从5v和高达6.3v相对于li金属的高电压下电解质对抗氧化的稳定性。基于上文知识,通过针
对所需的充电电压选择相对稳定的电解质和阳极材料,可使用浮动充电方法来研究阴极材料在高电压下的稳定性,其中可通过漏电流反映来自阴极材料的金属溶解。此外,在“nature comm.,4,2437(2013)”中,来自锂锰氧化物阴极的溶解锰以金属或金属合金的形式沉积在阳极的表面上,而沉积量可通过感应耦合电浆
‑
原子吸收光谱法(icp)检测。这种对阳极的icp实验也可用于研究基于锂钴氧化物的材料的金属溶解问题。因此,与icp测量相关联的浮动充电方法(以下称为“浮动实验(floating experiment)”)是评估基于锂钴氧化物的阴极材料在高电压和高温下的副反应和金属溶解的可行方法。
[0108]
在本发明研究中,进行浮动实验以评估阴极材料在高电压充电下以及在高温(50℃)下的稳定性。在一些实施方案中,所测试的电池构型是钮扣电池,所述钮扣电池组装如下:两个分隔件(来自sk innovation)位于正极(先前所述)与负石墨电极(mitsubishi mpg)之间。电解质是在ec/dm(1:2体积比)溶剂中的1m lipf6。对所制备的钮扣电池进行以下充电方案:首先在恒定电流模式下以c/20倍率渐减(taper)电流(其中1c=160mah/g)将钮扣电池充电至限定的上压(upper voltage)(4.45v相对于石墨),随后在50℃下恒定保持在4.45v的电压(cv模式)120小时。在浮动实验后,将钮扣型电池拆开。通过icp
‑
oes分析阳极和与阳极接触的分隔件,以用于金属溶解分析。
[0109]
x射线光电子光谱法(xps):xps测量是在得自ulvac
‑
phi(q2)的quantera sxm
tm
中进行。测量使用单色alkα辐射和100μm(100瓦特)的光点大小跨1200
×
500μm2的面积扫描而进行。测量角度θ为45
°
;在此设定下,信息深度为大约7nm。通过宽扫描测量,识别存在于表面处的元素。执行精确的窄扫描以测定精确的表面组成。浓度
‑
深度曲线测定通过交替测量和离子轰击(氩离子,v
i
=4kv,光栅3.6
×
3.6mm2,在sio2中的溅射速率:8.8nm/分钟。在其他材料中,溅射速率将为不同的)。使用标准灵敏度因子以将峰面积转化成原子浓度。因此,在绝对意义上(absolute sense)浓度可能会偏离实际值。在溅射深度曲线中,偏差可能由于优先溅射效应而较大。不同元素的检测极限一般为0.1at%;这意味着具有低于0.1at%的原子浓度的元素将无法通过xps观察到,且无法排除其以<0.1at%的浓度存在的状况。
[0110]
实施例1:
[0111]
此实施例将展示包括基于licoo2的粒子(此类基于licoo2的粒子具有电子绝缘表面且包括离子传导结晶li3po4)的材料具有与锂钴氧化物的目前最佳技术相比优异的电化学行为。
[0112]
样本制备:ti和mg掺杂的基于licoo2的材料为以量产规模使用1.075/0.9967/0.0008/0.0025的li/co/ti/mg的摩尔比的li2co3、co3o4、tio2和mgo的混合物而制备。将产物放置于陶瓷盘中,在990℃的温度下于空气中烧结10h。随后将产物压碎并分级,得到约28μm的中位数体积粒径(median volumetric particle size)d50,如图1所示。粉末的压制密度为3.90g/cm3。如此制备的样本称为核心_1。核心_1样本的sem图像显示于图2中(参考:1a)。
[0113]
接着,如下将1mol%li3po4施加在核心_1的表面上:将一定量的核心_1粉末以5:2重量比浸没于去离子水中,并持续搅拌。以1ml/min的速率同时逐滴添加1.02mol/l lih2po4的水溶液和2.04mol/l lioh的水溶液。随后在90℃下于干燥器中蒸发水。因此,lih2po4沉淀至li3po4粒子中,并沉积于核心_1粒子的表面上。如此制备的样本称为核心_2。核心_2样本的sem图像显示于图2(参考:1b)中,并显示出核心_2粒子被沉淀的li3po4薄片均匀覆盖,其中典型的长度尺寸为约500nm。
[0114]
接着,如下制备样本1a:将一定量的核心_2的粉末以(核心_2):co(co3o4)=0.8696:0.1304的摩尔比与精细的co3o4粉末(该精细的co3o4粉末具有典型的d10、d50和d90粒径体积(particle sizes in volume),根据累积体积粒径分布确定,此类粒径在d10<3μm、d50=3至5μm、且d90<10μm的范围内,可商购自umicore)混合,并且还添加mgo和tio2,以便获得0.9925/0.0025/0.0050的总体co/ti/mg摩尔组成。将97.74重量%的上文所述的混合物和2.26重量%的li2co3进一步共混在一起。将如此制备的共混物放入陶瓷坩埚中,在980℃下于空气中烧12h。随后将烧结产物压碎并分级,得到样本1a。样本1b以与样本1a类似的方式制备,不同之处在于添加2.63重量%li2co3。样本1c以与样本1a类似的方式制备,不同之处在于添加2.99重量%li2co3。样本1d以与样本1a类似的方式制备,不同之处在于添加3.17重量%li2co3。样本1e以与样本1a类似的方式制备,不同之处在于添加3.35重量%li2co3。样本1f以与样本1a类似的方式制备,不同之处在于添加3.71重量%li2co3。样本1g以与样本1a类似的方式制备,不同之处在于添加4.07重量%li2co3。
[0115]
核心_1以及样本1a、样本1c和样本1e的粒径分布显示于图1中,且其d5、d50和d90值于表1中给出。核心_1显示d50接近于28μm的对称粒径分布。实施例1a、实施例1c和实施例1e的特点在于双峰粒径分布,其中第一模式接近4μm且是由于精细co3o4粒子试剂的引入所致,而第二模式接近28μm,其归因于核心_1。在此类构型中;大粒子与精细粒子之间的模式比为约7。化学icp分析的结果列举于表2中。在目标mg、ti和p含量与实验mg、ti和p含量之间发现非常好的一致性。对于样本1a至样本1g,当所添加的li2co3的重量%增加时,li:co的比率和li:(co 3p)的比率两者均增加。样本1c、样本1d和样本1e的li:(co 3p)的比率接近于1。
[0116]
结果和讨论:
[0117]
fesem分析:
[0118]
图2显示核心_1、核心_2以及样本1c和样本1e在5000倍(5000
×
)放大倍率下的sem图像。核心_1表面是光滑的。核心_2图像显示li3po4薄片在粒子表面上的均匀沉淀和沉积。样本1c和样本1e的图像显示,在热处理之后粒子表面恢复光滑方面。在样本1c和样本1e的图像上所观察到的精细粒子源自于co3o4(d50约3μm)的添加。
[0119]
xrd分析:
[0120]
图3显示核心_1、核心_2以及样本1a、样本1b、样本1c、样本1e和样本1f的xrd图谱。为了清晰起见,衍射强度以对数标度显示。所有xrd图谱都由良好结晶的六方成层o3型licoo2相的典型反射主导,其中在r
‑
3m空间群中,晶格参数且如li3po4沉淀于核心_1粒子表面上之后所预期,核心_2的xrd图谱显示了在22.3和23.1的2θ度数处,附加存在的弱且宽的峰,所述峰为li3po4(120)和(101)反射的特征峰。针对所有样本1a至样本1g,观察到归因于li3po4相的良好限定的峰,且使用斜方晶格对li3po4相进行索引,该li3po4相具有pmnb空间群,参数且这些单位晶胞参数对应于li3po4的“γ”多形体。针对样本1a和样本1b;观察到尖晶石co3o4相的杂质峰。针对将较大量的li2co3添加至共混物中的样本1c至样本1g,未观察到此类尖晶石峰。这意味着,共混物中可得的锂不足以用于在样本1a和样本1b中将尖晶石co3o4试剂转化成o3型成层氧化物。至于样本1c,获得了尖晶石co3o4试剂变成o3型成层氧化物的完全转化;此观察符合
icp滴定数据,该数据显示样本1c具有接近于1.00的li:co的摩尔比和li:(co 3p)的摩尔比。
[0121]
传导性:
[0122]
表1列举核心_1、核心_2和样本1a至样本1g的导电性。核心_1和核心_2具有高于10
‑3s/cm的导电性且符合针对掺杂licoo2所报告的值。样本1c的传导性达最小值,且比核心_1和1g样本低3个数量级。此类显著的传导性下降对于基于licoo2的材料而言是意外的且非预期的。
[0123]
可溶性碱含量ph滴定:
[0124]
表1列举针对样本1a至样本1g通过ph滴定所测定的残余lioh和li2co3量(以重量%为单位)以及sbc。可溶性碱含量从样本1g至样本1c迅速减小,其中样本1c达到9.9μmol/g的最小值。lioh重量%遵循相同的行为,且1c的lioh是最小的,具有0.0002重量%的值。可溶性li2co3含量从样本1a至样本1d迅速减小,且随后0.3重量%至0.35重量%附近稳定。
[0125]
压制密度:
[0126]
表1列举核心_1和样本1a至样本1g的压制密度。样本1a至样本1g的压制密度比核心_1高约0.1g/cm3。此压制密度增加是由于粒径分布的具体修饰;即引入双峰特点以用较小粒子填充大核心_1粒子之间之间隙位点,因此增加装填密度。结晶学中熟知的是,设想具有半径“r”的球形粒子的均匀且理想的紧密装填;八面体间隙位点、四面体间隙位点和三角间隙位点仅为分别具有0.414
×
r、0.225
×
r和0.155
×
r的最大直径的较精细粒子可得的。将此类考虑施加至样本1a至样本1g;其是具有约14μm直径的大粒子(源自于核心_1)并具有约1.5至2.5μm直径的较精细粒子(源自于co3o4)的混合物;分别具有5.8μm、3.2μm和2.2μm的半径所有八面体间隙位点、四面体间隙位点和三角间隙位点对样本1a至样本1g的粒子都为可得的。结果填充间隙位点允许样本1a至样本1g相比于核心_1具有较大填充密度。
[0127]
xps:
[0128]
样本1c所检测到的元素列表显示表6中。一些有机c(峰接近284.8ev)存在于样本的表面处。这对于储存于环境空气中的样本而言是正常的。在795ev处的钴2p
1/2
xps峰是licoo2的co
3
离子的特征峰,且在xps at%分辨率和灵敏度范围内,排除了粒子表面处co
2
的显著存在。对于样本1c,观察到在约49.5ev处的解析良好的mg 2p xps峰;这对于在氧环境中的mg诸如mgo或包含mg的磷酸盐而言是典型的。根据共混物组成,预期的标称mg/co的原子比为0.0050/0.9925=0.0054。通过xps所测量的样本1c的粒子表面上的mg/co的原子比为1.4/9.4=0.1489,大于预期的共混物中标称mg/co的原子比的27倍。同样地,在接近458ev观察到解析良好的ti 2p峰。这些结合能量与六倍氧环境中仅存有ti
4
的状况非常一致,如在诸如tio2和li2tio3的化合物或包含ti的磷酸盐中所发现的。根据共混物组成,预期的标称ti/co的原子比为0.0025/0.9925=0.0025。通过xps所测量的样本1c的粒子表面上的ti/co原子比为2.0/9.4=0.2127,高于预期的标称ti/co的原子比约2个数量级。
[0129]
在约133.6ev观察到p 2p峰,该峰为
–
po
43
‑
基团的特征峰。此外,通过xps所测量的样本1c的粒子表面上的p/co原子比为2.1/9.4=0.2234,高于预期的标称p/co的原子比约1个数量级。通过xps深度曲线对样本1c监测ca、mg、na、p、s和ti对co的摩尔比随粒子深度而变动的演变,如图13所示。在样本1c的粒子表面下50nm处,s、mg、na和ca对co的摩尔比趋于“0”,表明这些元素的原子含量低于xps的检测极限。此外,接近450nm深度处,ti/co的摩尔
比减小且等于“0”,表明ti的原子含量低于xps的检测极限。预期的标称mg和ti对co的原子比与表面xps mg和ti对co的原子比之间的巨大差异表面,mg和ti的原子分布为不均匀的且为偏析的:样本1c的粒子表面相较于核心为mg和ti富集的,同时核心的元素仍存在。此类元素偏析解释了样本1c的电子绝缘行为。磷酸盐分布在0与50nm深度之间快速减小,且随后一致地减小直至600nm,材料的核心具有低于0.05mol/mol的p对co的摩尔比。快速的初始减小可能通过以下来解释:只有非常薄的磷酸盐化合物(诸如li3po4)层均匀地覆盖表面;典型的厚度小于20nm,且被认为在0.01nm至10nm的范围内。50nm与600nm之间的线性减小最可能是由于大的li3po4粒子附接于样本1c的表面上所造成,如通过sem和xrd所证明。
[0130]
钮扣电池:
[0131]
表3显示核心_1和核心_2以及样本1a至样本1g的钮扣电池性质。图4_1、图4_2、图4_3和图4_4分别显示核心_1、核心_2、样本1c和样本1g的详细的钮扣电池曲线。在各左侧图中,从右至左明显地给出循环1至循环6的倍率性能。在各中间图中,显示容量衰退,比较循环7与循环31和循环8与循环32,其中从右至左为循环7、循环8、循环31和循环32;不同之处在于,在图4_3中,循环顺序为7、31、8和32。在各右侧图中,显示循环稳定性,放电容量dq略微低于充电容量cq。核心_1显示在4.3v处的cq1、dq1和倍率性能以及在4.6v处的循环稳定性两者的目前最佳技术的电化学性能现状。这些性能是差的,不允许此类目前最佳技术材料带有相当好的性能地在4.6v下操作。经li3po4涂布的核心_2性能甚至更差,其具有大幅加速的4.6v的容量和能量衰退。样本1a至样本1g的电化学性能在许多方面中是显著的,且带来相比于基于锂钴氧化物的材料的目前最佳技术巨大的改善。从样本1g、样本1f、样本1e、样本1d至样本1c,电化学性能持续改善,其中cq_1、cq_7和3c倍率持续地增加,且在0.1c和1c处的qirr.、qfad.和efad.被最小化。样本1c至样本1a显示电化学性质的稳定化,并保持杰出的性能水平。图5显示dq_7和1c efad.随导电子传导性而变动的演变。样本1d的dq_7是最大的,其具有<10
‑4s/cm的传导性。1c efad.持续减小,且样本1c的1c efad.是最小的,其具有<10
‑5s/cm的传导性。
[0132]
实施例2:
[0133]
此实施例将展示包括基于licoo2的粒子(此类基于licoo2的粒子具有电子绝缘表面且包括离子传导结晶li3po4)的材料具有与仅电子绝缘的材料相比优异的电化学行为。
[0134]
样本制备:ti和mg掺杂的基于licoo2的材料为以量产规模使用1.060/0.9967/0.0008/0.0025的li/co/ti/mg的摩尔比的li2co3、co3o4、tio2、mgo的混合物而制备。将产物放置于陶瓷盘中,在990℃的温度下于空气中烧结10h。随后将产物压碎并分级,从而得到约18μm的中位数粒径体积d50(median particle size in volume d50)。如此制备的样本称为核心_3。图6a显示核心_3的sem图像。
[0135]
接着,如下将1.85mol%li3po4施加于核心_3的表面上:首先,将氢氧化锂单一水化物和磷酸(h3po4,由wako chem.ltd.生产)添加至去离子水中以提供10重量%lih2po4水溶液。将2公斤(2
‑
kilos)的co(核心_3)/co(co3o4)=0.8696/0.1304的摩尔比的核心_3粉末和co3o4(其中d50约3.5μm)材料的混合物放置于滚筒流体化涂布设备(tumbling fluidized coating apparatus)(来自powrex corp.的mp
‑
01mini)中。在90℃的热空气下将lih2po4的水溶液喷洒于粉状材料混合物的表面上,并在粉状材料混合物的表面上干燥。通过旋转片(300rpm)和0.4nm3/min的空气流来流体化该粉状材料混合物。将如此制备的溶液以5g/min
的恒定速率给料至喷雾嘴,且以60l/min的雾化空气进行喷雾。因为在喷雾期间,粉状材料混合物被热空气流体化,所以喷雾溶液的水性溶剂立即蒸发。如此制备的样本称为核心_4。核心_4的sem图像显示于图6b中,且显示核心_3粒子被“lipo
3”或“lih2po
4”的均匀膜均匀地覆盖。
[0136]
接着,如下制备样本2a。将核心_4材料的粉末与mgo、tio2和al2o3混合以获得0.9919/0.0035/0.0028/0.0018的总体co/ti/mg/al摩尔组成。接着,将95.48重量%的上文所述的混合物和4.52重量%的li2co3进一步共混在一起。将如此制备的共混物放入陶瓷坩埚中,在980℃下于空气中烧10h。随后将烧结产物压碎并分级,得到样本2a。图6c显示样本2a的sem图像。样本2a的icp数据列举于表2中。
[0137]
如下制备样本2b:将核心_3粉末和co3o4(其中d50约3.5μm)材料以co(核心_3)/co(co3o4)=0.8696/0.1304的摩尔比(当认为核心_3和co3o4的钴重量含量分别为60.21%和73.42%时,等效于0.8905/0.1095的重量比)混合。进一步添加mgo、tio2和al2o3以获得0.9919/0.0035/0.0028/0.0018的总体co/ti/mg/al的摩尔组成。mg、ti和al掺杂物的组成与样本2a相同。接着,将96.94重量%的上文所述的混合物和3.06重量%的li2co3进一步共混在一起。将如此制备的共混物放入陶瓷坩埚中,在980℃下于空气中烧10h。随后将烧结产物压碎并分级,得到样本2b。样本2b的icp数据列举于表2中;li:(co 3p)的摩尔比和li:(co 3p al mg ti)的摩尔比非常接近于样本2a,这使得得以可靠地比较其物理和电化学性质。
[0138]
图7显示样本2a和样本2b的xrd图谱。两个xrd图谱均显示典型的o3成层锂钴氧化物(空间群r
‑
3m)的反射。在两种样本中,均观察到痕量的co3o4尖晶石杂质,估计该co3o4尖晶石杂质低于0.5重量%。类似量的co3o4杂质的存在显示,两种样本中的锂含量非常类似,且两种样本均以良好控制的方式是略微li亚化学计量的(li
‑
sub
‑
stoichiometric)。样本2a的xrd图谱显示样本中良好结晶的li3po4相的存在。根据xrd分析作出结论,样本2b与样本2a的不同之处仅在于:不存在附接至样本2b表面的结晶li3po4。样本2a和样本2b的传导性分别为1.78
×
10
‑5s/cm和5.63
×
10
‑5s/cm。在钮扣电池0.1c和1c这两种情况下,能量衰退是优异的,样本2a衰退速率分别为每100个循环14.4%和33.8%,而样本2b衰退速率分别为每100个循环22.1%和46.5%。
[0139]
图8显示样本2a(下线)和样本2b(上线)的在t=0小时与t=120小时之间的浮动电流i(t)的演变。表5列举以mah/g为单位的浮动容量,该浮动容量通过对0与120小时之间的浮动电流进行积分来计算;以及co溶解,该co溶解通过对阳极和对与阳极接触的分隔件进行icp
‑
oes而测量。样本2a显示出相比于样本2b低6.2倍的浮动容量和低约100倍的co溶解。
[0140]
eds表面映像针对样本2a在30
×
40μm2表面上进行。图9a、图9b和图9c显示进行eds映像的样本表面。co和p元素的空间分布示于图9b和图9c中。图10显示所研究表面的映像和谱图(map sum spectrum)。富集磷的域似乎是分开的,但又致密地附接至锂钴氧化物粒子的表面。此sem/eds观察与xrd一致,且透露致密地附接在锂钴氧化物粒子表面上的li3po4域的存在。
[0141]
以与针对样本1c相同的方式对样本2a进行xps分析。样本2a的结果显示于表6和图14中。同样地,表面分析和深度曲线显示mg和ti原子分布为不均匀的且为偏析的:样本2a的粒子表面相较于核心为mg(直至25nm)和ti(直至200nm)富集的。磷酸盐分布在0与25nm深度之间快速减小,且随后一致地减小直至600nm,材料的核心具有低于0.05mol/mol的p对co的
摩尔比。快速的初始减小可能通过以下来解释:只有非常薄的磷酸盐化合物(诸如li3po4)层均匀地覆盖表面;典型的厚度被认为低于20nm,且可能在0.01nm至10nm的范围内。50nm与600nm之间的线性减小最可能是由于大的li3po4粒子附接于样本2a的表面上所造成,如通过sem、eds映射和xrd所证明。
[0142]
实施例3:
[0143]
此实施例将展示,施加至基于licoo2的粒子表面上的li3po4涂层的形态受退火温度的选择的影响。在低于850℃的温度下,基于licoo2的粒子覆盖有连续的li3po4层。在高于850℃的温度下,基于licoo2的粒子表面包括li3po4岛状物;根据岛状物中间的非常光滑的粒子表面推断,li3po4岛状物之间存在连续的保护膜。
[0144]
将2mol%li3po4施加于25μm的基于licoo2的核心的表面上。图11显示li3po4涂布之前(样本3a)和li3po4涂布之后(样本3ac)的粒子表面。随后将100g如此制备的混合物在700℃下于干燥空气中退火10h,并冷却至室温。将产物粉碎并分级,得到样本3b。以与样本3b类似的方式制备样本3c,不同之处在于退火温度设定成800℃。以与样本3b类似的方式制备样本3d,不同之处在于退火温度设定成900℃。以与样本3b类似的方式制备样本3e,不同之处在于退火温度设定成980℃。
[0145]
图12显示样本3a至样本3e的xrd图谱。表7概述了根据里特沃尔德精算法所获得的具有空间群r
‑
3m的o3成层相和li3po4相的晶格参数。样本3ac和样本3a至样本3e的sem图像显示于图11中。样本3ac粒子表面被li3po4薄片均匀且连续地覆盖。将热处理从700℃增加至980℃允许li3po4化合物结晶化并使其致密化附接至粒子表面直至发生聚结,同时形成岛状物。如先前实施例1中所示;基于岛状物中间的极光滑的粒子表面,可预期在li3po4岛状物形成之后,粒子表面上和li3po4岛状物之间存在li3po4的薄连续层。
[0146]
[0147]
[0148]
[0149]
[0150]
[0151]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。