1.本发明属于医药质量分析技术领域,具体涉及一种治疗乳腺增生、乳腺炎的药物组合物的检测方法,特别是涉及一种同时检测乳辟舒胶囊中多种有效成分的含量测定方法。
背景技术:
2.乳腺增生、乳腺炎是女性最常见的乳腺疾病,近年来该病的发病率呈逐年上升趋势,其中乳腺增生的发病率占乳腺疾病的首位;根据调查数据显示,我国患乳腺增生的女性逐年递增,中国女性乳腺增生发病率从70%递增到80%,乳腺增生占所有乳腺疾病增长率也增加到75%,在育龄女性中占40%。
3.目前治疗乳腺增生和乳腺炎的方法有很多,常用的西药如口服激素类药物或通过手术治疗,口服激素类药物虽然能够减轻一些症状,但很难达到彻底治愈的目的,药物副作用大,且有可能增加其癌变。中药治疗以疏肝解郁、散结化瘀为主,临床常用药有乳癖舒胶囊、乳癖消片及小金胶囊等,其中乳癖舒胶囊为西安千禾药业股份有限公司生产,其处方由丹参、瓜蒌、赤芍、土贝母、蒲公英、延胡索及柴胡7味药物组成,主要用于肝气郁结,毒瘀互阻所致的乳腺增生、乳腺炎。该品种因其疗效确切在临床试验过程中深受患者和医务工作者的认可,但是现行国家药品标准ybz05022017中仅对处方中丹参、土贝母和赤芍进行了单一成分的薄层鉴别,对赤芍中芍药苷进行了含量测定,未对处方中其他关键药味进行质量控制。专利cn201911250147.5公布了“一种治疗乳腺增生、乳腺炎的药物组合物乳癖舒胶囊的检测方法”但该专利也仅对处方中瓜蒌皮和蒲公英中共有有效成分槲皮素进行了含量测定并不能全面反映药品的质量情况,因此有必要继续完善该产品的检测方法,确保该产品的质量和临床疗效。
技术实现要素:
4.本发明的目的是克服现有技术的不足,提供一种同时检测乳辟舒胶囊中多种有效成分的含量检测方法,该方法对处方中土贝母中有效成分土贝母苷甲,柴胡中有效成分柴胡皂苷a、柴胡皂苷d,延胡索中有效成分延胡索乙素,丹参中有效成分丹参酮ⅱa、丹酚酸b,赤芍中有效成分芍药苷,共计7种有效成分同时进行测定。该检测方法能够弥补现有方法的不足,提供一种更为全面、有效的药物控制质量标准,进一步有效保障产品质量。
5.为实现上述发明目的,本发明采用如下技术方案:
6.一种同时检测乳辟舒胶囊中多种有效成分的含量检测方法,所述药物组合物由以下七味药材制备而成:丹参、瓜蒌皮、赤芍、土贝母、蒲公英、延胡索、柴胡;采用液相色谱方法同时测定土贝母中有效成分土贝母苷甲、柴胡中有效成分柴胡皂苷a、柴胡皂苷d、延胡索中有效成分延胡索乙素、丹参中有效成分丹参酮ⅱa、丹酚酸b、赤芍中有效成分芍药苷,其包含以下步骤:
7.(1)色谱条件与系统适用性试验
8.以十八烷基硅烷键合硅胶为填充剂;以有机溶剂为a相、一定浓度的稀酸水溶液为b相,梯度洗脱,所述有机溶剂为选自甲醇、乙腈或乙腈
‑
甲醇混合液,所述稀酸水溶液选自重量或体积百分浓度0.01%
‑
1.0%的磷酸、甲酸、冰乙酸或以上三种的混合;柱温10
‑
40℃;二极管阵列检测器(pda),测定土贝母苷甲、柴胡皂苷a和柴胡皂苷d检测波长210
‑
216nm;测定延胡索乙素检测波长275
‑
285nm;测定丹参酮ⅱa检测波长265
‑
275nm;测定丹酚酸b检测波长280
‑
290nm;芍药苷检测波长225
‑
235nm;流速为0.8ml/min
‑
1.2ml/min;进样体积为10
‑
20μl;理论板数均应不低于3000;
9.优选的,所述a相为乙腈,b相为体积百分浓度0.2%的磷酸水溶液;流速为0.85ml/min;柱温为20℃;测定土贝母苷甲、柴胡皂苷a和柴胡皂苷d检测波长为212nm;测定延胡索乙素、丹参酮ⅱa检测波长为275nm;测定丹酚酸b检测波长为286nm;测定芍药苷检测波长为230nm;理论板数均应不低于3000;
10.优选的,梯度洗脱时,梯度洗脱程序为:0~10min,20%a;10~25min,20~60%a;25~35min,60%a;35~50min,60~20%a;50~60min,20%a;具体梯度洗脱程序为:
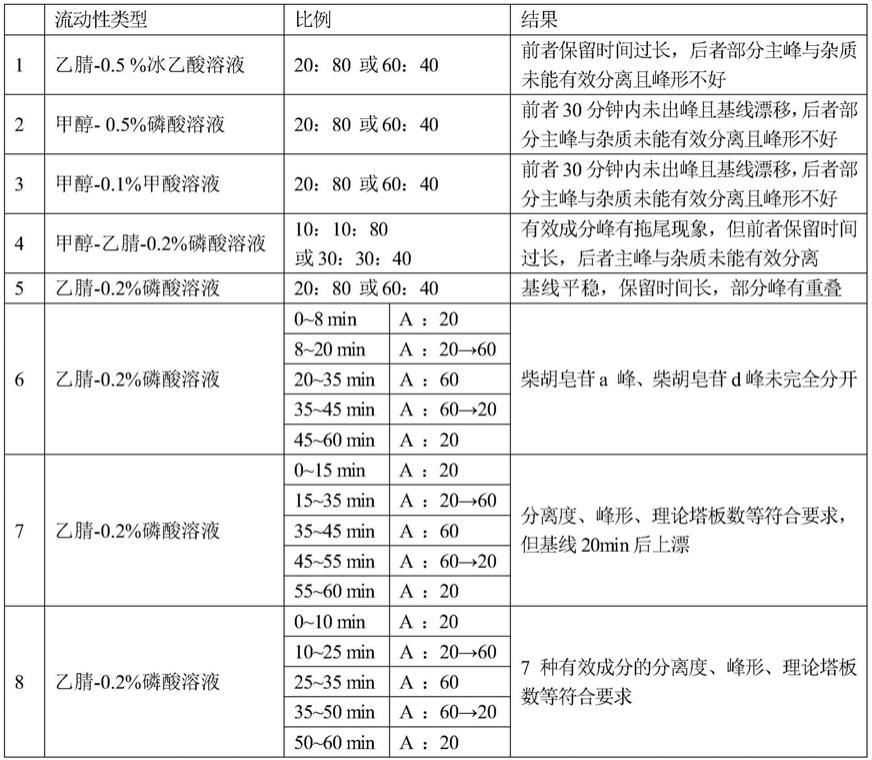
11.时间(分钟)流动相a(%)流动相b(%)0~10208010~2520
→
6080
→
4025~35604035~5060
→
2040
→
8050~602080
12.(2)对照品溶液的制备
13.精密称取各对照品适量,分别加80%甲醇制成含土贝母苷甲0.3mg/ml、柴胡皂苷a0.65mg/ml、柴胡皂苷d0.8mg/ml、延胡索乙素0.1mg/ml、丹参酮ⅱa 0.045mg/ml、丹酚酸b 0.2mg/ml、芍药苷0.7mg/ml的贮备液;依次精密吸取上述7种贮备液各1ml置于同一10ml棕色容量瓶中,加80%甲醇溶解并定容,摇匀,作为质量浓度分别为30、65、80、10、4.5、20、70μg/ml的混合对照品溶液。
14.(3)供试品溶液的制备
15.取乳癖舒胶囊内容物,研细,称取0.3
‑
1.0g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20
‑
50ml,称定重量,超声处理(250w,50khz)30
‑
50min,放冷,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
16.优选的,取乳癖舒胶囊内容物,研细,称取0.4g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20ml,称定重量,超声处理(250w,50khz)40min,放冷,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
17.(4)测定
18.分别精密吸取对照品溶液与供试品溶液各10
‑
20μl,注入高效液相色谱仪,测定,即得测定结果。
19.本发明中乳癖舒胶囊具体处方为:瓜蒌皮450g、土贝母150g、蒲公英450g、丹参225g、赤芍225g、延胡索135g、柴胡135g。
20.与现有技术相比,本发明具有以下有益的技术效果:
21.本发明乳癖舒胶囊的含量测定方法,在原质量标准ybz05022017仅对土贝母、丹参
和赤芍进行单一成分薄层鉴别,对赤芍进行芍药苷的含量测定;并未对其余药味进行有效成分测定,现行质量控制标准专属性差,不能有效保障产品的质量和疗效的基础上进一步完善该产品的检测方法,增加了对处方中土贝母的有效成分土贝母苷甲、柴胡的有效成分柴胡皂苷a、柴胡皂苷d、延胡索的有效成分延胡索乙素、丹参的有效成分丹参酮ⅱa、丹酚酸b、赤芍的有效成分芍药苷,共计7种有效成分同时进行含量测定的方法,采用波长切换技术进行检测,用二极管阵列检测器综合考虑不同有效成分最强吸收且杂质干扰较小的波长进行波长切换检测。可在一张色谱图中较好的对测定组分进行分离;去除干扰杂质,有利于组分含量准确计算,该检测方法灵敏度高、专属性强、准确度及重现性好,能够弥补现有方法的不足,同时实现一次测定多个有效成分,节约检验时间提升检测效率,有效保障产品的质量。
附图说明
22.下面结合附图对本发明作进一步的说明:
23.图1
‑
2为本发明在色谱条件下专属性试验色谱图,图中,a为混合对照品的色谱图,b为供试品的色谱图,c为缺赤芍的阴性样品色谱图,d为缺延胡索的阴性样品色谱图,e为缺丹参的阴性样品的色谱图,f为缺土贝母的阴性样品的色谱图,g为缺柴胡的阴性样品的色谱图;其中1为芍药苷峰、2为延胡索乙素峰、3为丹酚酸b峰、4为土贝母苷甲峰、5为柴胡皂苷a峰、6为柴胡皂苷d峰、7为丹参酮ⅱa峰。
24.图3
‑
4为7种有效成分的线性关系图。
具体实施方式
25.以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原辅料、试剂材料等,如无特殊说明,均为市售购买产品。
26.实施例1:乳癖舒胶囊的处方及制备方法
27.处方:瓜蒌皮450g、土贝母150g、蒲公英450g、丹参225g、赤芍225g、延胡索135g、柴胡135g
28.制法:以上七味,取延胡索粉碎成细粉;丹参、赤芍加乙醇回流提取二次,每次加5倍量乙醇,各提取2小时,合并乙醇提取液,滤过,滤液回收乙醇并浓缩至相对密度为1.33~1.36(60℃)的稠膏;瓜萎皮等其余四味,加水煎煮三次,第一次加10倍量水,煎煮2小时,第二次加8倍量水,煎煮1.5小时,第三次加6倍量水,煎煮1小时,合并煎液,滤过,滤液减压浓缩至相对密度为1.33~1.36(60℃)的稠膏,合并上述稠膏,加入延胡索细粉、二氧化硅12g、预胶化淀粉8g,搅匀,真空干燥,粉碎成细粉,装胶囊,制成1000粒,即得。
29.实施例2:检测波长的选择
30.通过扫描土贝母苷甲、柴胡皂苷a、柴胡皂苷d、延胡索乙素、丹参酮ⅱa、丹酚酸b、芍药苷7种对照品溶液的紫外吸收光谱,并参考《中国药典》2020年版一部可知,土贝母苷甲在214nm有最大吸收,柴胡皂苷a、柴胡皂苷d在210nm有最大吸收,延胡索乙素在280nm有最大吸收、丹参酮ⅱa在270nm有最大吸收,丹酚酸b在286nm有最大吸收,芍药苷在230nm有最
大吸收;因土贝母苷甲、柴胡皂苷a、柴胡皂苷d最大吸收波长相近,结合相关文献并综合考虑方法的简便性,最终将土贝母苷甲、柴胡皂苷a、柴胡皂苷d的检测波长确定为212nm;同理将延胡索乙素和丹参酮ⅱa的检测波长确定为275nm;丹酚酸b和芍药苷因其最大吸收波长与其他成分差异较大,综合考虑方法的含量灵敏度和准确度采用原最大吸收波长为检测波长。试验表明,采用二极管阵列检测器(pda)波长切换技术进行检测,用二极管阵列检测器综合考虑不同有效成分最强吸收且杂质干扰较小的波长进行波长切换检测。可在一张色谱图中较好的对测定组分进行分离;去除干扰杂质,有利于组分含量准确计算。
31.实施例3:流动相的选择
32.为使供试品中7种有效成分能得到有效的分离,同时峰形、理论塔板数等达到要求,预试验分别采用乙腈
‑
0.5%冰乙酸溶液、甲醇
‑
0.5%磷酸溶液、甲醇
‑
0.1%甲酸溶液、甲醇
‑
乙腈
‑
0.2%磷酸溶液、乙腈
‑
0.2%磷酸溶液等不同比例的流动性进行试验。结果表明,以乙腈
‑
0.2%磷酸水溶液线性梯度分离效果最好,基线最平稳,故最终选择流动相a相为乙腈,流动相b相为体积百分浓度0.2%的磷酸水溶液。详细结果见表1。
33.表1流动相选择试验结果
[0034][0035]
如表1的流动相选择试验结果表明:采用乙腈
‑
0.2%磷酸水溶液组成的流动相体系,在进行梯度洗脱的条件下分离效果最好,基线平稳,7种有效成分成分的色谱峰可完全分离,各组分出峰时间、峰型和分离度均符合要求,故该条件能满足分析测定的相关要求。
[0036]
实施例4:专属性研究
[0037]
4.1各阴性样品的制备:
[0038]
按样品的处方工艺和配方比例(实施例1),分别制备不含土贝母、丹参、延胡索、柴胡和赤芍的阴性样品;如丹参阴性样品的制备:取延胡索粉碎成细粉;赤芍加乙醇回流提取二次,每次加5倍量乙醇,各提取2小时,合并乙醇提取液,滤过,滤液回收乙醇并浓缩至相对密度为1.33~1.36(60℃)的稠膏;土贝母、瓜蒌皮、蒲公英、柴胡,加水煎煮三次,第一次加10倍量水,煎煮2小时,第二次加8倍量水,煎煮1.5小时,第三次加6倍量水,煎煮1小时,合并煎液,滤过,滤液减压浓缩至相对密度为1.33~1.36(60℃)的稠膏,合并上述稠膏,加入延胡索细粉、二氧化硅12g、预胶化淀粉8g,搅匀,真空干燥,粉碎成细粉,即得。
[0039]
4.2阴性样品溶液的制备:
[0040]
分别称取各阴性样品0.4g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20ml,称定重量,超声处理(250w,50khz)40min,放冷,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0041]
4.3供试品溶液的制备:
[0042]
试验方法:取同一批号胶囊内容物,研细,称取0.4g,精密称定,置具塞锥形瓶中,精密加提取溶剂20ml,称定重量,加热回流或超声提取,放冷,补足重量,滤过,取续滤液,即得;分别精密吸取对照品溶液与供试品溶液各10μl,注入高效液相色谱仪,测定,即得测定结果。结果详见表2。
[0043]
方法1:精密加甲醇20ml,精密称定,超声提取30min,放冷补足重量,过滤;
[0044]
方法2:精密加80%甲醇20ml,精密称定,超声提取30min,放冷补足重量,过滤;
[0045]
方法3:精密加60%甲醇20ml,精密称定,超声提取40min,放冷补足重量,过滤;
[0046]
方法4:精密加80%甲醇20ml,精密称定,加热回流提取60min,放冷补足重量,过滤;
[0047]
方法5:精密加80%甲醇20ml,精密称定,加热回流提取90min,放冷补足重量,过滤;
[0048]
方法6:精密加80%甲醇20ml,精密称定,超声提取40min,放冷补足重量,过滤;
[0049]
方法7:精密加80%甲醇20ml,精密称定,超声提取50min,放冷补足重量,过滤;
[0050]
方法8:精密加80%甲醇30ml,精密称定,超声提取60min,放冷补足重量,过滤;
[0051]
表2供试品制备方法的选择试验结果
[0052][0053]
结果表明:采用方法1时丹酚酸b提取不充分,峰面积明显低于其他方法;采用方法2时各有效成分能得到有效提取,但峰面积略低于方法6,表明超声提取30min有效成分未能
100%转移;采用方法3时因甲醇浓度较低各有效成分峰面积明显较低,说明有效成分转移率较低;采用方法4时和方法5时丹参酮ⅱa峰面积较小,可能因丹参酮ⅱa在受热过程中存在损失;采用方法7和方法8时峰面积与采用方法6时无显著性差异,但其超声提取较长;故确定最佳的供试品溶液的制备方法为:取乳癖舒胶囊内容物,研细,称取0.4g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20ml,称定重量,超声处理(250w,50khz)40min,放冷,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
[0054]
4.4色谱条件与系统适用性试验
[0055]
以zorbax eclipse xdb c 18柱(250mm
×
4.6mm,5μm)为色谱柱;按表1中确定的流动相,以流速为0.9ml/min;柱温为20℃;土贝母苷甲、柴胡皂苷a和柴胡皂苷d测定的检测波长为212nm;延胡索乙素、丹参酮ⅱa测定的检测波长为275nm;丹酚酸b测定的检测波长为286nm;芍药苷测定的检测波长为230nm;流速为0.9ml/min;进样体积为10μl;理论板均应不低于3000;
[0056]
对照品溶液的制备:
[0057]
精密称取各对照品适量,分别加80%甲醇制成含土贝母苷甲0.3142mg/ml、柴胡皂苷a0.6644mg/ml、柴胡皂苷d0.8104mg/ml、延胡索乙素0.1016mg/ml、丹参酮ⅱa0.0446mg/ml、丹酚酸b0.2224mg/ml、芍药苷0.7086mg/ml的贮备液;依次精密吸取上述7种贮备液各1ml置于同一10ml棕色容量瓶中,加80%甲醇溶解并定容,摇匀,作为质量浓度分别为31.42,66.44,81.04,10.16,4.46,22.24,70.86μg/ml的混合对照品溶液。
[0058]
测定:
[0059]
分别精密吸取各阴性样品溶液、混合对照品溶液、供试品溶液各10μl,注入perkinelmer altus a
‑
10型高效液相色谱仪(pda检测器,empower工作站,珀金埃尔默企业管理上海有限公司),测定。
[0060]
结果显示:供试品溶液色谱图中,在7种对照品色谱峰相应的位置上有相同的色谱峰;阴性无干扰,且分离度和理论板数均符合要求;表明该色谱条件专属性良好。具体见下附图1
‑
2。
[0061]
实施例5:重复性试验
[0062]
5.1色谱条件与系统适用性试验
[0063]
同实施例4
[0064]
5.2精密吸取同一供试品溶液10μl,注入液相色谱仪,重复进样6次,计算峰面积rsd。4.3结果显示,7种有效成分峰面积的rsd值分别为0.18%、0.28%、0.18%、0.31%、0.20%、0.34%,均小于2.0%;表明精密度良好。具体结果见下表3。
[0065]
表3:重复性实验7种有效成分峰面积数据表
[0066]
[0067]
实施例6:线性范围
[0068]
6.1色谱条件与系统适用性试验
[0069]
同实施例4
[0070]
6.2分别精密吸取各对照品贮备液1.3ml置于10ml棕色量瓶中用80%甲醇定容,得混合对照品溶液,再分别精密吸取上述混合对照品溶液各2μl、5μl、8μl、10μl、15μl、20μl、按实施例3所述色谱条件进行检测,记录色谱图,以峰面积(y)对浓度(x)进行线性回归分析,绘制线性关系图,报告线性方程。
[0071]
结果见表4(7种成分的标准曲线方程和相关系数及线性范围),图3
‑
4为7种成分的线性关系图。
[0072]
表4:7种成分的标准曲线方程和相关系数及线性范围
[0073]
成分名称回归方程r线性范围(μg/ml)芍药苷y=2.19e 05x
‑
8.05e 040.9918.42
‑
184.24延胡索乙素y=7.20e 04x
‑
2.91e 040.992.64
‑
264.16丹酚酸by=2.16e 04x
‑
6.78e 0315.78
‑
57.82土贝母苷甲y=3.77e 04x
‑
1.17e 040.998.17
‑
81.69柴胡皂苷ay=1.40e 05x
‑
6.15e 040.9917.27
‑
172.74柴胡皂苷dy=2.17e 05x
‑
1.23e 05121.07
‑
316.06丹参酮ⅱay=2.72e 03x
‑
8.32e 0211.16
‑
11.60
[0074]
实施例7:准确度(回收率)
[0075]
7.1色谱条件与系统适用性试验
[0076]
同实施例4
[0077]
7.2试品溶液的制备:
[0078]
取乳癖舒胶囊内容物,研细,分别称取0.2g样品各6份,精密称定,置20ml棕色量瓶中,再分别精密吸取芍药苷0.7086mg/ml的贮备液2ml、延胡索乙素0.1016mg/ml的贮备液0.5ml、丹酚酸b 0.2224mg/ml的贮备液5ml、土贝母苷甲0.3142mg/ml的贮备液3ml、柴胡皂苷a0.6644mg/ml的贮备液0.2ml、柴胡皂苷d0.8104 mg/ml的贮备液0.2ml、丹参酮ⅱa 0.0446mg/ml的贮备液5ml,置于上述棕色量瓶中加80%甲醇适量,超声处理(250w,50khz)40min,放冷,用80%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
[0079]
测定:分别精密吸取对照品溶液、各10μl,注入高效液相色谱仪,测得土贝母苷甲、柴胡皂苷a、柴胡皂苷d、延胡索乙素、丹参酮ⅱa、丹酚酸b、芍药苷的回收率和rsd%,结果见表4,标准要求:回收率要求85%~110%,回收率rsd<4.0%;
[0080]
实验结果表明该方法准确度良好。结果见表5。
[0081]
回收率%=(c
‑
a)/b
×
100%,
[0082]
式中a为供试品所含被测成分量;b为加入对照品量;c为实测值。
[0083]
表5回收率试验数据表
[0084][0085]
实施例8:中间精密度
[0086]
8.1色谱条件与系统适用性试验
[0087]
同实施例4
[0088]
8.2在同一实验室,由不同实验人员a和b在不同日期,使用不同色谱仪、进行实验,计算各有效成分含量。结果见表6,结果表明发明专利方法精密度良好。
[0089]
表6:中间精密度试验数据表
[0090][0091]
实施例9:耐用性
[0092]
9.1色谱条件与系统适用性试验
[0093]
同实施例4
[0094]
9.2取同一供试品溶液,室温下放置,分别于0、4、8、12、24小时,精密吸取10μl注入高效液相色谱仪;结果见表6,结果表明,供试品溶液24小时内保峰面积的rsd最大值仅为0.19%,远小于标准2.0%;结果见表7,表明试品溶液在室温条件下36h内稳定。
[0095]
表7:稳定性试验数据表
[0096][0097][0098]
综上所述:本发明乳癖舒胶囊中多种有效成分的含量测定方法,从专属性、重复、准确度(回收率)、中间精密度、耐用性等方面验证了本方法的科学性。通过对处方中5味药材中7中有效成分含量的测定,进一步提升了产品质量保证,保证了乳辟舒胶囊的安全性和有效性,弥补了现有技术的不足。
[0099]
实施例10:稳定性试验
[0100]
10.1色谱条件与系统适用性试验同实施例4
[0101]
10.2取乳辟舒胶囊市售样品(批号:20191001、20191002、20191003,西安千禾药业
股份有限公司生产)进行加速和室温长期稳定性实验。加速实验条件:将样品放置在相对湿度为75%
±
5%,温度为40℃
±
2℃的稳定性试验箱中进行6个月加速实验考察;室温实验条件:将样品放置在相对湿度60%
±
5%,温度25℃
±
2℃的稳定性试验箱中进行长期24个月的实验考察。试验结果见表8。
[0102]
表8:乳癖舒胶囊加速及长期稳定性试验数据
[0103][0104][0105]
实验结果表明,加速试验6个月和长期实验24个月,所检测的有效成分芍药苷、延胡索乙素、土贝母苷甲、柴胡皂苷a、柴胡皂苷d、丹酚酸b、丹参酮ⅱa含量均未发生显著性变化,符合要求,说明该检测方法,能有效控制药物的质量,可考虑纳入质量标准中。另,从连续三批售产品(批号:20191001、20191002、20191003)24个月末的检测结果来看,芍药苷的平均含量为3.21mg/粒、延胡索乙素的平均含量为0.11mg/粒、土贝母苷甲的平均含量为
1.91mg/粒、柴胡皂苷a的平均含量为0.25mg/粒、柴胡皂苷d的平均含量为0.34mg/粒mg/ml、丹酚酸b的平均含量为2.04mg//粒、丹参酮ⅱa的平均含量为0.54mg/粒;考虑到不同原料之间的差异及生产及检验过程种的误差,建议可将含量标准定为:含芍药苷≥2.75mg/粒、延胡索乙素≥0.10mg/粒、土贝母苷甲≥1.65mg/粒、柴胡皂苷a≥0.24mg/粒、柴胡皂苷d≥0.30mg/粒mg/ml、丹酚酸b≥1.76/粒、丹参酮ⅱa≥0.48mg/粒。
[0106]
以上给出的实施例是实现本发明较优的例子,本发明不限于上述实施例。本领域的技术人员根据本发明技术方案的技术特征所做出的任何非本质的添加、替换,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。