转录中介体med31基因及其互作转录因子在提高三角褐指藻中岩藻黄素含量的应用
技术领域
1.本发明属于植物基因工程领域,尤其是涉及转录中介体med31基因、互作转录因子及其编码蛋白和应用。
背景技术:
2.近年来,海洋资源的利用越来越受到重视。海洋硅藻在全球碳循环中发挥着重要作用。三角褐指藻也逐渐产业化生产岩藻黄素,岩藻黄素是一种类胡萝卜素,具有抗氧化、抗炎、抗肥胖、抗糖尿病和抗癌等多种生物学活性,在食品、医药和美容等领域具有广阔的开发前景,因此具有重要的研究意义。
3.生物有机体的遗传信息,是以基因的形式储藏于dna分子上。转录就是将dna序列在由rna聚合酶和转录辅助因子组成的转录复合体的催化下产生与之互补rna序列的过程。通过转录遗传信息得以从dna传递到rna。转录是分子生物学中心法则中最重要的步骤之一,是基因表达的第一步。因此转录几乎是生物体中每个生物学过程的前提条件。
4.原核生物中的转录是在胞浆中进行的,并且蛋白的翻译过程也与之同时进行。在原核生物转录过程中,转录起始因子σ因子能够识别模板dna上的起始位点,它和rna聚合酶结合在起始位点上,形成起始全酶dna-复合物,从而起始转录。原核生物转录调控中的激活蛋白或者是抑制蛋白起作用的方式是直接的,即直接作用于rna聚合酶。真核生物中的转录调控更为精细复杂,但两者的基本构成是一致的,即都有rna聚合酶的存在。但与原核生物不同,真核生物转录的起始必须由转录因子首先识别启动子序列进而介导rna聚合酶与启动子区的结合。真核生物中的转录因子包括基因特异的转录因子和一些通用转录因子(generaltranscriptionfactor,gtf)。此外,真核转录和原核转录最突出的差异还在于,转录因子和rna聚合酶之间的这种信号传递不是直接的。在真核生物中,两者之间存在着多种中间的组分,包括染色质和中介体,它们代表了维系转录调节蛋白和rna聚合酶之间新的一层介质。也正因为如此,真核生物中才能够进行更为精细复杂的转录调控。在基因表达时,rna聚合酶ii(pol ii)以及通用转录因子gtfs(tfiia,tfiib,tfiid,tfiie,tfiif,tfiih)都会被招募到靶标基因的启动子上,形成预起始复合物(pre-initiation complex,pic),进而起始转录。控制真核基因转录的蛋白除了pol ii和各通用转录因子外,还包括了各种与靶基因的启动子和增强子序列特异结合的激活因子和抑制因子,以及各种辅因子,它们在转录因子和预起始复合物之间发挥桥梁作用或酶活催化作用。作为桥梁的中介体是真核生物特有的,并且参与了几乎所有pol ii介导的转录调控。
5.中介体最初是在酵母中由kornberg教授实验室发现的。一直以来人们在研究转录调控时发现一个现象,即过表达一个转录激活子反而会抑制另一转录激活子激活下游的转录,人们把这一现象叫做squelching,意思就是说两个转录激活子可能在竞争性地结合某种转录必需的因子。人们想当然的认为这种转录必需因子就是一些通用的转录因子或者就是rna聚合酶ii本身的一些亚基。这种想法在某些实验系统中被证实,但是在用酵母粗提物
进行体外实验的时候这种想法受到了挑战。一方面,添加任一通用转录因子或是rnapol ii本身都不能减轻squelching;另一方面,添加小份酵母粗提物却能够缓解squelching。于是人们就把这种小份酵母粗提物中的活性物质叫做中介体。
6.由于中介体庞大的分子量,自身内部构成易变化以及整个复合物在体内含量较低等因素,中介体的结构一直没有得到很好的解析。约80%的中介体结构还没有解析出来,中介体和rna聚合酶互作的细节也知之甚少。因此在未来通过结构生物学去解析中介体的结构能够更好地揭示中介体的功能从而让我们对基因转录调控的机制有更深的认识。中介体不仅仅是作为联系转录因子和rna聚合酶ii转录机器的桥梁,它参与了和转录相关的多个生物学过程。中介体可以是转录激活子,也可以是转录抑制子或者就是一类通用转录因子。目前国内外均尚未有报道转录中介体med31基因编辑三角褐指藻对产生岩藻黄素有一定影响的相关研究报道。
技术实现要素:
7.本发明所要解决的技术问题是提供一种转录中介体med31基因及其互作转录因子在提高三角褐指藻中岩藻黄素含量的应用,转录中介体med31基因及其互作转录因子可显著提高三角褐指藻中岩藻黄素含量。
8.本发明解决上述技术问题所采用的技术方案为:一种转录中介体med31基因在提高三角褐指藻中岩藻黄素含量的应用,该基因核苷酸序列如seq id no:1所示。
9.上述转录中介体med31在提高三角褐指藻中岩藻黄素含量的应用,该蛋白氨基酸序列如seq id no:2所示。
10.上述转录中介体med31基因的强互作转录因子,所述的强互作转录因子为myb2r1基因。
11.上述强互作转录因子myb2r1基因在提高三角褐指藻中岩藻黄素含量的应用。
12.上述强互作转录因子myb2r1基因在提高三角褐指藻中岩藻黄素含量的应用,该基因核苷酸序列如seq id no:3所示。
13.与现有技术相比,本发明的优点在于:
14.1、首次证实了通过筛选三角褐指藻的基因库,找到了转录中介体med31,对其进行crispr基因敲除后,发现med31突变株在岩藻黄素合成有表型变化;
15.2、进行了更深入的探究,以med31为诱饵在三角褐指藻cdna文库中筛选互作的转录因子,选择在岩藻黄素合成有表型的转录因子14143(myb2r1),研究med31协同转录因子调控岩藻黄素合成机制,为了解三角褐指藻的岩藻黄素合成提供更深层次的依据,为产业化生产岩藻黄素提供基础。
附图说明
16.图1为基因编辑藻株的敲除结果与wt细胞和含有靶向突变细胞的菌落的序列色谱图。(a)两种基因编辑藻株的敲除鉴定情况;(b)野生型测序产生的序列色谱图;(c)med31-2测序产生的序列色谱图;(d)med31-7测序产生的序列色谱图;
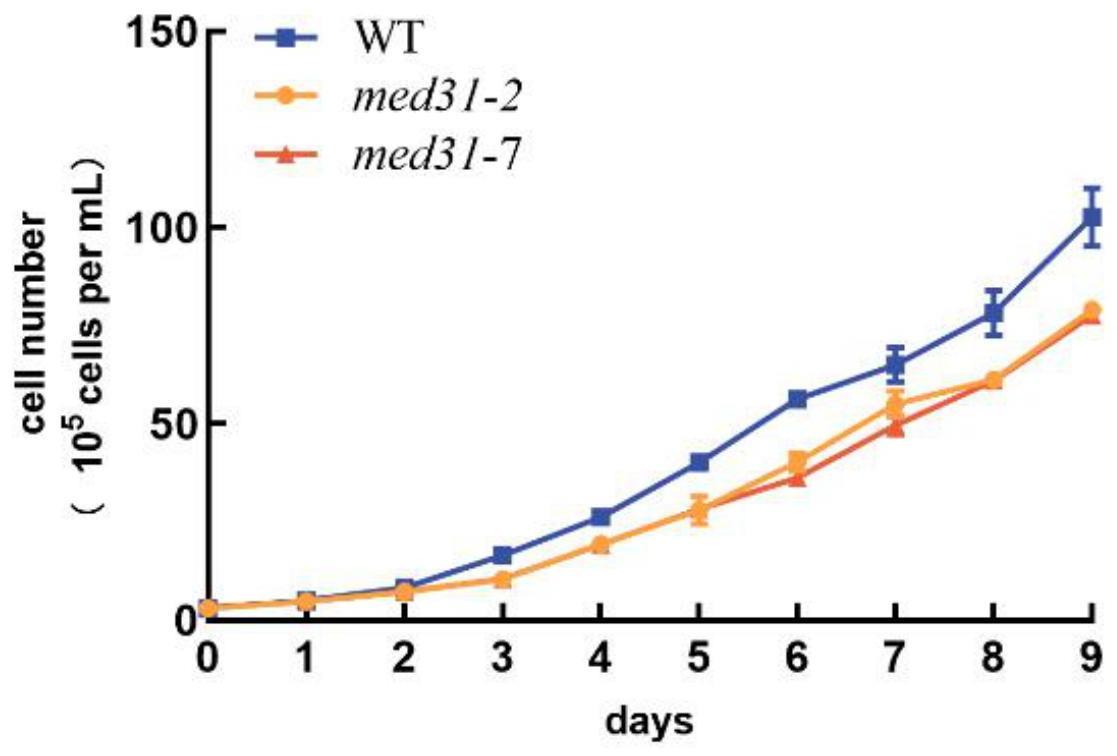
17.图2为两种med31基因编辑藻株和野生wt藻的生长曲线;
18.图3为两种med31基因编辑藻株和wt的岩藻黄素含量*表示实验组与wt组间差异显
著(*:p《0.05);
19.图4为两种med31基因编辑藻株和wt的岩藻黄素合成基因表达量。(a)zds表达量;(b)zep3表达量;(c)crtiso3表达量;(d)pds2表达量。*表示实验组与wt组间差异显著(*:p《0.05,**:p《0.01,***:p《0.001);
20.图5为酵母双杂实验,共筛选到了300互作蛋白,其中转录因子为15个;
21.图6为酵母验证实验,med31-bd与互作因子myb2r1-ad的酵母双杂结果;
22.图7为myb2r1过表达转基因藻与wt的myb2r1基因的表达量,*表示实验组与对照组差异显著(*:p《0.05,****:p《0.0001)
23.图8为myb2r1过表达转基因藻与wt的生长曲线;
24.图9为myb2r1过表达转基因藻与wt的岩藻黄素含量,*表示实验组与wt组间差异显著(**:p《0.01)。
具体实施方式
25.以下结合附图实施例对本发明作进一步详细描述。
26.具体实施例一
27.转录中介体mde31基因克隆与序列分析
28.1、利用rna提取试剂盒(plantrnakit)提取对数期三角褐指藻(藻浓度为1
×
106cells/ml)总rna,以提取的rna为反应物,参考primescripttm rtreagent kit(货号为rr037a)的说明书进行反转录,得到cdna模板。
29.2、根据med31的基因序列设计引物:
30.上游引物序列:5
’‑
atgtcggaaggggaaccgac-3’,
31.下游引物序列:5
’‑
tcaattgttcaccgccgactc-3’;
32.3、pcr扩增:通过pcr扩增得到mde31基因扩增产物,pcr扩增的反应体系为:0.5μlcdna,10μl2
×
primestarmax premix,上、下游引物各0.5μl,8.5μlddh2o;pcr扩增程序为:变性98℃10s,退火55℃15s,延伸,72℃1min,30个循环;
33.4、将pcr扩增产物用1%琼脂糖凝胶电泳纯化回收后与pmd19-t载体连接,经pcr进一步验证后测序,获得mde31基因,其核苷酸序列如seq id no:1所示:atgtcggaaggggaaccgaccaagcccttggcgccgacgaatcctttgccggaaaaccggttcgagctggaactggaattcgttcaggcgctcgcatcgccggcgtatctacactttttggccacgtcgagagccgaagaagacggcaagctttttctgcaagattcttccttccaacagtatcttcgctatcttttcgatacgtggagccgcccggaatacgcgaggtttttgtcgtatcctcacgctctgtactttctcgagttgctgattgagaagcccactgtcttgaaggaatggagtttgccagcctttcggaacttttgtcaccggcaacaatttttatcctggcaacatcgccacgagtctctctacggtaagggaacagtccccgctgctatggacccgaagatcgacggtactacttctcccgtcgtacctagagcagacgaagagtcggcggtgaacaattga;
34.med31其氨基酸序列如seq id no:2所示:
35.msegeptkplaptnplpenrfelelefvqalaspaylhflatsraeedgklflqdssfq
36.qylrylfdtwsrpeyarflsyphalyflelliekptvlkewslpafrnfchrqqflsw qhrheslygkgtvpaamdpkidgttspvvpradeesavnn。
37.具体实施例二
38.一、构建med31敲除载体
39.使用pptpuc3-cas9-sgrna(addgene id:109219)载体作为crispr/cas9的表达盒,构建了2个med31的敲除载体(crispr-med31-1/2),具体步骤如下:
40.1、选定待敲除的med31目标基因,使用在线sgrna设计软件,获得两组sgrna序列,序列如表1所示:
41.表1两组sgrna序列
[0042][0043]
2、将步骤1获得的sgrna序列添加酶切位点并合成单链引物,单链引物序列如下表2所示,表2med31的两对sgrna单链引物序列
[0044][0045][0046]
3、将步骤2配对的单链引物退火反应得到对应的双链dna片段,反应体系为10
×
buffer,1μl;10μm med31-fw,1μl,10μm med31-rv 1μl,用ddh2o补足至10μl,将反应体系混匀,在85℃程序反应10min,室温冷却90min,得到双链dna。
[0047]
4、将步骤3得到的2个双链dna分别与pptpuc3-cas9-sgrna线性质粒用t4连接酶进行连接,16℃连接1h,连接体系为1μl的t410
×
buffer,4μl的pptpuc3-cas9-sgrna线性质粒,3.5μl的双链dna,0.5μl的t4 dna连接酶,用水补齐至10μl,得到2个重组载体,命名为med31-sgrna重组载体;其中线性质粒制备方法如下:取1μl 1μg/μl的pptpuc3-cas9-sgrna载体(109219)加入5μl 10
×
buffer,1μl限制性内切酶,用ddh2o补足至50μl,将反应体系混匀,在37℃恒温培养箱中反应1h,随后进行回收和浓度检测,得到pptpuc3-cas9-sgrna线性质粒。
[0048]
5、med31-sgrna重组载体进行大肠杆菌转化,具体步骤如下
[0049]
(1)取10μl步骤4得到的2个med31-sgrna重组载体分别加入到大肠杆菌dh5α感受态细胞中,放置冰上30min;轻轻摇菌,于42℃水浴60s进行热激,后迅速冰上静置3-5min;
[0050]
(2)然后各自加入400μl不含抗生素的lb液体培养基并轻轻混匀,然后在37℃摇床中220rpm振荡培养1h;其中lb液体培养基组成:酵母提取物5g/l,胰蛋白胨10g/l,氯化钠10g/l,溶剂为水;
[0051]
(3)取步骤(2)中得到混合物100μl分别加入到含有氨苄(amp
)抗生素的lb固体培养基上,均匀涂布,直到菌液在平板上干透为止,封口膜密封,做好标记,放在37℃恒温培养箱中过夜培养12-16h;
[0052]
(4)将过夜培养的平板从培养箱中取出,观察平板上菌落情况;如有单菌落,在超净工作台上各挑取8个单菌落分别于装有1ml含有氨苄(amp
)抗生素的lb液体培养基中培养,在37℃恒温培养摇床中220rpm振荡培养3h后,分别进行菌液pcr鉴定,取pptpuc3-cas9-sgrna载体上的一段序列作为反向鉴定引物(crispr-rv:caggaaacagctatgacc),将表2中med31-fw为正向鉴定引物,进行pcr鉴定。反应体系和反应程序见下表3和表4,
[0053]
表3菌液pcr体系
[0054][0055]
表4菌液pcr程序
[0056][0057]
将得到的pcr产物进行凝胶电泳查看结果,取有阳性条带的样品进行扩大培养(100μl菌液和4ml的氨苄(amp
)抗生素的lb液体培养基)并使用plasmid mini kitⅱ质粒提取试剂盒提取质粒,得到两个med31基因敲除载体。
[0058]
二、med31基因敲除藻株的制备
[0059]
1、选用econⅰ分别对两个med31基因敲除载体进行线性化酶切,酶切体系为:5μl的10
×
buffer,3μl的econⅰ,6μg的med31基因敲除载体,用水补齐至50μl,酶切处理3小时后,用胶回收提取试剂盒(gel extraction kit)进行纯化,酶切回收产物,得到两个med31基因敲除载体线性质粒。
[0060]
2、三角褐指藻与med31基因敲除载体线性质粒进行电转化,具体步骤如下:
[0061]
(1)将三角褐指藻培养到对数期1
×
106cells/ml,1500
×
g在4℃离心10min,弃上清;将沉淀用1ml 375mm的山梨醇(无菌,冰上预冷)清洗细胞3次,将清洗后的细胞用100μl 375mm山梨醇重悬,得到重悬溶液最终密度2
×
109cells/ml;
[0062]
(2)将100μl重悬溶液、4μg(0.2μg/μl)步骤1纯化的med31基因敲除载体线性质粒、40μg(10μg/μl)鲑鱼精子dna(高温煮沸10min)混合在一起,冰上孵育10min,然后转移到0.2cm经冰上预冷的电穿孔电击杯中,设置参数:500v场强、25μf电容和400ω并联电阻,进
行电击转化;
[0063]
(3)电击转化后,立即将细胞转移到含有10ml的f/2液体培养基中,在弱光下(约30μmol/m2·
s)孵育24h以恢复后,转移到正常光照条件下培养24h;
[0064]
(4)将步骤(3)培养得到的藻液,于1500
×
g,4℃,离心10min,弃上清,用600μl f/2液体培养基重悬,取200μl加到含有博来霉素的f/2培养基固体平板(1.2%的琼脂)上,正常培养条件培养12-14d。
[0065]
3、med31基因敲除藻株的筛选及鉴定
[0066]
(1)挑取博来霉素抗性板上长出来的单克隆藻落,在含有博来霉素的f/2固体培养基上划3-5mm的短线,正常条件下培养至长出藻落,取一半藻落,加入20μl藻裂解液,100℃水浴10min后,取藻裂解产物作为模板进行藻液pcr鉴定,其中藻裂解液配方为:1%np40,10.00mm tris,0.14mm nacl,5.00mm kcl;
[0067]
(2)设计藻液鉴定引物为:med31-ko-fw:gtcgatgtcgtcatggtcggat;med31-ko-rw ttctcaatcagcaactcgagaa,
[0068]
表5反应体系
[0069][0070]
表6反应条件
[0071][0072]
将pcr扩增产物进行测序分析,选取成功敲除med31基因的阳性转化子,选择其中敲除杂合的藻株在f/2液体培养基培养后稀释涂布进行新一轮的筛选,重复pcr和测序步骤直至筛选出2株纯合转化株,命名为med31基因敲除藻株。
[0073]
结果如图1所示,med31-2纯合转化株测序结果如图1(c)所示,med31-7纯合转化株的测序结果如图1(d)所示,野生型(没有进行基因改造的原生三角褐指藻)的测序结果如图1(b)所示,由图1(a)可知,med31-2纯合转化株相对于野生型缺失了6bp;图1(b)可知med31-7纯合转化株相对于野生型缺失了15bp。
[0074]
具体实施例三
[0075]
1、生长曲线测定
[0076]
将具体实施例二得到的med31基因敲除藻株在含博来霉素的f/2液体培养基中正
常条件下培养至对数期后,用不含博来霉素的f/2液体培养基清洗三遍后,接种到50ml f/2液体培养基中,以野生型为对照,均调整初始浓度为3
×
105cells/ml,在正常条件下培养。每天选择固定的时间通过光学显微镜计数藻细胞数量确定藻细胞密度,重复三次,直至培养至对数期。
[0077]
图2为med31基因敲除藻株生长曲线,分别连续9天测定了med31基因敲除藻株和野生型(wt)细胞浓度,得到了图2生长曲线。从图中可以得出,在初始密度一致的情况下,从第三天开始,med31基因敲除藻株的细胞数量低于野生型,在第九天时该现象最显著,可见敲除med31后,三角褐指藻的生长量会降低。
[0078]
2、岩藻黄素提取及测定
[0079]
将在对数期的med31基因敲除藻株和野生型三角褐指藻冷冻干燥48h后,使用90%乙醇、料液比1:10(g/ml)、并在40℃通过超声1h提取岩藻黄素。过滤后取上清液(0.22μm)用于hplc分析。全程在黑暗条件下进行。由g1312a二元泵、g1367b自动进样器、g1315d pda检测器和g1316a柱温箱组成的agilent 1200hplc系统(agilent technologies,american)用于岩藻黄素定量。流动相,即甲醇和水,以0.7ml
·
min-1
的流速洗脱在35℃,ymc类胡萝卜素柱(250mm长
×
4.6mm内径;5μm粒径;waters,american)用于在以下梯度程序下进行分离:甲醇从90%增加到100%持续20min,保持在100%接下来的5min,降低到90%5min,然后保持在90%5min。注入样品溶液(10μl),并在445nm处记录色谱图。基于浓度范围为0.5-50μg
·
ml-1
的校准曲线对岩藻黄素进行定量。
[0080]
图3为分别测定了med31基因敲除藻株和野生型在细胞密度为1.5x10
6 cells/ml的岩藻黄素含量,从图3中可以看出,med31-7的岩藻黄素含量比wt下降了0.13倍,med31-2的岩藻黄素比wt下降了0.14倍,实验数据表明,将三角褐指藻中的med31基因敲除导致岩藻黄素的含量降低,med31基因编辑藻在岩藻黄素的合成中起到了正调控的作用。注:柱子上的星号表示统计分析上有显著差异(t检验,p《0.05),wt表示野生型三角褐指藻。
[0081]
3、岩藻黄素合成关键基因表达量检测
[0082]
选取并合成岩藻黄素合成基因zds-like、zep3、crtiso3、pds2的引物(引物序列见表4)。分别使用med31基因敲除藻株med31-2、med31-7和野生型三角褐指藻所提取的rna反转录的cdna为模板,进行real-time pcr。定量试剂使用的是taq pro universal sybr qpcrmaster mix(vazyme,q712),反应体系和反应程序见表7和表8
[0083]
表7荧光定量引物序列
[0084][0085]
表8qpcr反应体系
[0086][0087]
表9qpcr反应程序
[0088][0089]
结果如图4所示,在med31基因敲除藻株中,岩藻黄素的4种关键基因zds、zep3、pds2、crtiso3的表达量降低,因此我们可以证明,med31可以调控岩藻黄素合成基因的表达,并且是正向调控。
[0090]
具体实施例四
[0091]
利用酵母双杂交系统(y2h)证实
[0092]
1、使用pgbkt7载体作为诱饵空载体,将med31的全长序列构建到空载体上,得到pgbkt7-med31重组载体;
[0093]
2、将pgbkt7-med31重组载体与y2h酵母感受态细胞菌种通过热激转化,筛选阳性克隆,得到y2h-pgbkt7-med31重组菌;
[0094]
3、通过酵母转化试剂盒将y2h-pgbkt7-med31重组菌制成y2h-pgbkt7-med31重组菌感受态细胞,将酵母文库(购于北京桓世伟光生物科技发展有限公司)转化进y2h-pgbkt7-med31重组菌感受态细胞进行筛选,挑选蓝色菌落经培养获得酵母单菌落;
[0095]
4、互作蛋白确定
[0096]
将酵母单菌落挑至相应的四缺液体培养基(sd-ade-his-leu-trp培养基)中,30℃振荡培养1d。将菌液在100℃水浴下裂解10min后进行菌液pcr鉴定,酵母鉴定正向引物序列:5
’‑
tttaatacgactcactatagggcga-3’;酵母鉴定反向引物序列:5
’‑
agatggtgcacgatgcacag-3’;酵母测序引物序列:5
’‑
tttaatacgactcactatagggcga-3’。将pcr产物进行测序,再于ncbi网站进行blast比对,获得蛋白的信息,共筛选到了300个左右的互作蛋白,其中转录因子共15个。图5为酵母双杂交实验进行酵母筛库得到的15个转录因子,选择与mde31有强互作的转录因子(结合频次更高)类myb家族myb2r1(locus tag:phatrdraft_14143,312bp)进行研究。
[0097]
进一步对要研究的转录因子myb2r1进行了二次互作验证,结果如图6所示,证明了med31和myb2r1之间存在互作关系,并且可能共同调控某条通路。
[0098]
具体实施例五
[0099]
转录因子myb2r1基因的克隆方法,包括如下步骤:
[0100]
1、提取三角褐指藻总rna并反转录成cdna作为模板;
[0101]
2、根据myb2r1的基因序列设计引物:
[0102]
上游引物序列:5
’‑
atgtggaccaaggaagaagacg-3’,
[0103]
下游引物序列:5
’‑
tcattcacgttcgtactgtcgg-3’;
[0104]
3、pcr扩增:通过pcr扩增得到myb2r1基因扩增产物,pcr扩增的反应体系为:0.5μl cdna,10μl 2
×
primestar max premix(pcr扩增试剂盒:2
×
primestar max premix购自宝成生物公司),上、下游引物各0.5μl,8.5μl ddh2o;pcr扩增程序为:变性98℃10s,退火55℃15s,延伸,72℃1min,30个循环;
[0105]
4、将pcr扩增产物用1%琼脂糖凝胶电泳纯化回收后与pmd19-t载体连接,经pcr进一步验证后测序,获得myb2r1基因,其核苷酸序列如seq id no:3所示:atgtggaccaaggaagaagacgccattctgctcaaaattgtgcaagggatgcaaatgcccatgaagtggagtgttgtcgcacaaaacttacacgatcgtacgggaaagcagtgtcgcgagcgctacgtcaatcatctcaatccccgtctcaaggtcacggactggaatccggtcgaagactccaccatatttcacctttacaacactatcggtagccactgggcaaaaatgtccaaggtcatccccggacgcacggacaacggcatcaagaatcgcttccataatctccgccgacagtacgaacgtgaatga。
[0106]
具体实施例六
[0107]
上述转录因子myb2r1过表达重组藻构建方法,包括如下步骤:
[0108]
1、根据myb2r1的基因序列和ppha-t1载体信息设计引物:上游引物序列:5
’‑
tgtctgccgtttcgagaattcatgtggaccaaggaagaagacg-3’,下游引物序列:5
’‑
ttagtcgatgatatcgaattctcattcacgttcgtactgtcgg-3’,引物两端分别包括载体的序列作为同源臂(划线处);
[0109]
2、pcr扩增的反应体系为:0.5μlt载体,10μl 2
×
primestarmax premix(pcr扩增试剂盒:2
×
primestar max premix购自宝成生物公司),上、下游引物各0.5μl,8.5μl ddh2o;pcr扩增程序为:变性98℃10s,退火55℃15s,延伸,72℃1min,30个循环;pcr扩增产物用1%琼脂糖凝胶电泳纯化回收;
[0110]
3、载体构建:将载体ppha-t1使用限制性内切酶ecorⅰ进行酶切以获得线性化质粒,在37℃恒温培养箱中反应3h,反应完毕后进行回收和浓度检测。
[0111]
4、使用试剂盒clonexpress ii one step cloning kit将步骤(2)纯化回收的pcr产物和步骤(3)得到的线性化载体进行同源重组反应,将myb2r1基因构建进ppha-t1载体中得到ppha-t1-myb2r1重组质粒;
[0112]
5、三角褐指藻转化:将重组质粒ppha-t1-myb2r1重组质粒通过电转化的方法转化进三角褐指藻中,在低光强(30μmol.m-2
.s-1
)下孵育24h,再转移到正常光强下培养24h,然后3000g离心10min收集细胞并用0.6ml f/2培养液重悬该细胞,平均涂布于3个含有75μg/ml的博来霉素的f/2固体培养基上,15-25d后出现藻落,挑取博来霉素抗性板上长出来的初步阳性藻,在含有博来霉素的f/2固体培养基上划3-5mm的短线,正常条件下培养至长出藻落,取一半藻落,加入20μl藻裂解液,100℃水浴10min后作为模板进行藻液pcr,跑胶出现条带后,初步说明转基因成功,鉴定引物如下:myb2r1-f:gaagaagacgccattctgctc;myb2r1-r:cacgttcgtactgtcggcgg。
[0113]
6、myb2r1过表达阳性藻鉴定
[0114]
将转基因成功的藻接种到50ml f/2液体培养基中,培养至对数期后,离心收集藻细胞,提取rna,反转录成cdna后,qpcr检测myb2r1自身表达量,myb2r1荧光定量引物序列如
下:myb2r1-qpcr-fw:5'-atgcccatgaagtggagtgt-3';
[0115]
myb2r1-qpcr-rv:5'-gacggggattgagatgattg-3'。
[0116]
如图7所示myb2r1过表达转基因藻的自身表达量升高。为了研究myb2r1在岩藻黄素合成中的作用,生成了过表达myb2r1的转基因三角褐指藻藻株,并通过qrt-pcr证实其中三个株系的myb2r1表达量显著上调。
[0117]
具体实施例七
[0118]
1、myb2r1过表达转基因藻的生长曲线测定
[0119]
将myb2r1过表达转基因藻在含博来霉素的f/2液体培养基中正常条件下培养至对数期后,用不含博来霉素的培养基清洗三遍后,接种到50ml f/2液体培养基中,以野生型为对照,均调整初始浓度为3
×
105cells/ml,在正常条件下培养。每天选择固定的时间通过光学显微镜计数藻细胞数量确定藻细胞密度,重复三次,直至培养至对数期。
[0120]
图8为myb2r1过表达转基因藻株的生长曲线,分别连续9天测定了myb2r1过表达转基因藻株和野生型(wt)的细胞浓度,得到了生长曲线如图8所示。从图中可以得出,在初始密度一致的情况下,从第五天开始,myb2r1过表达转基因藻株的细胞数量高于野生型,在第九天时该现象最显著,可见过表达myb2r1后,三角褐指藻的生长量会升高。
[0121]
2、myb2r1过表达转基因藻的岩藻黄素含量测定
[0122]
将在对数期的myb2r1过表达转基因藻株和野生型三角褐指藻冷冻干燥48h后,使用90%乙醇、料液比1:10(g/ml)、并在40℃通过超声1h提取岩藻黄素。过滤后取上清液(0.22μm)用于hplc分析。全程在黑暗条件下进行。由g1312a二元泵、g1367b自动进样器、g1315d pda检测器和g1316a柱温箱组成的agilent 1200hplc系统(agilent technologies,american)用于岩藻黄素定量。流动相,即甲醇和水,以0.7ml
·
min-1
的流速洗脱在35℃,ymc类胡萝卜素柱(250mm长
×
4.6mm内径;5μm粒径;waters,american)用于在以下梯度程序下进行分离:甲醇从90%增加到100%持续20min,保持在100%接下来的5min,降低到90%5min,然后保持在90%5min。注入样品溶液(10μl),并在445nm处记录色谱图。基于浓度范围为0.5-50μg/ml的校准曲线对岩藻黄素进行定量。
[0123]
结果如图9所示,测定myb2r1过表达转基因藻和wt的岩藻黄素含量,与wt相比较可得,myb2r1过表达转基因藻的岩藻黄素含量提高了0.43-0.53倍。表明,myb2r1基因表达水平的提高可以引起岩藻黄素含量的提高,说明myb2r1在岩藻黄素的合成途径中起到了正向调控的作用。
[0124]
上述说明并非对本发明的限制,本发明也并不限于上述举例。本技术领域的普通技术人员在本发明的实质范围内,做出的变化、改型、添加或替换,也应属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。