一种通过mitsunobu缩合法合成恩曲他滨的方法
技术领域
1.本发明属于药物化学技术领域,具体涉及一种通过mitsunobu缩合法合成恩曲他滨的方法。
背景技术:
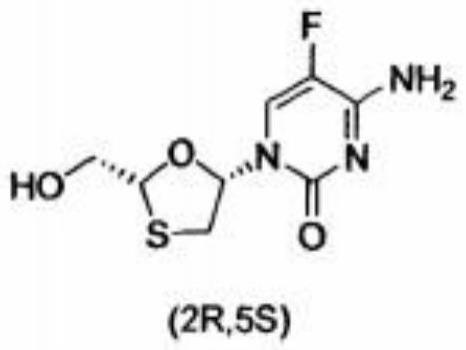
2.恩曲他滨(emtricitabine,ftc),化学名为4-氨基-5-氟-1-[(2r,5s)-2-羟甲基-1,3-氧硫杂环戊烷-5-基]-2(1h)-嘧啶酮,是由美国gilead siences公司研制成功的新型核苷类逆转录酶抑制剂,属抗病毒类药物。2003年经美国fda批准上市,用于治疗由人类免疫缺陷病毒(hiv)感染引起的艾滋病,以及乙型肝炎病毒(hbv)引起的乙型肝炎。自上市以来,临床证明ftc的抗病毒失败率低,治疗效果好,是治疗艾滋病和乙型肝炎最成功的药物之一。
[0003][0004]
恩曲他滨为l,3-氧硫杂环核苷类似物,分子结构中含有两个手性中心,有4个光学异构体,但恩曲他滨是单一(2r,5s)光学结构的药物。
[0005]
目前国内外合成恩曲他滨的方法有三种基本类型:
[0006]
(1)通过手性柱拆分光学异构体获得恩曲他滨,1992年,choi在合成氟胞嘧啶的l,3-氧硫杂环化合物后,经手性柱拆分后得到恩曲他滨。由于拆分恩曲他滨光学过程复杂,拆分规模有限,严重限制了其开发利用。
[0007]
(2)通过酶拆分光学异构体获得恩曲他滨。1996年,dionne等合成氟胞嘧啶的l,3-氧硫杂环化合物后,将其形成脱磷酸盐,再经5-核酸酶选择性脱磷酸得恩曲他滨。酶拆分能得到高纯度的恩曲他滨,且可以进行规模生产,但该方法的缺陷是5-核酸酶的制备烦琐,导致成本增加。
[0008]
(3)通过先合成手性中问体,再诱导合成恩曲他滨。2002年,宫平以水合乙醛酸为原料,与噻烷、1-薄荷醇反应形成手性中间体,再与碱基选择性偶合,还原后通过硅胶柱分离得到恩曲他滨。该方法合成步骤少,条件简单,但总收率不高。
技术实现要素:
[0009]
为了解决现有技术的不足,本发明提供了一种通过mitsunobu缩合法合成恩曲他滨的方法。关键的合成步骤为:(2r,5r)-5-羟基-1,3-氧硫杂环戊烷-2-羧酸1-孟基酯(i)和n4位保护的5-氟胞嘧啶(ii)经mitusnobu反应缩合,接着硼氢化钠还原,最后酸性条件下脱除保护基,得到恩曲他滨。其中:缩合步骤中仲醇手性翻转,为立体选择性反应,避免了异构体的产生和分离,步骤简洁,收率高。通过采用该方法得到的恩曲他滨,成本低,操作简便,
反应条件温和,表明了该合成方法的优越性和简便性。
[0010]
本发明的技术方案是:一种通过mitsunobu缩合法合成恩曲他滨的方法,反应方程式表示如下:
[0011][0012]
其中:tr为三苯甲基;boc为叔丁氧羰基。
[0013]
包括以下步骤:
[0014]
第一步:(2r,5r)-5-羟基-1,3-氧硫杂环戊烷-2-羧酸1-孟基酯(i)与n4位保护的5-氟胞嘧啶(ii),在有机溶剂中采用mitsunobu反应缩合,得到中间体(iii);
[0015]
第二步:中间体(iii)在醇溶剂中,在还原剂存在下反应,得到中间体(iv);
[0016]
第三步:中间体(iv)在酸性条件下,脱除保护基,得到恩曲他滨。
[0017]
进一步地,在上述技术方案中,化合物i与化合物ii摩尔比为1:1-0.5。
[0018]
进一步地,在上述技术方案中,mitsunobu反应采用ph3p/diad或ph3p/dead组合试剂;其中ph3p与diad/dead摩尔比为1:1。
[0019]
进一步地,在上述技术方案中,化合物ii与mitsunobu试剂摩尔比为1:1.2-1.5。
[0020]
进一步地,在上述技术方案中,所述有机溶剂选自四氢呋喃、乙腈、氯仿、二甲基亚砜、乙酸乙酯、二氯甲烷中的一种或多种。
[0021]
进一步地,在上述技术方案中,还原剂选自硼氢化钠、硼氢化锂或硼氢化钾。
[0022]
进一步地,在上述技术方案中,醇溶剂选自甲醇或乙醇。
[0023]
进一步地,在上述技术方案中,第三步脱保护采用氯化氢/醇溶液、乙酰氯/醇或三氟乙酸。
[0024]
通过采用该方法得到恩曲他滨,成本低,操作简便,反应条件温和,表明了该合成方法的优越性和简便性。
具体实施方式
[0025]
下面结合实施例对本发明做详细说明。
[0026]
实施例1
[0027]
将(2r,5r)-5-羟基-1,3-氧硫杂环戊烷-2-羧酸1-孟基酯(i)(3.00g,10.4mmol)、n4-三苯甲基-5-氟胞嘧啶(iia,3.86g,10.4mmol)和三苯基膦(3.27g,12.5mmol)加入四氢呋喃(200ml)。控温0-10℃条件下,滴加dead(1.96ml,12.5mmol)。滴加完毕,温度升至室温搅拌12h。接着升温至40℃再搅拌3h。
[0028]
反应温度降至室温,四氢呋喃(100ml)稀释。有机相水(2
×
100ml)洗涤后,再次四氢呋喃(100ml)反萃取水层。硫酸钠干燥,过滤,将溶剂和低沸点化合物用旋转蒸发器和真空泵除去。得到的粗产品悬浮在四氢呋喃静置,产生白色固体,减压抽滤,采用体积比1:1乙醚和正己烷洗涤,将得到的滤液旋转蒸发仪浓缩,得到的黄色油状物质溶解于二氯甲烷(10ml)中,然后柱层析纯化后得到中间体iiia 5.66g,收率85%。1h nmr(500mhz,dmso-d6)δ8.02(d,j=6.80hz,1h),7.74(brs,1h),7.25-7.33(m,15h),6.24(t,j=4.60hz,1h),5.46(s,1h),3.52(dd,j=4.42hz,1h),3.12(dd,j=4.46hz,1h),1.02-2.05(m,10h),0.93(dd,j=6.82hz,6h),0.78(d,j=6.82hz,3h).
13
c nmr(125mhz,dmso-d6)δ169.6,158.0,150.1,129.2,126.1,126.3,127.1,128.6,129.2,89.8,80.2,78.6,76.0,46.3,35.8,34.3,31.2,26.6,23.0,22.2,21.6,20.4,16.3.
[0029]
实施例2
[0030]
在250ml圆底烧瓶中,加入中间体iiia(2.0g,3.12mmol)和无水乙醇15ml,滴加含有25%氢氧化钠(0.04ml)/硼氢化钠(0.353g,9.36mmol)水溶液(4ml),混合物室温下搅拌反应4h,静置分液,上层盐酸调ph=4-5,氢氧化钠中和,甲苯提取除去有机物三次,蒸干剩余溶剂,残余物中加入无水乙醇,加热至60-70℃,趁热过滤除去无机盐后,减压浓缩,乙酸乙酯重结晶,得到中间体iva 1.23g,收率81%。1h nmr(500mhz,dmso-d6)δ8.04(d,j=6.80hz,1h),7.71(brs,1h),7.22-7.30(m,15h),6.26(t,j=4.60hz,1h),5.82(brs,1h),5.42(t,j=5.11hz,1h),4.74-4.81(dd,j=4.38hz,2h),4.16-4.18(dd,j=4.65hz,2h).
13
c nmr(125mhz,dmso-d6)δ169.2,158.1,150.3,129.4,129.2,128.2,127.2,126.3,126.1,87.3,62.8,36.5.
[0031]
实施例3
[0032]
将中间体iva(2.0g,4.09mmol)加入甲醇(50ml)中,滴加tfa(0.61ml,8.18mmol),加热至60℃反应4h,饱和碳酸氢钠溶液中和,减压浓缩,乙酸乙酯重结晶,得到恩曲他滨0.93g,收率92%。1h nmr(500mhz,dmso-d6)δ8.20(d,j=6.80hz,1h),7.72(brs,1h),7.45(brs,1h),6.14(t,j=4.60hz,1h),5.16(m,1h),4.74-4.81(m,1h),4.16-4.18(dd,j=4.65hz,1h),3.76(dt,1h),3.42(dd,j=4.46hz,1h),3.13(dd,j=4.46hz,1h).
13
c nmr(125mhz,dmso-d6)δ158.3,153.2,137.3,135.4,126.3,87.3,62.8,36.9.
[0033]
实施例4
[0034]
将(2r,5r)-5-羟基-1,3-氧硫杂环戊烷-2-羧酸1-孟基酯(i)(3.00g,10.4mmol)、n4-boc-5-氟胞嘧啶iib(2.38g,10.4mmol)和三苯基膦(3.27g,12.5mmol)加入四氢呋喃(200ml)。控温0-10℃温度条件下滴加dead(1.96ml,12.5mmol)。滴加完毕,升至室温搅拌12h;随后升温至40℃再搅拌3h。反应温度降至室温,四氢呋喃(100ml)稀释。有机相水(2
×
100ml)洗涤,再次四氢呋喃(100ml)反萃取水层。硫酸钠干燥,过滤,将溶剂和低沸点化合物用旋转蒸发器和真空泵除去。得到的粗产品悬浮在四氢呋喃静置,产生白色固体,减压抽滤,体积比1:1乙醚和正己烷洗涤,将得到的滤液旋转蒸发仪浓缩,得到的残留黄色油状物
质溶解于二氯甲烷(10ml)中,柱层析纯化后得到白色结晶性固体(产量4.57g,产率88%)。1h nmr(500mhz,dmso-d6)δ8.05(d,j=6.80hz,1h),7.78(brs,1h),6.26(t,j=4.58hz,1h),5.42(s,1h),4.13-4.18(dd,j=4.65hz,2h),1.38(s,9h),1.02-2.05(m,10h),0.89(dd,j=6.81hz,6h),0.81(d,j=6.80hz,3h).
13
c nmr(125mhz,dmso-d6)δ171.0,161.2,158.0,156.3,153.2,121.0,102.5,98.2,89.8,78.6,47.2,40.9,34.3,31.2,26.0,23.7,22.2,21.0,20.7.
[0035]
实施例5
[0036]
在250ml圆底烧瓶中,加入中间体iiib(2.0g,4mmol)和无水乙醇15ml,滴加含有25%氢氧化钠(0.03ml)/硼氢化钠(0.275g,7.30mmol)水溶液(4ml),混合物在室温下搅拌反应4h,静置分液,上层盐酸调ph=4-5,氢氧化钠中和,甲苯提取除去有机物三次,蒸干剩余溶剂,残余物中加入无水乙醇,加热至60-70℃,趁热过滤除去无机盐,减压浓缩,乙酸乙酯重结晶,得到中间体ivb 1.08g,收率78%。1h nmr(500mhz,dmso-d6)δ8.03(d,j=6.80hz,1h),7.65(brs,1h),6.24(t,j=4.60hz,1h),5.81(brs,1h),5.39(t,j=5.10hz,1h),4.71-4.78(dd,j=4.35hz,2h),4.12-4.15(dd,j=4.65hz,2h),1.38(s,9h).
13
c nmr(125mhz,dm so-d6)δ162.1,157.5,152.1,121.2,109.7,98.2,87.6,84.1,66.3,29.8,27.4.
[0037]
实施例6:
[0038]
将中间体ivb(2.0g,5.76mmol)加入甲醇(50ml)中,滴加tfa(0.85ml,11.5mmol),加热至60℃反应2h,饱和碳酸氢钠溶液中和,减压浓缩,乙酸乙酯重结晶,得到恩曲他滨1.27g,收率90%。
[0039]
以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。