1.本发明属于有机合成技术领域,具体涉及一种沙库比曲中间体的酶催化制备方法。
背景技术:
2.沙库巴曲缬沙坦钠片在临床上主要是用于治疗射血分数降低的慢性心力衰竭的成人患者,属于心脑血管类药物。沙库巴曲缬沙坦钠片是由沙库巴曲和缬沙坦两种成分以1:1摩尔比例结合而成的盐复合物晶体,可同时抑制脑啡肽酶和阻断at1受体(血管紧张素ii 1型受体)。其中沙库必曲是其中脑啡肽酶(nep)抑制剂组成部分,其结构如下:
[0003][0004]
专利wo2008031567中报道一种沙库必曲中间体的制备方法,主要步骤如下:
[0005][0006]
该专利采用手性化合物作为原料构建联苯结构,再与乙氧甲酰基亚乙基三苯基膦对接,原材料结构复杂且成本高,制备过程中原子消耗大,环保三废压力更大。
技术实现要素:
[0007]
为解决上述问题,本发明公开了一种沙库比曲中间体的酶催化制备方法,以4-卤代联苯与环氧卤代丙烷为原料对接,经氧化、酶水解、转氨、再次氧化后备用,以2-卤代丙酸乙酯与硫叶立德试剂对接,经过氧化后与另一片段对接合成沙库比曲中间体,本发明的原料简单易得,同时与现有技术工艺相比,工序简单、反应条件温和、生产成本低、三废排放少,对环境友好。
[0008]
为达到上述目的,本发明的技术方案如下:
[0009]
一种沙库比曲中间体的酶催化制备方法,其合成路线如下:
[0010][0011]
其中:x为卤素;y为卤素或-or5,其中r5为c1~c6的烷基;z为卤素;r1,r2,r3,r4彼此分别为独立的s、o、n、p、c、n-r6、p-r6或c-r6,其中r6为c1~c6的烷基或芳基;
[0012]
具体制备步骤如下:
[0013]
(1)以4-卤代联苯与环氧卤代丙烷为原料,通过格式反应合成化合物1;
[0014]
(2)将步骤(1)制得的化合物1通过氧化剂氧化后得到化合物2;
[0015]
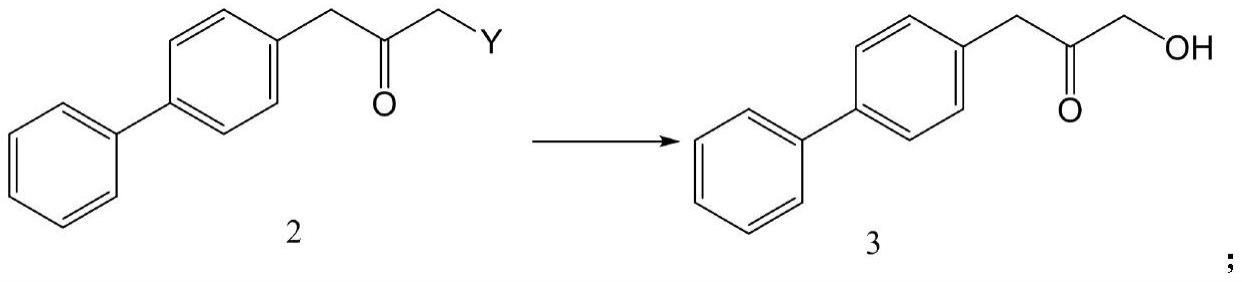
(3)将步骤(2)制得的化合物2经酶催化水解得到化合物3;
[0016]
(4)将步骤(3)制得的化合物3通过转氨酶反应构建手性分子,合成化合物4;
[0017]
(5)将步骤(4)制得的化合物4通过氧化剂氧化后得到化合物5;
[0018]
(6)将2-卤代丙酸乙酯与硫叶立德试剂对接得到化合物6;
[0019]
(7)将步骤(6)制得的化合物6经过氧化剂氧化得到化合物7;
[0020]
(8)将步骤(5)制得的化合物5在碱催化下与步骤(7)制得的化合物7对接,合成得到化合物8。
[0021]
作为本发明的一种改进,所述步骤(1)中,4-卤代联苯与环氧卤代丙烷的摩尔比为1:1~2,格式反应的催化剂为卤代亚铜。
[0022]
作为本发明的一种改进,所述步骤(2)中,化合物1与氧化剂的摩尔比为1:1~2,氧化剂为次氯酸钠或次氯酸钙。
[0023]
作为本发明的一种改进,所述步骤(3)中,化合物2与水解酶的质量比为1:2~5,反应温度30~35℃。
[0024]
作为本发明的一种改进,所述步骤(4)的具体步骤为:以转氨酶为催化剂,在辅酶plp、氨基供体、缓冲液和助溶剂存在的条件下,催化步骤(3)制得的化合物3得到化合物4,所述氨基供体为异丙胺或丙氨酸。
[0025]
作为本发明的一种改进,所述步骤(4)中,化合物3和转氨酶的质量比为1:1~10,转氨酶与辅酶plp的质量比为10~100:1,化合物3与助溶剂的质量体积比为1:5~10。
[0026]
作为本发明的一种改进,所述步骤(5)中,化合物4与氧化剂的摩尔比为1:1~2,氧化剂为次氯酸钠或次氯酸钙。
[0027]
作为本发明的一种改进,所述步骤(6)中,2-卤代丙酸乙酯与硫叶立德试剂的摩尔比为1:0.6~1,硫叶立德试剂为中的任一种。
[0028]
作为本发明的一种改进,所述步骤(7)中,化合物6与氧化剂的摩尔比为1:2~5,氧化剂为过氧化氢、臭氧和mcpba中的任一种。
[0029]
作为本发明的一种改进,所述步骤(8)中,化合物5与化合物7的摩尔比为1:1~2,碱为碱金属碳酸盐、碱金属碳酸氢盐、碱金属氢氧化物和碱金属醇盐中的任一种,反应温度为-60~-50℃。
[0030]
本发明的有益效果为:
[0031]
(1)本发明引入硫叶立德试剂作为离去基团,更容易得到目标化合物;利用空间位阻效应,手性选择性高,反应简单,原料便宜。
[0032]
(2)本发明采用转氨酶催化构建分子中的手性中心,条件更加温和,手性选择性更好,原料利用率提高,避免了手性拆分或者手性还原过程,更利于工业化生产。
[0033]
(3)本发明采用非手性卤代丙烷作为原料,物料成本较其他路线更低。
[0034]
(4)化合物3稳定性比化合物2更好,后续转氨反应更加稳定。
[0035]
实施方式
[0036]
下面结合具体实施方式,进一步阐明本发明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。
[0037]
实施例1:化合物1的合成
[0038][0039]
向反应器中加入1.16g 5mmol 4-溴联苯、2.88g 120mmol镁粉与5ml四氢呋喃搅拌混合,加入0.01g碘粒引发,补加20ml四氢呋喃。控温40~45℃,向反应器中滴加4-溴联苯(22.14g,95mmol)和30ml四氢呋喃混合溶液,滴加完毕后保温反应3h。
[0040]
反应结束后,降温至0℃,加入0.2g碘化亚铜,降温至-15~-10℃,滴加9.25g 100mmol环氧氯丙烷和10ml四氢呋喃混合溶液,保温反应2h。
[0041]
反应结束,升至室温,向反应液中加入4m盐酸调酸,体系分层,有机相回收溶剂,向残余物中加入乙醇和水重结晶,干燥后得到化合物1(23.27g,94.3mol),收率94.3%。
[0042]
实施例2:化合物2的合成
[0043][0044]
向反应器中加入20g 81.06mmol化合物1、6.81g 81.06mmol碳酸氢钠、1.93g 16.21mmol溴化钾、0.04g 0.259mmoltempo和200ml dcm,搅拌降温,降温至8℃时开始滴加75.43g81.06mmol次氯酸钠溶液,控制过程温度15℃以下,约1h滴完,滴完后搅拌反应30min。
[0045]
反应液分层,水相用40mldcm萃取一次,合并dcm层,水洗一次,dcm层旋干,得到类白色固体化合物2(19.56g,79.92mmol),收率98.6%。
[0046]
实施例3:化合物3的合成
[0047][0048]
向反应器中加入10g 40.86mmol化合物2、30ml dmso和100ml水,搅拌下加入30g水解酶液和3.42g 40.86mmol碳酸氢钠,控制温度30~35℃间反应6h。
[0049]
反应完全后,加入乙酸乙酯,加热至60℃后分液,水相用热的乙酸乙酯萃取一次,合并乙酸乙酯相,热水洗涤一次,将有机相降温至0~10℃后抽滤,得到白色固体化合物3(9.05g,39.99mmol),收率97.88%。
[0050]
实施例4:转氨酶的制备
[0051]
将转氨酶基因进行全合成后设计上下游引物f、r(如seq id no:2、seq id no:3所示)进行pcr扩增,扩增后纯化扩增产物并通过双酶切(ecor i,hind iii)将目的基因载入质粒pet-28a,构建好重组载体后通过转化技术导入e.coli bl21(de3)中,涂布在含卡那霉素的lb平板中,放入37℃培养箱中过夜,将长出来的单菌落进行质粒提取和测序,最终获得含生物酶基因的重组工程菌:escherichia coli工程菌tran-001。
[0052]
引物名称序列编号fggaattcatgcagaaacagcgtactactaseqidno:2rcccaagcttcgccagaccacgagctseqidno:3
[0053]
将表达转氨酶omega-transaminase-01的escherichia coli工程菌tran-001,接种至种子液中(lb培养基),接种量为培养基体积的1~5%,37℃的条件下,220rpm摇床中过夜培养后,作为种子液。将新鲜的种子液接入tb培养基中发酵培养,37℃的条件下,220rpm摇床中培养至od
600
在0.6~0.8时向培养基中加入iptg,iptg的浓度为0.5mm,再于18℃诱导
16h。发酵液诱导结束后,于4℃,12000rpm离心5min,收集菌体。用ph7.0,0.2m的pb缓冲液重洗后重悬,超声破碎后冻干,获得冻干酶粉,即转氨酶omega-transaminase-01酶粉。
[0054]
seq id no.1:
[0055][0056][0057]
实施例5:化合物4的合成
[0058][0059]
向反应器中加入10g 44.19mmol化合物3,80mldmso(助溶剂),室温搅拌溶解,开启氮气保护。
[0060]
在另一反应器中加入15ml水、3g异丙胺,混合均匀,降温至10℃以下,缓慢加入4g浓盐酸,控制ph在10.0~11.0。将上述溶液投入至第一步反应器中。再向第一步反应釜中投入2.5g的plp,水浴升至45℃。
[0061]
将转氨酶50g omega-transaminase-01酶粉在30℃水浴中解冻,投入至第一步反应器中。再配制40%异丙胺水溶液20ml备用。
[0062]
保温45℃搅拌反应。用40%的异丙胺水溶液调节反应液ph,使其维持在8.70-9.10(ph计测定)之间,反应约7h后,中控反应效果;反应结束后,向反应液中加入适量浓盐酸,调节反应液ph至2.00。保温搅拌1h。
[0063]
降温在30℃以下,向反应液中加入10g硅藻土,搅拌30min以上。压滤,滤完后用20g纯水洗涤滤渣。
[0064]
合并续滤液,加入150mldcm,搅拌30min。静置分层。水层在搅拌和稍冷却下用25%的氢氧化钠水溶液调ph至10.00-10.50,缓慢加入9.64g 44.19mmol(boc)2o,反应2h,反应结束后加入150ml二氯甲烷,搅拌30min,静置分层。
[0065]
有机层回收干溶剂,得化合物4固体(139.15g,42.55mmol),收率96.3%。
[0066]
实施例6:化合物5的合成
[0067][0068]
向反应器中加入20g 61.08mmol化合物4、5.13g 61.08mmol碳酸氢钠、1.45g 12.22mmol溴化钾、0.03g 0.195mmol tempo和200ml dcm,搅拌降温,降温至8℃时开始滴加71.05g 61.08mmol次氯酸钠溶液,控制过程温度15℃以下,约1h滴完,滴完后搅拌反应30min。
[0069]
反应液分层,水相用40mldcm萃取一次,合并dcm层,水洗一次,dcm层旋干,得到类白色固体化合物5(19.61g,60.26mmol),收率98.7%。
[0070]
实施例7:化合物6的合成
[0071][0072]
向反应器中依次投入6.6g 50mmol 5-甲基-1,3,4-噻二唑-2-硫醇、8.19g 60mmol 2-卤代丙酸乙酯、10.36g 75mmol碳酸钾、20mldmf,氮气真空置换5次,后氮气保护。搅拌升温至55-60℃,在此温度下保温反应6小时,后升温至90℃反应14小时,tlc中控至5-甲基-1,3,4-噻二唑-2-硫醇消失。
[0073]
降温至室温,向内加45ml水,搅拌10分钟转入分液漏斗,用50ml甲苯萃取三次,合并有机相,有机相用45ml水洗一次,有机相回收干,得化合物6(10.76g,46.3mmol),收率92.6%。
[0074]
实施例8:化合物7的合成
[0075][0076]
向反应器中加入11.62g 50mmol化合物6,再加入50ml异丙醇,6g 5mmol四水钼酸铵,搅拌下滴加150mmol双氧水,滴加过程通过冰水浴控制温度不超30℃,约1-1.5小时滴完,后室温搅拌,tlc中控至化合物6完全消失,通常需反应72小时。
[0077]
室温过滤,所得固体用50ml水淋洗,滤至无液体流出。
[0078]
向固体中加入150ml水,室温搅拌4小时,室温过滤。固体于鼓风干燥箱50℃烘至恒重。得化合物7固体(12.17g,46.05mmol),收率92.1%。
[0079]
实施例9:化合物8的合成
[0080][0081]
向反应器中依次投入9.76g 30mmol化合物5、9.52g 36mmol化合物7,氮气真空置换5次,后氮气保护下向内加入25ml dcm,搅拌下降温至-60℃。将8.4g 75mol叔丁醇钾溶于叔丁醇后(呈糊状),一次性加入反应瓶。于-60~-50℃搅拌,反应需4-5小时。
[0082]
向体系中加入盐酸中和,加入水和乙酸乙酯洗涤萃取,有机层浓缩干,得到化合物8固体(11.39g,27.81mmol),收率92.7%,纯度99.1%。
[0083]
实施例10:化合物1的合成
[0084]
其他条件同实施例1,仅改变4-卤代联苯与环氧卤代丙烷的摩尔比详见表1。
[0085]
表1实施例1、10的不同条件与结果
[0086]
实施例n1:n2反应温度/℃反应时间/h收率/%11:1-15~-10294.3101:2-15~-10292.3
[0087]
其中:n1为4-卤代联苯的摩尔量,n2为环氧卤代丙烷的摩尔量。
[0088]
实施例11-12:化合物2的合成
[0089]
其他条件同实施例2,仅改变化合物1与氧化剂的摩尔比,氧化剂种类,详见表2。
[0090]
表2实施例2,11-12的不同条件与结果
[0091]
实施例n
化合物1
:n
氧化剂
氧化剂种类收率/%21:1次氯酸钠98.6111:2次氯酸钠96.7121:1次氯酸钙97.1
[0092]
实施例13-14:化合物3的合成
[0093]
其他条件同实施例3,仅改变化合物2和水解酶质量比,详见表3。
[0094]
表3实施例3、13-14的不同条件与结果
[0095]
实施例m
化合物2
:m
水解酶
反应温度/℃收率/%31:330~3597.8131:230~3595.4141:530~3597.2
[0096]
实施例15-20:化合物4的合成
[0097]
其他条件同实施例5,改变反应中各物料投料比,详见表4。
[0098]
表4实施例5、15-20的不同条件与结果
[0099]
实施例m
化合物3
:m
转氨酶m转氨酶
:m
辅酶plpm化合物3
:v
助溶剂
收率/%51:520:1896.3151:120:1891.7161:1020:1893.8171:510:1890.1
181:5100:1892.8191:520:1894.9201:520:11092.9
[0100]
实施例21-22:化合物5的合成
[0101]
其他条件同实施例6,仅改变化合物4与氧化剂的摩尔比,氧化剂种类,详见表5。
[0102]
表5实施例6、21-22的不同条件与结果
[0103]
实施例n
化合物4
:n
氧化剂
氧化剂种类收率/%61:1次氯酸钠98.7211:2次氯酸钠97.3221:1次氯酸钙96.6
[0104]
实施例23-24:化合物6的合成
[0105]
其他条件同实施例7,仅改变2-卤代丙酸乙酯与硫叶立德试剂的摩尔比,详见表6。
[0106]
表6实施例7、23-24的不同条件与结果
[0107]
实施例n3:n4收率/%71:0.8392.6231:0.688.8241:191.4
[0108]
其中:n3为2-卤代丙酸乙酯的摩尔量,n4为硫叶立德试剂的摩尔量。
[0109]
实施例25-26:化合物7的合成
[0110]
其他条件同实施例8,仅改变化合物6与氧化剂的摩尔比,详见表7。
[0111]
表7实施例8、25-26的不同条件与结果
[0112]
实施例n
化合物6
:n
氧化剂
收率/%81:392.1251:290.6261:587.3
[0113]
实施例27-30:化合物8的合成
[0114]
其他条件同实施例9,改变化合物5与化合物7的摩尔比,反应温度,详见表7
[0115]
表7实施例8、27-30的不同条件与结果
[0116]
实施例n
化合物5
:n
化合物7
反应温度/℃收率/%纯度/%91:1.2-60~-5092.799.1271:1-60~-5089.198.9281:2-60~-5090/898.5291:1.2-70~-6081/197.1301:1.2-50~-4083.496.9
[0117]
需要说明的是,上述仅仅是本发明的较佳实施例,并非用来限定本发明的保护范围,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,在上述实施例的基础上还可以做出若干改进和润饰,这些改进和润饰均落入本发明权利要求书的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。