1.本发明提供了一种用于合成ε-己内酯的磺酸功能化杂多酸离子液体催化剂及其制备方法,并能达到良好的催化效果以及优异的产品收率。
技术背景
2.近年来,随着环保要求的提高,人们对可生物降解无毒材料的需求越来越大。聚己内酯(pcl)因其环保性能,广泛用于制备可降解塑料和高附加值包装材料,其单体ε-己内酯是一种用途广泛的化学中间体,通过合成反应能够给合成物带来优异的化学性能,在材料领域具有广泛的应用。利用双氧水间接氧化法制备ε-己内酯是目前主流的工艺方法,但反应过程对催化剂的酸度和种类要求十分严格,传统的固体酸催化剂由于活性组分单一且易流失无法满足工业化生产需求。功能化杂多酸离子液体不仅融合了杂多酸和离子液体的性能优势,并且可以通过引入特定官能团来调节酸性强度以实现更高的催化活性和选择性,近年来在催化领域备受人们关注。
3.ε-己内酯单体的应用价值非常高,目前ε-己内酯在国内市场上供不应求。由环己酮氧化法制备ε-己内酯是目前工业化生产ε-己内酯最可靠的工艺方法。由传统的固体酸以及液体酸催化剂来催化环己酮制备ε-己内酯工艺虽比较成熟但还是存在如催化剂活性低、寿命短等问题。同时该工艺由于反应时间过长、产物的收率低等原因无法满足工业化生产要求,并且该催化剂在使用过后,活性组分钨的含量流失比较严重,导致催化剂的重复利用效果不理想。杂多酸因具有独特的物理化学性质和催化性能在环己酮氧化法过程中表现出广阔的应用前景。但由于杂多酸本身在反应过程中易与体系互溶,导致催化剂难以分离与回收。而杂多酸离子液体是一类结构可调节,酸度易控制的固体催化剂,结合ε-己内酯合成反应的特点,发明设计出催化活性高且活性组分稳定高效可重复使用的杂多酸离子液体催化剂对双氧水间接氧化法制备ε-己内酯具有十分重要的意义,在工业上有实际的应用价值。
技术实现要素:
4.本发明所要解决的技术问题是:采用磺酸功能化杂多酸离子液体催化剂代替传统固体酸及液体酸通过双氧水间接氧化法制备ε-己内酯,以解决传统催化剂活性低以及活性组分易流失等问题,在达到优异催化效果的同时尽可能提升催化剂使用稳定性。
5.本发明解决其技术问题采用以下的技术方案:
6.一种磺酸功能化杂多酸离子液体催化剂,所述磺酸功能化杂多酸离子液体催化剂的分子式为[mimps]3pw
12
o4或[nhso]3pw
12o40
。
[0007]
一种磺酸功能化杂多酸离子液体催化剂的用途,所述磺酸功能化杂多酸离子液体催化剂的分子式为[mimps]3pw
12
o4或[nhso]3pw
12o40
;所述磺酸功能化杂多酸离子液体催化剂用于催化合成ε-己内酯。
[0008]
进一步的改进,所述磺酸功能化杂多酸离子液体催化剂用于催化合成ε-己内酯的
方法如下:
[0009]
将52.85-65.19重量份有机酸、25-35重量份带水剂和0.7-0.8重量份磺酸功能化杂多酸离子液体催化剂加入反应容器中,然后滴加0.3重量份的稳定剂,然后调节反应容器至真空度为45-99kpa的负压条件下加热到40-70℃,再加入25重量份50%质量分数的h2o2,边搅拌边保持带水剂回流,连续脱水后得到过氧乙酸反应液,然后向过氧乙酸反应液中逐渐滴入5.4重量份的环己酮,并调整反应温度40-80℃,保温0.5-3h,即反应得到ε-己内酯。
[0010]
进一步的改进,所述有机酸为苯甲酸、氯乙酸、乙酸、丙酸中的一种或任意组合。
[0011]
进一步的改进,所述带水剂为环己烷、乙酸丁酯或丙酸乙酯。
[0012]
进一步的改进,所述连续脱水的时间为2-6h,优选为4h。
[0013]
进一步的改进,所述稳定剂为2-甲基吡啶。
[0014]
一种磺酸功能化杂多酸离子液体催化剂的制备方法,所述磺酸功能化杂多酸离子液体催化剂的分子式为[mimps]3pw
12
o4,制备方法如下:
[0015]
取0.1mol1,3-丙烷磺内酯溶于100ml乙酸乙酯中,然后滴加0.1moln-甲基咪唑,50℃下匀速搅拌3h,过滤得到白色沉淀,白色沉淀采用乙酸乙酯洗涤后加入真空干燥箱干燥得到离子液体前驱体mimps;分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中得到磷钨酸水溶液,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体mimps中,滴加完成后,在25℃的室温条件下搅拌24h,反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[mimps]3pw
12o40
。
[0016]
一种磺酸功能化杂多酸离子液体催化剂的制备方法,所述磺酸功能化杂多酸离子液体催化剂的分子式为[nhso]3pw
12o40
,制备方法如下:
[0017]
取0.05mol1,3-丙烷磺内酯溶于50ml极性溶剂中,然后滴加0.05moln-甲基咪唑,50℃下匀速搅拌3h,过滤得到白色沉淀,白色沉淀采用乙酸乙酯洗涤后加入真空干燥箱干燥得到得到离子液体前驱体nhso,分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中得到磷钨酸水溶液,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体nhso中,滴加完成后,在25℃的室温条件下搅拌24h,反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[nhso]3pw
12o40
。
[0018]
进一步的改进,所述极性溶剂为乙腈或有机酯,有机酯包括乙酸乙酯。
[0019]
本发明与现有技术相比具有以下主要的优点:
[0020]
(1)实用性强:
[0021]
本发明制备的磺酸功能化杂多酸离子液体不仅融合了杂多酸和离子液体的性能优势,并且可以通过引入特定官能团磷钨酸,并且由该酸来调节酸性强度以实现更高的催化活性和选择性。采用双氧水间接氧化法合成ε-己内酯的收率和选择性分别可到达93.26%和99.73%。并且催化剂回收简便,复用性强。
[0022]
(2)ε-己内酯制备工艺简洁,生产周期短:
[0023]
在传统的固体酸或液体酸催化合成ε-己内酯精馏工艺过程中,过氧酸产生阶段和脱水过程需要10小时左右,而过氧酸氧化环己酮反应过程需要5小时左右,生产周期过长。而本发明中磺酸功能化杂多酸离子液体催化第一步过氧酸的合成仅需4小时左右,且制备
ε-己内酯过程仅需1小时左右,极大地缩短了生产周期,提高了生产效率。
[0024]
(3)产品经济收益高:
[0025]
本发明首次提出将制备的磺酸功能化杂多酸离子液体应用于双氧水间接氧化法制备ε-己内酯。发明设计出的多酸离子液体催化剂具有催化活性高且活性组分稳定高效可重复使用等优势,对双氧水间接氧化法制备ε-己内酯具有十分重要的意义,在工业上有实际的应用价值。此工艺方法技术的开发不仅能在技术上填补国内的空白,且具有巨大的经济前景。
[0026]
总之,本发明采用磺酸功能化杂多酸离子液体代替传统固体酸或液体酸应用到ε-己内酯的合成过程中,在反应过程中能够达到优异的催化效果和可观的产品收率,而且整个生产工艺简洁、生产周期短、产品收率高,具有巨大的经济前景。
具体实施方式
[0027]
(1)将1,3-丙烷磺内酯溶于极性溶剂中,然后将n-甲基咪唑/1-(3-氨基丙基)咪唑缓慢滴加到其中,在30-70℃下匀速搅拌2-6h。反应结束后,所得白色沉淀经极性溶剂洗涤3次后过滤,80℃真空干燥4h,得到离子液体前驱体mimps/nhso;按照摩尔比为3比1的比例分别称取mimps/nhso和纯磷钨酸各溶于等量的去离子水中,通过离子交换将其质子交换成磺酸功能化离子液体的阳离子,即将两种溶液在搅拌下缓慢混合,随后在25℃下匀速搅拌24h。反应结束后,对所得产物进行旋蒸、极性溶剂洗涤数次,于80℃真空状态下干燥6h,得到两种功能化杂多酸离子液体催化剂[mimps]3pw
12
o4和[nhso]3pw
12o40
。(2)在催化剂[mimps]3pw
12
o4/[nhso]3pw
12o40
和稳定剂的作用下,以质量分数为50%的双氧水作为氧化剂将有机酸氧化为过氧有机酸,随后将环己酮滴入过氧有机酸溶液中,在40-80℃下反应0.5-3h时间得到质量浓度为5%~10%的ε-己内酯溶液,最后通过精馏得到ε-己内酯。杂多酸离子液体催化剂经简单的过滤分离、溶剂洗涤以及干燥进行回收。
[0028]
下面结合具体实施例对本发明提供的上述方法作进一步描述,只为说明本发明的技术构思及特点,但不构成对本发明的任何限制。
[0029]
实施例1
[0030]
称取1,3-丙烷磺内酯(12.21g,0.1mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(9.02g,0.1mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[mimps]3pw
12o40
。称取52.85g乙酸、35g乙酸丁酯和0.8g杂多酸离子液体催化剂[mimps]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在20.4kpa的负压条件下由水浴锅将混合液加热到40℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧乙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到40℃左右,随后维持此温度匀速搅拌0.5h。取样进行分析环己酮转化率可到达91.25%,ε-己内酯收率可达到
78.28%,选择性可达到85.79%。
[0031]
实施例2
[0032]
称取1,3-丙烷磺内酯(12.21g,0.1mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(9.02g,0.1mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[mimps]3pw
12o40
。称取52.85g乙酸、35g乙酸丁酯和0.8g杂多酸离子液体催化剂[mimps]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在合适的负压条件下26.2kpa由水浴锅将混合液加热到50℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧乙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到60℃左右,随后维持此温度匀速搅拌1h。取样进行分析环己酮转化率可到达94.62%,ε-己内酯收率可达到82.68%,选择性可达到87.38%。
[0033]
实施例3
[0034]
称取1,3-丙烷磺内酯(12.21g,0.1mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(9.02g,0.1mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[mimps]3pw
12o40
。称取52.85g乙酸、35g乙酸丁酯和0.8g杂多酸离子液体催化剂[mimps]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在25kpa的负压条件下由水浴锅将混合液加热到65℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧乙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到70℃左右,随后维持此温度匀速搅拌1h。取样进行分析环己酮转化率可到达93.51%,ε-己内酯收率可达到93.26%,选择性可达到99.73%。
[0035]
实施例4
[0036]
称取1,3-丙烷磺内酯(12.21g,0.1mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(9.02g,0.1mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[mimps]3pw
12o40
。称取52.85g乙酸、35g乙酸丁酯和0.8g杂多酸
离子液体催化剂[mimps]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在25.5kpa的负压条件下由水浴锅将混合液加热到70℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧乙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到70℃左右,随后维持此温度匀速搅拌2h。取样进行分析环己酮转化率可到达86.94%,ε-己内酯收率可达到75.41%,选择性可达到86.74%。
[0037]
实施例5
[0038]
称取1,3-丙烷磺内酯(12.21g,0.1mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(9.02g,0.1mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[mimps]3pw
12o40
。称取52.85g乙酸、35g乙酸丁酯和0.8g杂多酸离子液体催化剂[mimps]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在22kpa的负压条件下由水浴锅将混合液加热到70℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧乙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到80℃左右,随后维持此温度匀速搅拌3h。取样进行分析环己酮转化率可到达93.57%,ε-己内酯收率可达到65.79%,选择性可达到70.31%。
[0039]
实施例6
[0040]
称取1,3-丙烷磺内酯(6.11g,0.05mol)溶于50ml乙酸乙酯中,然后将1-(3-氨基丙基)咪唑(6.26g,0.05mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体nhso。分别称取0.1mol磷钨酸、0.3molnhso各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[nhso]3pw
12o40
。称取65.19g丙酸、25g环己烷和0.7g杂多酸离子液体催化剂[nhso]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在20.4kpa的负压条件下由水浴锅将混合液加热到40℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧丙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到40℃左右,随后维持此温度匀速搅拌0.5h。取样进行分析环己酮转化率可到达72.76%,ε-己内酯收率可达到49.95%,选择性可达到65.90%。
[0041]
实施例7
[0042]
称取1,3-丙烷磺内酯(6.11g,0.05mol)溶于50ml乙酸乙酯中,然后将1-(3-氨基丙
基)咪唑(6.26g,0.05mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体nhso。分别称取0.1mol磷钨酸、0.3molnhso各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[nhso]3pw
12o40
。称取65.19g丙酸、25g环己烷和0.7g杂多酸离子液体催化剂[nhso]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在26.2kpa的负压条件下由水浴锅将混合液加热到60℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧丙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到60℃左右,随后维持此温度匀速搅拌1h。取样进行分析环己酮转化率可到达75.25%,ε-己内酯收率可达到55.50%,选择性可达到73.75%。
[0043]
实施例8
[0044]
称取1,3-丙烷磺内酯(6.11g,0.05mol)溶于50ml乙酸乙酯中,然后将1-(3-氨基丙基)咪唑(6.26g,0.05mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体nhso。分别称取0.1mol磷钨酸、0.3molnhso各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[nhso]3pw
12o40
。称取65.19g丙酸、25g环己烷和0.7g杂多酸离子液体催化剂[nhso]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在25kpa负压条件下由水浴锅将混合液加热到65℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧丙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到70℃左右,随后维持此温度匀速搅拌1h。取样进行分析环己酮转化率可到达89.54%,ε-己内酯收率可达到83.36%,选择性可达到93.10%。
[0045]
实施例9
[0046]
称取1,3-丙烷磺内酯(6.11g,0.05mol)溶于50ml乙酸乙酯中,然后将1-(3-氨基丙基)咪唑(6.26g,0.05mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体nhso。分别称取0.1mol磷钨酸、0.3molnhso各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[nhso]3pw
12o40
。称取65.19g丙酸、25g环己烷和0.7g杂多酸离子液体催化剂[nhso]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在25.5kpa负压条件下由水浴锅将混合液加热到70℃并保持稳定,同时通过恒压漏斗向其中
加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧丙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到70℃左右,随后维持此温度匀速搅拌2h。取样进行分析环己酮转化率可到达79.60%,ε-己内酯收率可达到62.60%,选择性可达到78.64%。
[0047]
实施例10
[0048]
称取1,3-丙烷磺内酯(6.11g,0.05mol)溶于50ml乙酸乙酯中,然后将1-(3-氨基丙基)咪唑(6.26g,0.05mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体nhso。分别称取0.1mol磷钨酸、0.3molnhso各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥6h,所得白色固体即为[nhso]3pw
12o40
。称取65.19g丙酸、25g环己烷和0.7g杂多酸离子液体催化剂[nhso]3pw
12o40
,将其依次加入到500ml三口烧瓶(带有温度计、分水器和回流冷凝管)中,再向其中滴入0.3g2-甲基吡啶稳定剂,随后调整体系的真空度,在22kpa负压条件下由水浴锅将混合液加热到70℃并保持稳定,同时通过恒压漏斗向其中加入25g50%(质量分数)的h2o2,边搅拌边保持带水剂回流,经过4h的连续脱水后,由恒压漏斗中向含过氧丙酸反应液中缓慢滴入5.4g环己酮,调整水浴锅温度到80℃左右,随后维持此温度匀速搅拌3h。取样进行分析环己酮转化率可到达93.25%,ε-己内酯收率可达到54.31%,选择性可达到58.24%。
[0049]
对比例1
[0050]
称取1,3-丙烷磺内酯(24.42g,0.2mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(18.04g,0.2mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥24h,仍然未能得到白色固体,物料仍处于粘稠状态。
[0051]
对比例2
[0052]
称取1,3-丙烷磺内酯(3.05g,0.025mol)溶于100ml乙酸乙酯中,然后将n-甲基咪唑(3.13g,0.025mol)缓慢滴加到其中,之后在50℃下匀速搅拌3h。反应结束后,所得白色沉淀经乙酸乙酯洗涤3次后过滤,放入80℃的真空干燥箱中干燥4h,得到离子液体前驱体mimps。分别称取0.1mol磷钨酸、0.3molmimps各溶于20ml去离子水中,随后在搅拌下将磷钨酸水溶液以每分钟30-35滴的速率缓慢加入到离子液体前驱体水溶液中,滴加完成后,在25℃的室温条件下搅拌24h。反应结束后,所得产物经旋蒸、乙酸乙酯洗涤数次后置于真空干燥箱中80℃干燥24h,仍然未能得到白色固体,物料仍处于粘稠状态。
[0053]
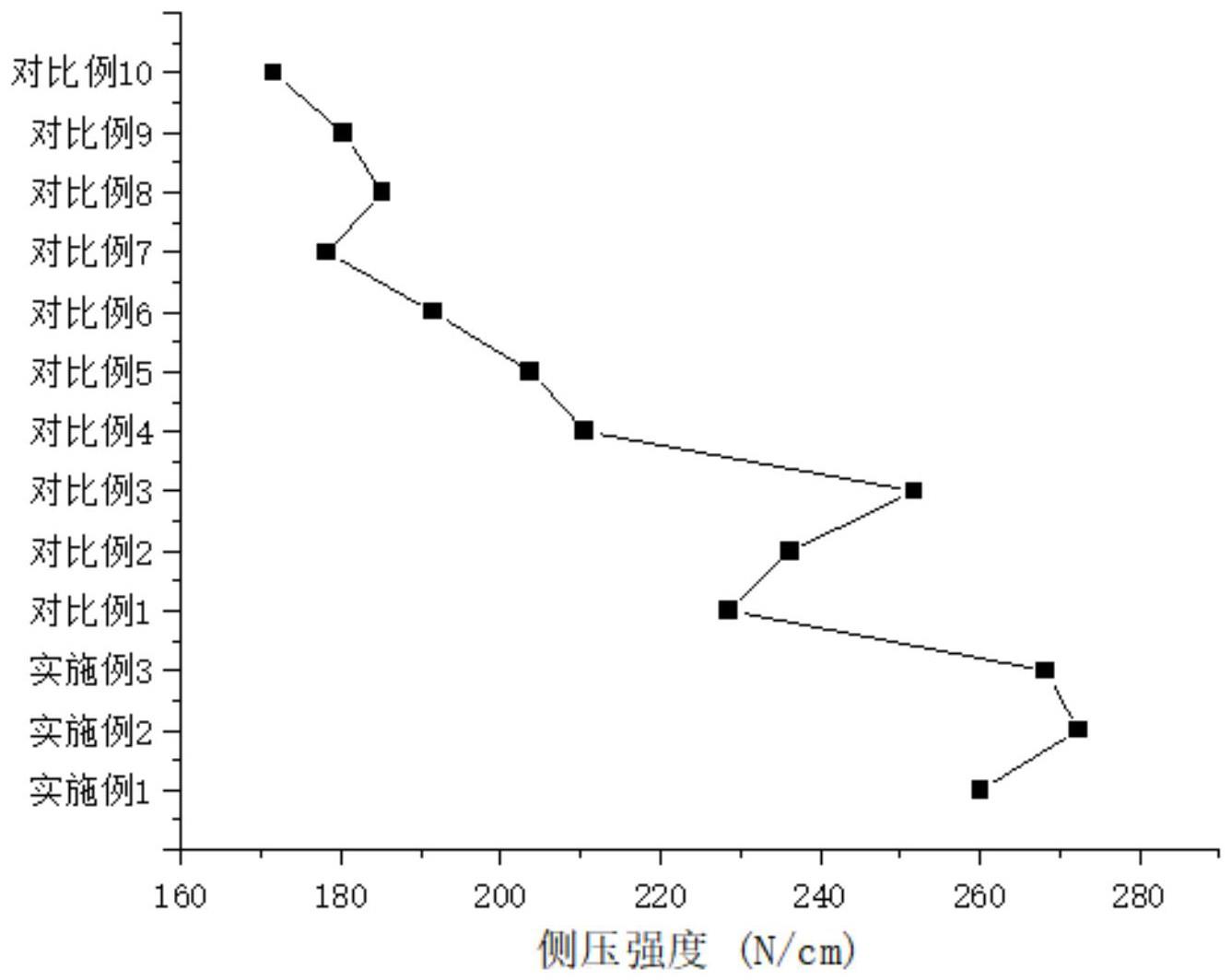
从实施例1-10可以得出,当1,3-丙烷磺内酯和n-甲基咪唑用量范围均为0.05mol-0.1mol时,制得的杂多酸离子液体催化剂具有良好的催化环己酮转化为ε-己内酯的催化效率;而其用量降低或升高后则难以得到固体产物,这是由于1,3-丙烷磺内酯和n-甲基咪唑在进行环杂化反应时,物料过多和过低都会造成两个环状物质在杂化时出现杂化基团闭合
或者基团脱落的现象;当物料过少时,环状物质经过开环杂化,裸露出来的阳离子基团因杂化键能过低引发整体的脱落,导致最终制备出来的前驱体阳离子活性过低,无法与阴离子进行很好的结合,影响了整体液体离子的性能。当物料过多时,在杂化的过程中裸露出来的阳离子基团会与剩余未反应的带负电荷的基团形成离子键,导致杂化基团出现假性闭合,最终无法裸露出过多的阳离子基团,导致前驱体阳离子活性过低。
[0054]
此外从实施例1-5和实施例6-10可以得出,1,3-丙烷磺内酯和n-甲基咪唑用量为0.1mol时的催化效果明显优于用量为0.05mol时,这是由于1,3-丙烷磺内酯与n-甲基咪唑的反应主要为环状物质之间的杂化反应,随着两种物质用量的增加相对应的杂化的程度也会上升,前驱体mimps在制备时的物料用量要大于前驱体nhso制备时的物料用量,相应的mimps这一前驱体的环状物质杂化程度更高,其阳离子的活性更高,能够结合更多的阴离子,通过阴阳离子之间的极性作用力使得最终的[nhso]3pw
12o40
具有更高的催化活性和选择性,而nhso这一前驱体的阳离子活性稍低,最终制备而成的离子液体催化剂[nhso]3pw
12o40
的催化活性和选择性偏低。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。