黑皮质素受体激动剂化合物的晶型iii及其制备方法
技术领域
1.相关申请的交叉引用
2.本技术要求于2020年10月29日提交的韩国专利申请第10-2020-0142398号的优先权权益,该韩国专利申请的全部公开内容作为本说明书的一部分并入。
3.技术领域
4.本发明涉及一种对黑皮质素受体表现出优异的激动剂活性的新化合物的晶型iii、其制备方法以及包含其的药物组合物。
背景技术:
5.瘦素蛋白是一种由脂肪细胞分泌的激素,其分泌量随着体脂含量的增加而增加。它调节下丘脑产生的各种神经肽的功能,从而调节各种体内功能,包括食欲、体脂含量和能量代谢(schwartz等人,nature 404,661-671(2000))。控制食欲和体重的瘦素蛋白信号转导是通过调节许多下游因子来进行,其中最具代表性的是黑皮质素、刺鼠相关肽(agrp)和神经肽y(npy)激素。
6.当体内热量过多导致血液中瘦素浓度增加时,垂体腺中促阿片黑皮素(pomc)蛋白激素的分泌增加,agrp和npy的产生减少。小肽激素α-黑素细胞刺激激素(msh)由pomc神经元产生。该激素是次级神经元的黑皮质素-4受体(mc4r)的激动剂,最终导致食欲下降。同时,当瘦素浓度因热量不足而降低时,mc4r拮抗剂agrp的表达增加,npy的表达也增加,最终增强食欲。即,根据瘦素的变化,α-msh激素和agrp激素充当mc4r的激动剂和拮抗剂,因此参与食欲控制。
7.除mc4r以外,α-msh激素还结合三种mcr亚型以诱导各种生理反应。迄今已经鉴定了五种mcr亚型。其中,已知mc1r主要在皮肤细胞中表达并参与调节黑色素沉着(皮肤色素沉着)。mc2r主要在肾上腺中表达,已知参与糖皮质激素的产生。其配体只有来源于pomc的促肾上腺皮质激素(acth)。主要在中枢神经系统中表达的mc3r和mc4r参与调节食欲、能量代谢和体脂储存效率,而已知在各种组织中表达的mc5r调节外分泌功能(wikberg等人,pharm res 42(5)393-420(2000))。特别地,激活mc4r受体诱导食欲下降和能量代谢增加,因此具有有效减轻体重的效果。因此,已经被证明是抗肥胖药物开发的主要作用点(综述:wikberg,eur.j.pharmacol 375,295-310(1999));wikberg等人,pharm res 42(5)393-420(2000);douglas等人,eur j pharm 450,93-109(2002);o'rahilly等人,nature med 10,351-352(2004))。
8.mc4r在控制食欲和体重中的作用已通过在刺鼠蛋白异常表达动物模型(agouti小鼠)中的实验得到了初步证明。在agouti小鼠的情况下,发现由于基因突变之故,刺鼠蛋白在中枢神经系统中以高浓度表达,并在下丘脑中充当mc4r的拮抗剂,从而导致肥胖(yen,tt等人,faseb j.8,479-488(1994);lu d.等人,nature 371,799-802(1994))。随后的研究结果表明类似于实际刺鼠蛋白的刺鼠相关肽(agrp)在下丘脑神经中表达,已知这些肽是mc4r拮抗剂并参与控制食欲(shutter等人,genes dev.,11,593-602(1997);ollman等人,
science 278,135-138(1997))。
9.对动物脑内施用体内mc4r激动剂α-msh产生了降低食欲的效果。当用mc4r拮抗剂shu9119(肽)或hs014(肽)治疗所述动物时,观察到食欲再次增加(kask等人,biochem.biophys.res.comm.245,90-93(1998))。此外,在使用美拉诺坦ii(mtii,ac-nle-c[asp-his-dphe-arg-trp-lys]-nh2)及其类似激动剂hp228的动物研究中,在脑内、腹膜内或皮下施用后,发现了抑制食欲、减轻体重和增加能量代谢等功效。(thiele t.e.等人,am j physiol 274(1pt 2),r248-54(1998);lee m.d.等人,faseb j 12,a552(1998);murphy b.等人,j appl physiol 89,273-82(2000))。相比之下,向动物施用代表性shu9119表现出显著且持续的采食量和体重增加,从而提供了mcr激动剂可能是抗肥胖剂的药理学证据。施用mtii后明显表现出来的食欲降低效果在mc4r敲除(ko)小鼠中没有观察到。该实验结果再次证明食欲降低效果主要通过激活mc4r来实现(marsh等人,nat genet 21,119-122(1999))。
[0010]
作用于中枢神经系统的食欲抑制剂是迄今开发的抗肥胖药物中的主要类型。其中,大部分是调节神经递质作用的药物。实例包括去甲肾上腺素剂(芬特明(phentermine)和马吲哚(mazindol))、血清素能剂、氟西汀(fluoxetine)和西布曲明(sibutramine)等。然而,除了食欲抑制以外,神经递质调节剂还通过许多亚型受体对各种生理作用具有广泛影响。因此,所述调节剂缺乏对每一种亚型的选择性,因此主要缺点是它们在长期施用时会伴随各种副作用。
[0011]
另一方面,黑皮质素激动剂是神经肽,而不是神经递质。鉴于在mc4r基因ko小鼠中,除能量代谢外的所有功能都正常,因此黑皮质素作为作用点的优势在于它们仅会通过食欲抑制来诱导体重减轻,而不会影响其它生理功能。特别地,该受体是g蛋白偶联受体(gpcr),属于迄今开发的新药作用点中最成功的类别。因此,所述作用点与现有作用点有很大的不同,因为它相对容易确保对亚型受体的选择性。
[0012]
作为利用黑皮质素受体作为作用点的实例,国际公开第wo 2008/007930号和第wo 2010/056022号公开了作为黑皮质素受体激动剂的化合物。
[0013]
此外,本发明的发明人已经进行了广泛的研究,并发明了一种具有优异的激动剂活性的下式1的新化合物,所述化合物对黑皮质素受体,特别是黑皮质素-4受体(mc4r)具有选择性,以及一种制备所述化合物的方法(申请号kr 10-2019-0141649(于2019年11月7日提交)):
[0014]
[式1]
[0015]
[0016]
其中r1是c
2-c5烷基。
[0017]
同时,药物活性成分的晶体结构往往会影响药物的化学稳定性。不同的结晶条件和储存条件可能导致化合物晶体结构的变化,有时还会伴随产生其它形式的晶型。一般来说,无定形药物产品不具有规则的晶体结构,而且往往存在其它缺陷,如产品稳定性差、粒径较小、过滤困难、容易结块、流动性差。因此,有必要改善产品的各项物理性能。因此,有必要研究单一化合物的具有高纯度和良好化学稳定性的晶体结构。
[0018]
[现有技术文献]
[0019]
[专利文献]
[0020]
(专利文献1)国际专利申请公开号wo 2008/007930
[0021]
(专利文献2)国际专利申请公开号wo 2010/056022
技术实现要素:
[0022]
技术问题
[0023]
本发明的一个方面提供了一种具有优异的激动剂活性的对黑皮质素受体,特别是黑皮质素-4受体(mc4r)具有选择性的新化合物的稳定晶型,以及制备所述晶型的方法。
[0024]
本发明的另一个方面提供了一种包含所述新化合物的稳定晶型的药物组合物。
[0025]
技术方案
[0026]
根据本发明的一个方面,提供了一种下式1化合物、其药学上可接受的盐或溶剂合物的晶型iii,其中x射线粉末衍射图案具有选自具有以下衍射角(2θ值)的峰的3个以上、5个以上、7个以上、9个以上或10个以上特征峰:6.62
±
0.2
°
、7.44
±
0.2
°
、9.18
±
0.2
°
、9.89
±
0.2
°
、10.83
±
0.2
°
、11.42
±
0.2
°
、12.92
±
0.2
°
、14.61
±
0.2
°
、15.36
±
0.2
°
、15.79
±
0.2
°
、15.95
±
0.2
°
、17.37
±
0.2
°
、18.20
±
0.2
°
、18.99
±
0.2
°
、19.34
±
0.2
°
、19.69
±
0.2
°
、20.40
±
0.2
°
、21.66
±
0.2
°
、21.98
±
0.2
°
、22.45
±
0.2
°
、22.85
±
0.2
°
、24.66
±
0.2
°
、25.52
±
0.2
°
、26.55
±
0.2
°
、28.08
±
0.2
°
、29.31
±
0.2
°
以及29.54
±
0.2
°
,
[0027]
[式1]
[0028][0029]
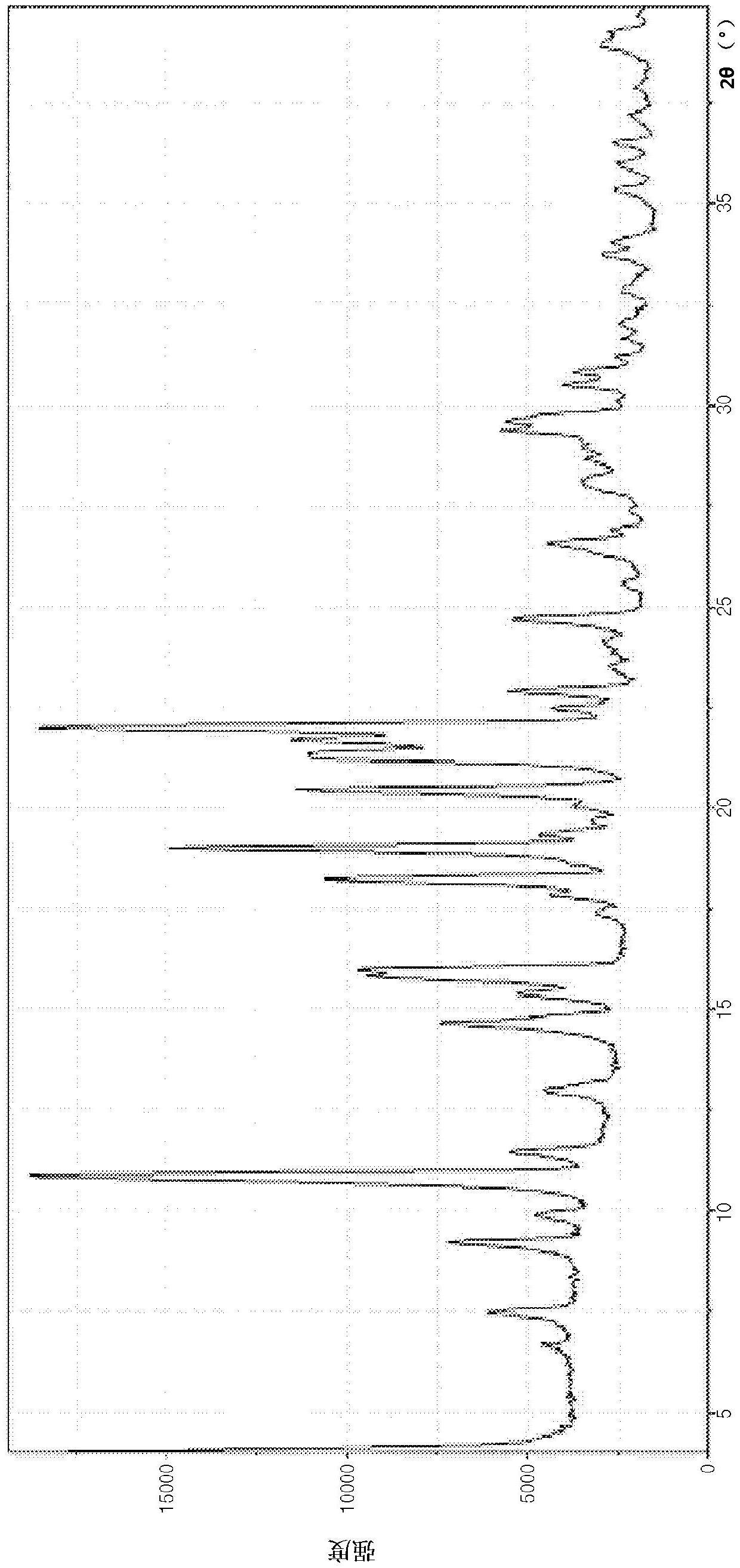
其中r1是c
2-c5烷基。
[0030]
由于式1化合物可以具有不对称的碳中心和不对称的轴或不对称的平面,因此它可以作为顺式或反式异构体、r或s异构体、外消旋体、非对映体混合物和个别非对映体存在,所有这些都在式1化合物的范围内。
[0031]
在本说明书中,除非另有说明,否则使用式1化合物来包括所有式1化合物、其药学
上可接受的盐、异构体和溶剂合物。
[0032]
在根据本发明的一个实施方式中,在式1中,r1是c2至c5烷基。在根据本发明的另一个实施方式中,在式1中,r1是直链或支链c2至c5烷基,例如乙基、正丙基、异丙基、正丁基、异丁基、仲丁基或叔丁基。
[0033]
在根据本发明的另一个实施方式中,在式1中,r1是c2或c3烷基。在根据本发明的另一个实施方式中,在式1中,r1是直链或支链c2或c3烷基,例如乙基、正丙基或异丙基。
[0034]
在根据本发明的一个实施方式中,所述药学上可接受的盐包括但不限于酸加成盐,所述酸加成盐由以下酸形成:无机酸,如盐酸、硫酸、硝酸、磷酸、氢溴酸和氢碘酸;有机羧酸,如酒石酸、甲酸、柠檬酸、乙酸、三氯乙酸、三氟乙酸、葡萄糖酸、苯甲酸、乳酸、富马酸和马来酸;或磺酸,如甲磺酸、苯磺酸、对甲苯磺酸或萘磺酸。
[0035]
在根据本发明的一个实施方式中,所述溶剂合物可以包括水合物;以及与有机溶剂如甲醇、乙醇、2-丙醇、1,2-丙二醇、1,3-丙二醇、正丁醇、1,4-丁二醇、叔丁醇、乙酸、丙酮、乙酸丁酯、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸叔丁酯、乙酸异丁酯、甲乙酮、2-戊酮、四氢呋喃、乙腈、三氯甲烷、甲苯及其混合物的溶剂合物。
[0036]
在根据本发明的一个实施方式中,所述晶型iii可以是式1化合物的药学上可接受的盐的晶型。
[0037]
所述式1化合物的药学上可接受的盐可以是下式2的盐酸盐化合物:
[0038]
[式2]
[0039][0040]
其中r2是c
2-c5烷基。
[0041]
在根据本发明的另一个实施方式中,所述式1化合物的药学上可接受的盐可以是下式3的n-((3s,5s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-5-(吗啉-4-羰基)吡咯烷-3-基)-n-((1s,4r)-4-甲基环己基)异丁酰胺盐酸盐。
[0042]
[式3]
[0043][0044]
在根据本发明的另一个实施方式中,所述晶型iii可以是式1化合物的药学上可接受的盐的溶剂合物,具体来说是水合物的晶型。
[0045]
更具体地,所述晶型iii可以是式1化合物的盐酸盐的水合物的晶型。
[0046]
在根据本发明的一个实施方式中,所述晶型iii可以是下式4化合物的晶型。
[0047]
[式4]
[0048][0049]
如通过x射线粉末衍射(xrd)所分析,根据本发明的晶型iii显示出选自具有以下2θ值的峰的3个以上、5个以上、7个以上、9个以上或10个以上特征峰:6.62
±
0.2
°
、7.44
±
0.2
°
、9.18
±
0.2
°
、9.89
±
0.2
°
、10.83
±
0.2
°
、11.42
±
0.2
°
、12.92
±
0.2
°
、14.61
±
0.2
°
、15.36
±
0.2
°
、15.79
±
0.2
°
、15.95
±
0.2
°
、17.37
±
0.2
°
、18.20
±
0.2
°
、18.99
±
0.2
°
、19.34
±
0.2
°
、19.69
±
0.2
°
、20.40
±
0.2
°
、21.66
±
0.2
°
、21.98
±
0.2
°
、22.45
±
0.2
°
、22.85
±
0.2
°
、24.66
±
0.2
°
、25.52
±
0.2
°
、26.55
±
0.2
°
、28.08
±
0.2
°
、29.31
±
0.2
°
以及29.54
±
0.2
°
。
[0050]
在根据本发明的一个实施方式中,所述晶型iii可以具有图4所示的xrd图案。
[0051]
在根据本发明的晶型iii的差示扫描量热法(dsc)曲线中,两个吸热峰出现在40℃至170℃,由于分解导致的吸热峰出现在220℃以上。
[0052]
在根据本发明的一个实施方式中,所述晶型iii可以具有图5所示的dsc曲线。
[0053]
在热重分析(tga)曲线中,根据本发明的晶型iii在加热至160℃以下的温度时可能存在15%以下,例如1%至15%、1%至10%、5%至10%、或7%的重量损失。
[0054]
在根据本发明的一个实施方式中,所述晶型iii可以具有图6所示的tga曲线。
[0055]
稳定性测试结果(hplc)表明根据本发明的晶型iii在加速条件(40℃,75% rh)和苛性条件(80℃)下表现出化学稳定性达4周。因此,可见根据本发明的晶型iii对热和湿度
稳定。
[0056]
在本说明书中,x射线衍射(xrd)分析示出了使用panalytical x'pert pro mpd系统(malvern panalytical有限公司)进行的结果。
[0057]
差示扫描量热法(dsc)分析示出了使用dsc1(mettler-toledo控股公司)进行的结果。
[0058]
热重分析(tga)示出了使用tga/dsc 1(mettler-toledo控股公司)进行的结果。
[0059]
稳定性分析示出了使用hplc(agilent technologies公司)进行的结果。
[0060]
所述晶型iii的纯度可以比式1的粗化合物、式1的无定形化合物或式1化合物的其它晶型更高,并且可以在物理和化学上更稳定。
[0061]
此外,与已知的黑皮质素-4受体激动剂相比,式1化合物的晶型iii对黑皮质素-4受体的激动能力和对疾病如肥胖、糖尿病、炎症、勃起功能障碍等的预防或治疗效果可能更加优异。然而,本发明的效果不限于此。
[0062]
在另一个方面,本发明提供了一种制备所述晶型iii的方法,所述方法包括以下步骤:通过将式1化合物溶解在结晶溶剂中来制备混合溶液;以及从所述混合溶液获得晶体。
[0063]
首先,将由式1表示的化合物溶解在结晶溶剂中。
[0064]
用于制备晶型iii的式1化合物可以是式1化合物、其盐、其异构体或其溶剂合物。
[0065]
式1化合物可以通过申请号kr 10-2019-0141649(于2019年11月7日提交)的说明书所述的制备方法来获得。
[0066]
结晶溶剂可以不受特别限制地使用,只要它是化合物结晶的合适溶剂即可。在一个实施方式中,结晶溶剂可以包括水和极性非质子有机溶剂的混合物。
[0067]
极性非质子有机溶剂可以包括乙酸乙酯、甲基异丁基酮、二甲亚砜、四氢呋喃、丙酮、二甲基甲酰胺、乙腈或其混合物。
[0068]
在根据本发明的一个实施方式中,所述极性非质子有机溶剂可以包括乙酸乙酯。
[0069]
在根据本发明的一个实施方式中,所述结晶溶剂可以是水和所述极性非质子有机溶剂以15:1至1:15,具体地以10:1至1:10、8:1至1:8、1:1至1:10、1:3至1:8、1:5至1:7、1:6.5至1:6.8、或1:6.7的体积比混合的混合溶剂。
[0070]
对于1g式1化合物,可以使用0.5至5ml、0.7至3ml、0.8至2ml、0.9至1.5ml、1至1.5ml、1.0至1.3ml、或1.15ml的结晶溶剂。
[0071]
将式1化合物溶解在结晶溶剂中可以在30℃至85℃,具体地在35℃至80℃、40℃至75℃、45℃至70℃、50℃至65℃、或60℃下不搅拌或随搅拌进行。
[0072]
在根据本发明的一个实施方式中,通过对于1g式1化合物使用1ml etoac和0.15ml蒸馏水可以获得式1化合物在60℃已经溶解在其中的混合溶液。
[0073]
接下来,所述方法包括从式1化合物已经溶解在其中的混合溶液获得晶体的步骤。所述晶体可以例如通过冷却溶液、通过向溶液滴加酸以形成沉淀物、通过蒸发溶剂、通过加入反溶剂以实现过饱和或通过使用如浆液转化等方法来获得。
[0074]
在根据本发明的一个实施方式中,所述结晶步骤可以包括冷却所述混合溶液。可以进行冷却,使得已经滴加了所述酸的混合溶液的温度变为0℃至5℃。具体地,可以进行冷却,使得所述混合溶液的温度变为0℃至3℃。
[0075]
此外,结晶步骤可以包括搅拌所述混合溶液。所述搅拌可以通过已知的手段进行,
搅拌时间为例如但不限于15小时至50小时(包括),具体地,15小时至50小时、15小时至45小时、15小时至40小时、15小时至35小时、20小时至30小时、23小时至28小时、或25小时。
[0076]
在根据本发明的另一个实施方式中,可以对通过冷却所述混合溶液并且在保持冷却的所述混合溶液的温度的同时搅拌而形成的沉淀物进行过滤和洗涤,以获得晶体。
[0077]
在根据本发明的另一个实施方式中,所述方法还可以包括在冷却所述混合溶液之前、之后或同时,或者在搅拌所述混合溶液之前、之后或同时向所述混合溶液中加入非极性有机溶剂的步骤。可以通过加入非极性有机溶剂以增加结晶粒子的产率来提高所获得的晶型iii的收率或产生稳定性,但本发明不限于此。所述非极性有机溶剂可以不受特别限制地使用,只要它是具有非极性性质的有机溶剂即可,但可以使用例如己烷、庚烷、环己烷、四氯化碳、苯、三氯甲烷等。在根据本发明的一个实施方式中,所述方法可以包括在从所述混合溶液中结晶的过程中向溶液中加入庚烷的步骤。
[0078]
如上获得的晶型iii的纯度可以比式1的粗化合物、式1的无定形化合物或式1的任何其它晶型更高,并且可以在物理和化学上更稳定。然而,本发明的效果不限于此。
[0079]
在另一个方面,本发明提供了一种药物组合物,所述药物组合物包含:(i)所述晶型iii;和(ii)药学上可接受的载体。
[0080]
根据本发明的晶型iii对黑皮质素受体,特别是黑皮质素-4受体(mc4r)表现出优异的激动作用。因此,本发明可以提供一种用于激动黑皮质素受体的药物组合物,所述组合物含有上述晶型iii作为活性成分。具体地,所述药物组合物可以是用于激动黑皮质素-4受体的功能的组合物。
[0081]
此外,由于所述药物组合物可以表现出预防或治疗肥胖、糖尿病、炎症和勃起功能障碍的优异效果,因此它可以是用于预防或治疗肥胖、糖尿病、炎症或勃起功能障碍的组合物。然而,本发明的用途不限于所述疾病。
[0082]
如本文所用,“载体”是指有助于将化合物引入细胞或组织中的化合物。
[0083]
当出于临床目的施用本发明的晶型iii时,以单次剂量或分次剂量施用于宿主的总日剂量可以优选在0.01至10mg/kg体重的范围内。然而,个别患者的具体剂量水平可以取决于所使用的具体化合物、患者的体重、性别、健康状况、饮食、药物的施用时间、施用方法、排泄率、药物组合和疾病的严重程度等等。
[0084]
本发明的晶型iii可以按需要通过任何途径施用。例如,本发明的无定形化合物可以通过注射或口服施用。
[0085]
本发明的药物组合物可以是各种口服剂型,如片剂、丸剂、粉剂、胶囊、颗粒、糖浆或乳剂,或肠胃外剂型,如用于肌肉内、静脉内或皮下施用的注射制剂。
[0086]
注射用制剂可以根据已知技术,使用合适的分散剂、润湿剂、悬浮剂或赋形剂来制备。
[0087]
可用于本发明的药物制剂的赋形剂包括但不限于甜味剂、粘合剂、加溶剂、增溶剂、润湿剂、乳化剂、等渗剂、吸附剂、崩解剂、抗氧化剂、防腐剂、润滑剂、填充剂、芳香剂等。例如,作为赋形剂,可以使用乳糖、葡萄糖、蔗糖、甘露醇、山梨醇、纤维素、甘氨酸、二氧化硅、硅酸镁铝、淀粉、明胶、黄蓍树胶、海藻酸、海藻酸钠、甲基纤维素、羧甲基纤维素钠、水、乙醇、聚乙二醇、聚乙烯吡咯烷酮、氯化钠、氯化钙、橙香精、草莓香精、香草调味剂等。
[0088]
当本发明的药物组合物为口服剂型时,所使用的载体的实例可以包括但不限于纤
维素、硅酸钙、玉米淀粉、乳糖、蔗糖、葡萄糖、磷酸钙、硬脂酸、硬脂酸镁、硬脂酸钙、明胶、滑石等。
[0089]
当本发明的药物组合物为可注射制剂形式时,载体的实例可以包括但不限于水、生理盐水、葡萄糖水溶液、类糖水溶液、醇、乙二醇、醚、油、脂肪酸、脂肪酸酯、甘油酯等。
[0090]
在另一个方面,提供了一种如上所述的晶型iii以用于激动黑皮质素受体,特别是黑皮质素-4受体(mc4r)的功能。
[0091]
在一个实施方式中,提供了一种如上所述的晶型iii以用于治疗或预防肥胖、糖尿病、炎症或勃起功能障碍。
[0092]
在另一个方面,提供了一种用于激动黑皮质素受体,特别是黑皮质素-4受体(mc4r)的功能的方法,所述方法包括向受试者施用上述晶型iii的步骤。
[0093]
在另一个方面,提供了一种用于治疗肥胖、糖尿病、炎症或勃起功能障碍的方法,所述方法包括向受试者施用上述晶型iii的步骤。
[0094]
有益效果
[0095]
根据本发明的晶型iii对黑皮质素受体,特别是黑皮质素-4受体(mc4r)表现出了优异的激动作用,因此可以有效地用于预防或治疗肥胖、糖尿病、炎症和勃起功能障碍。
[0096]
根据本发明的晶型iii对黑皮质素-4受体表现出了中靶效应,从而表现出体重减轻和饮食减少效果,而不影响焦虑和抑郁。此外,它可以在没有任何安全问题,如人ether-a-go-go相关基因(herg)抑制或诱变的副作用的情况下施用。
[0097]
此外,根据本发明的晶型iii的纯度、收率、物理和化学稳定性比式1的粗化合物、式1的无定形化合物或式1的任何其它晶型更加优异。
[0098]
具体地,所述晶型iii的溶解度、储存稳定性和生产稳定性可以优于式1的粗化合物、式1的无定形化合物或式1的任何其它晶型。
附图说明
[0099]
图1是制备例4的xrd结果的图。
[0100]
图2是制备例4的dsc结果的图。
[0101]
图3是制备例4的tga结果的图。
[0102]
图4是实施例1的xrd结果的图。
[0103]
图5是实施例1的dsc结果的图。
[0104]
图6是实施例1的tga结果的图。
具体实施方式
[0105]
下文将通过制备例和实施例更详细地描述本发明。然而,这些实施例仅仅说明本发明,本发明的范围不限于此。
[0106]
制备例1:(2s,4s)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸甲酯盐酸盐的制备
[0107][0108]
通过以下步骤a、b、c、d和e获得了标题化合物。
[0109]
步骤a:(2s,4s)-4-偶氮吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯的制备
[0110]
在氮气下将(2s,4r)-4-((甲基磺酰基)氧基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(48.5g,150mmol)溶解在n,n'-二甲基甲酰胺(250ml)中,加入叠氮化钠(19.5g,300ml)。在80℃搅拌16小时后,减压浓缩反应溶剂,加入水,用乙酸乙酯萃取两次。用氯化钠水溶液和水洗涤有机层,经无水硫酸镁干燥,并过滤。减压浓缩滤液,得到粗物质(2s,4s)-4-偶氮吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(39.59g,98%),未进行纯化便用于下一步骤。
[0111]
ms[m h]=271(m 1)
[0112]1h nmr(400mhz,cd3od)δ4.43-4.37(m,1h),4.35-4.27(br,1h),3.77(s,1.8h),3.76(s,1.2h),3.73-3.66(m,1h),3.44-3.38(m,1h),2.63-2.49(m,1h),2.19-2.11(m,1h),1.50(s,4.5h),1.44(s,4.5h)
[0113]
步骤b:(2s,4s)-4-氨基吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯的制备
[0114]
将上述步骤a得到的(2s,4s)-4-偶氮吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(24.59g,91.0mmol)溶解在四氢呋喃(180ml)中,在0℃缓慢加入1m三甲基膦四氢呋喃溶液(109.2ml,109.2mmol)。在该温度下搅拌1小时后,室温搅拌混合物3小时。减压浓缩反应溶剂后,加入二氯甲烷(100ml)和水(150ml),并将混合物搅拌约30分钟。进行层分离,再一次用二氯甲烷萃取,有机层经无水硫酸镁干燥并过滤。减压浓缩滤液,得到粗物质(2s,4s)-4-氨基吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(20.62g,93%),未进行纯化便用于下一步骤。
[0115]
ms[m h]=245(m 1)
[0116]1h nmr(400mhz,cd3od)δ4.27(m,1h),3.77(s,1.8h),3.76(s,1.2h),3.75-3.67(m,1h),3.50-3.42(m,1h),3.22-3.17(m,1h),2.58-2.47(m,1h),1.82-1.71(m,1h),1.48(s,4.5h),1.42(s,4.5h)
[0117]
步骤c:(2s,4s)-4-(((1s,4r)-4-甲基环己基)氨基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯的制备
[0118]
将上述步骤b得到的(2s,4s)-4-氨基吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(20.62g,84.4mmol)溶解在二氯乙烷(150ml)中,加入4-甲基环己酮(9.5ml,101.3mmol)。在0℃加入三乙酰氧基硼氢化钠(26.8g,126.6mmol),室温搅拌混合物16小时。减压浓缩反应溶剂,加入水,用乙酸乙酯萃取两次。用氯化钠水溶液洗涤有机层,经无水硫酸镁干燥,并过滤。减压浓缩滤液并通过柱色谱法进行纯化,得到(2s,4s)-4-(((1s,4r)-4-甲基环己基)氨基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(22.9g,80%)。
[0119]
ms[m h]=341(m 1)
[0120]1h nmr(400mhz,cd3od)δ4.26(m,1h),3.76(s,1.8h),3.75(s,1.2h),3.78-3.71
(m,1h),3.49-3.40(m,1h),3.22-3.16(m,1h),2.69-2.60(br,1h),2.58-2.46(m,1h),1.87-1.77(m,1h),1.73-1.63(m,1h),1.62-1.35(m,8h),1.48(s,4.5h),1.42(s,4.5h),0.96(d,3h)
[0121]
步骤d:(2s,4s)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯的制备
[0122]
将上述步骤c得到的(2s,4s)-4-(((1s,4r)-4-甲基环己基)氨基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(37.29g,109.5mmol)溶解在二氯甲烷(500ml)中,加入三乙胺(61.1ml,438.1mmol),然后在0℃缓慢加入异丁酰氯(11.7ml,219mmol)。室温搅拌16小时后,减压浓缩反应溶剂,加入碳酸氢钠水溶液,用乙酸乙酯萃取两次。用氯化钠水溶液和水洗涤有机层,经无水硫酸镁干燥,并过滤。减压浓缩滤液,并通过柱色谱法进行纯化,得到(2s,4s)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(38.79g,86%)。
[0123]
ms[m h]=411(m 1)
[0124]1h nmr(400mhz,cd3od)δ4.27(m,1h),3.76(s,1.8h),3.75(s,1.2h),3.78-3.72(m,1h),3.50-3.41(m,1h),3.33-3.14(m,1h),2.69-2.60(m,2h),2.57-2.43(m,1h),1.87-1.79(m,1h),1.70-1.61(m,1h),1.60-1.32(m,8h),1.47(s,4.5h),1.41(s,4.5h),1.10(dd,6h),0.99(d,3h)
[0125]
步骤e:(2s,4s)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸甲酯盐酸盐的制备
[0126]
将上述步骤d得到的(2s,4s)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-1,2-二甲酸1-(叔丁基)酯2-甲酯(34.0g,82.8mmol)溶解在二氯甲烷(200ml)中,在0℃加入4n盐酸在1,4-二烷溶液中的溶液(82.8ml,331.3mmol)。室温搅拌6小时后,减压浓缩反应溶剂,得到粗物质(28.7g,99%),未进行纯化便用于下一步骤。
[0127]
ms[m h]=311(m 1)
[0128]
制备例2:(3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-甲酸的制备
[0129][0130]
根据国际专利公开号wo 2004/092126所述的方法获得标题化合物。
[0131]
ms[m h]=282(m 1)
[0132]1h nmr(400mhz,cd3od)δ7.43-7.33(m,4h),3.90-3.69(m,3h),3.59(dd,j=11.2,10.0hz,1h),3.29(dd,j=11.2,11.2hz,1h),3.18-3.09(m,1h),1.44(s,9h)
[0133]
制备例3:n-((3s,5s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-5-(吗啉-4-羰基)吡咯烷-3-基)-n-((1s,4r)-4-甲基环己基)异丁酰胺的制备
[0134][0135]
通过以下步骤a、b和c获得标题化合物。
[0136]
步骤a:(2s,4s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸甲酯的制备
[0137]
将制备例1得到的(2s,4s)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸甲酯盐酸盐(28.7g,82.73mmol)、制备例2得到的(3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-甲酸(24.5g,86.87mmol)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(22.2g,115.83mmol)和1-羟基苯并三唑水合物(15.7g,115.83mmol)溶解在n,n'-二甲基甲酰胺(400ml)中,缓慢加入n,n'-二异丙基乙胺(72.0ml,413.66mmol)。室温搅拌16小时后,减压浓缩反应溶剂,加入0.5n氢氧化钠水溶液,用乙酸乙酯萃取两次。有机层用氯化钠水溶液和水洗涤两次,经无水硫酸镁干燥,并过滤。减压浓缩滤液,并通过柱色谱法进行纯化,得到(2s,4s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸甲酯(41.19g,87%)。
[0138]
ms[m h]=575(m 1)
[0139]
步骤b:(2s,4s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸的制备
[0140]
将上述步骤a得到的(2s,4s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸甲酯(39.4g,68.62mmol)溶解在甲醇(450ml)中,然后加入6n氢氧化钠水溶液(57.2ml,343.09mmol)。室温搅拌16小时并用6n盐酸水溶液将ph调节至约5后,减压浓缩反应溶液。将浓缩物溶解在二氯甲烷中后,通过滤纸滤出不溶性固体。减压浓缩滤液,得到粗标题化合物(38.4g,99%),未进行纯化便用于下一步骤。
[0141]
ms[m h]=561(m 1)
[0142]
步骤c:n-((3s,5s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-5-(吗啉-4-羰基)吡咯烷-3-基)-n-((1s,4r)-4-甲基环己基)异丁酰胺的制备
[0143]
将上述步骤b得到的(2s,4s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-4-(n-((1s,4r)-4-甲基环己基)异丁酰胺基)吡咯烷-2-甲酸(38.4g,68.60mmol)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(18.4g,96.04mmol)和1-羟基苯并三唑水合物(13.0g,96.04mmol)溶解在n,n'-二甲基甲酰胺(200ml)中,然后依次缓慢加入吗啉(5.9ml,68.80mmol)和n,n'-二异丙基乙胺(59.7ml,343.02mmol)。室温搅拌16小时后,减压浓缩反应溶液,加入0.5n氢氧化钠水溶液,用乙酸乙酯萃取两次。有机层用氯化钠水溶液和水洗涤两次,经无水硫酸镁干燥,并过滤。减压浓缩滤液,并通过柱色谱法进行纯化,得到n-((3s,5s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-5-(吗啉-4-羰基)吡咯
烷-3-基)-n-((1s,4r)-4-甲基环己基)异丁酰胺(37.05g,86%)。
[0144]
ms[m h]=630(m 1)
[0145]
制备例4:n-((3s,5s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-5-(吗啉-4-羰基)吡咯烷-3-基)-n-((1s,4r)-4-甲基环己基)异丁酰胺盐酸盐的无定形化合物的制备
[0146][0147]
基于1g上述制备例3制备的化合物(mc70),使用19ml mbte在25℃溶解所述化合物(mc70)。溶解完毕后,加入1ml庚烷,然后将混合物冷却至-5℃至0℃。达到规定温度后,滴加1当量4m hcl/etoac,将混合物搅拌约90分钟并过滤,得到标题化合物(mc71)。
[0148]
(收率:约90%)
[0149]
制备例4的化合物的xrd(图1)、dsc(图2)和tga(图3)分析结果分别示于图1至图3中。分析结果证实了它是无定形化合物。xrd、dsc和tga分析方法各自如下面针对实施例1的实验例所述。
[0150]
实施例1.
[0151]
n-((3s,5s)-1-((3s,4r)-1-(叔丁基)-4-(4-氯苯基)吡咯烷-3-羰基)-5-(吗啉-4-羰基)吡咯烷-3-基)-n-((1s,4r)-4-甲基环己基)异丁酰胺盐酸盐的水合物的晶型iii的制备
[0152][0153]
在60℃下将1g上述制备例4制备的化合物(mc71)溶解在1ml etoac和0.15ml蒸馏水中。溶解完毕后,将混合物冷却至3℃并在保持所述温度的同时用电子搅拌器搅拌25小时,然后在氮气压力下进行过滤,以获得标题晶型iii。(收率:约80%)
[0154]
比较例1
[0155]
在室温下将1g上述制备例4制备的化合物(mc71)溶解在1ml etoac中。溶解完毕后,用电子搅拌器搅拌混合物约43小时,但未获得与实施例1相同的晶体。
[0156]
实验例1.xrd评估
[0157]
使用配备有单色辐射源和ni滤光片作为固态检测器的panalytical x'pert pro mpd系统,通过以下方法获得粉末xrd衍射图案。
[0158]
在玻璃样品容器中压缩约20至30mg样品以使样品具有平坦表面,将仪器的发生器设置为45kv(加速电压)和40ma(灯丝发射),然后在反射模式(非自旋)下进行测量。在步长为0.026
°
和每步时间为51秒的条件下,在4
°
至40
°
的范围内测量了布拉格角(2θ)。
[0159]
所获得的晶型iii的xrd测量结果示于图4中。
[0160]
如图4所示的谱图可见,根据本发明的晶型iii在6.62
°
、7.44
°
、9.18
°
、9.89
°
、10.83
°
、11.42
°
、12.92
°
、14.61
°
、15.36
°
、15.79
°
、15.95
°
、17.37
°
、18.20
°
、18.99
°
、19.34
°
、19.69
°
、20.40
°
、21.66
°
、21.98
°
、22.45
°
、22.85
°
、24.66
°
、25.52
°
、26.55
°
、28.08
°
、29.31
°
以及29.54
°
处表现出特征峰(2θ)。xrd的具体值示于下表1中。
[0161]
[表1]
[0162]
2θ相对强度(i/i0)6.6273827.44104829.18105539.89783910.832727211.42860612.92788414.611336415.36915615.791568915.951551317.37571518.201733018.992427119.34818319.69603420.401933521.661988121.982965322.45795222.85945924.66930125.52460526.55825228.08673929.31973329.549744
[0163]
实验例2.差示扫描量热法(dsc)
[0164]
使用mettler toledo dsc1系统测量dsc。称取2-5mg样品,放入40μl a1坩埚(带一
个针孔盖的平底铝锅)中,打一个针孔。然后,在样品以10℃/min的速率从25℃加热到350℃的同时进行dsc测量。在测量期间,以70ml/min的速率向仪器内部供应氮气,以防止氧气和其它气体流入。使用软件stare进行数据收集和评估。
[0165]
所获得的晶型iii的dsc测量结果示于图5中。
[0166]
如图5可见,对于晶型iii,在约42.3℃(起始)观察到第一吸热峰,在约124.7℃(起始)观察到第二吸热峰。在约220℃之后,出现了由于分解导致的吸热峰。温度值有
±
5℃的误差。
[0167]
实验例3.热重分析(tga)
[0168]
使用mettler toledo tga/dsc 1模块测量tga。称取约4-8mg样品,放入100μl al坩埚(平底铝坩埚)中。然后,在样品以10℃/min的速率从30℃加热到350℃的同时进行tga测量。在测量期间,以80ml/min的速率向仪器内部供应氮气,以防止氧气和其它气体流入。使用软件stare进行数据收集和评估。
[0169]
所获得的晶型iii的tga测量结果示于图6中。
[0170]
如图6可见,对于晶型iii,在低于100℃的温度下观察到约2.4%的重量损失。此后,在110℃至150℃下出现约4.8%的重量损失。在约220℃之后,出现了由于分解导致的重量损失。温度值有
±
5℃的误差。
[0171]
实验例4.稳定性评估
[0172]
将约10-30mg样品以开放状态在加速条件(40℃,75% rh)下或以密封状态在80℃烘箱中在苛性条件下储存4周。为了比较所述样品与室温储存的样品,通过下表2所示的方法进行hplc分析。
[0173]
[表2]
[0174][0175]
评估所获得的晶型iii的稳定性的结果示于下表中。
[0176]
[表3]
[0177][0178]
如上表3可见,根据本发明的晶型iii在加速条件(40℃,75% rh)和苛性条件(80℃)下显示出化学稳定性达4周。因此,证实了根据本发明的晶型iii对热和湿度显示出优异的稳定性。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。