1.本发明涉及医药化学领域,具体涉及一种纤溶酶抑制活性的化合物、其制备方法及在制药领域的应用。
背景技术:
2.纤溶酶是一种蛋白水解酶,可降解纤维蛋白。当组织受损造成血管破裂时,会触发止血机制:血管收缩,血小板栓塞形成,凝结过程启动,最终形成稳定的血纤蛋白。与此同时,由于血纤蛋白的沉积,纤溶系统被激活,该系统在血纤蛋白的形成和裂解之间保持平衡,在修复受损血管壁的过程中,发挥维持血管畅通并重塑受损组织的作用(tengborn l,m,berntorp e.thromb res.2015 feb;135(2):231-42)。
3.纤溶系统包括纤溶酶原,组织型纤溶酶原激活物(tpa)和尿激酶型纤溶酶原激活物(upa)。纤溶酶原与纤维蛋白表面的赖氨酸残基结合,通过从内皮细胞释放的活化剂(即tpa)转化为纤溶酶。纤维蛋白溶解抑制可用于治疗出血。抗纤溶药的使用可以减少心脏手术,创伤,骨科手术,实体器官移植,妇产科,神经外科和非外科疾病中的失血(ng w,jerath a,m.anaesthesiol intensive ther. 2015;47(4):339-50)。1950年初,人们发现赖氨酸氨基酸抑制了纤溶酶原的活化,但是作用太弱,无法用于治疗纤维蛋白溶解性出血病。1953年,shosuke okamoto等研究显示几种巯基和氨基碳酸具有抗血浆蛋白的作用,并发现赖氨酸的合成衍生物ε-氨基己酸(eaca)对纤溶酶原具有很强的抑制作用。eaca已在临床上被广泛使用,但是除了轻微的胃肠道副作用如恶心之外,还需要较大剂量。1962年,4-氨基-甲基
‑ꢀ
环己烷-碳酸(amcha)被发现,该化合物包含两个立体异构体,进一步研究表明其反式形式(反式-4
‑ꢀ
氨基甲基环己烷甲酸,即氨甲环酸,txa)具有抗纤维蛋白溶解能力,活性是eaca的10倍左右,并且被证明具有更强的耐受性(tengborn l,m,berntorp e.thromb res.2015 feb;135(2):231-42)。
4.氨甲环酸是一种合成的赖氨酸衍生物和抗纤溶剂,能与纤溶酶原形成可逆的复合物。通过与纤溶酶原结合,阻断纤溶酶原及纤溶酶重链与纤维蛋白赖氨酸残基的相互作用,从而阻止纤溶酶原与纤维蛋白表面的结合,进而延缓纤溶。氨甲环酸已被批准用于治疗严重的月经出血和各种外科出血性疾病,是目前临床上最常用的止血药物。然而,大量文献报道显示,氨甲环酸口服后容易产生胃肠道,如恶心,呕吐,腹泻和消化不良等不良反应,且其给药剂量较大,患者用药后可能引发癫痫等并发症。
5.其他同类止血药物,如氨基己酸,存在人体内排泄较快,止血效果弱、作用持续的时间短且毒性反应较多等问题,当用量过多时可形成血栓,限制了在有血栓形成倾向或有血栓性血管疾病病史者及肾功能不全者上的应用。氨甲苯酸机制同氨基己酸,作用较氨基己酸强4~5倍。对一般慢性渗血效果较显著,但对创伤出血以及癌症出血无止血作用。此外,用量过大也可促进血栓形成。在心脏搭桥手术中常用的止血药物抑肽酶也因可诱发肾衰竭、心肌梗死、心力衰竭等原因,于2008年被fda从市场撤回。
6.其他机制的止血药物,如作用于血管的卡巴克络,反复使用可诱发癫痫;促进凝血
过程的止血药凝血酶,仅可应用于胃肠道出血或局部出血。
7.鉴于临床上可选择的止血药物十分有限,在使用剂量、临床适应症等方面或多或少存在一定缺陷,且现有同类型药物均存在用药剂量大、不良反应多,易引发癫痫等并发症等问题,有必要开发一种新的止血药物,以更好的满足临床需求。
技术实现要素:
8.本发明的目的之一是提供一种式ⅰ所示的化合物,其药学上可接受的盐、水合物、异构体、前药及它们的混合物:
[0009][0010]
其中,x为ch或n;
[0011]
r1、r2、r3各自独立的选自氢或卤素;具体地,r1、r2、r3可各自独立选自h、f、cl、br、i。
[0012]
在某些具体的实施方案中,x为ch。
[0013]
在某些具体的实施方案中,x为n。
[0014]
在某些具体的实施方案中,r1、r2、r3各自独立的选自h或f。
[0015]
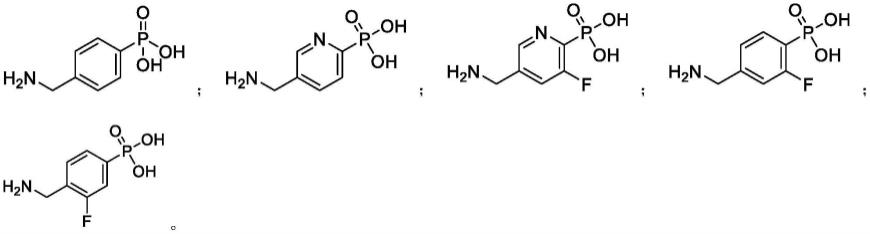
在某些具体的实施方案中,本发明提供如下化合物,其药学上可接受的盐、水合物、异构体、前药及它们的混合物:
[0016][0017]
本发明的另一目的在于提供一种药物组合物,包含至少一种前述的化合物,或其药学上可接受的盐、水合物、异构体、前药及混合物,和至少一种药学上可接受的辅料。
[0018]
本发明的另一目的是提供一种前述化合物或其药学上可接受的盐、水合物、异构体、前药及混合物、或药物组合物,用于制备药物的用途。所述药物具有凝血、止血的治疗活性,可用于纤溶亢进所致异常出血,外科手术和术后出血等。
[0019]
本发明的另一目的是提供一种治疗和/或缓解出血疾病或病症的方法,尤其是治疗和/或缓解纤溶亢进所致异常出血,外科手术和术后出血等出血疾病的方法,包括给予所需要的患者一种或多种前述的药物组合物或式ⅰ化合物或其药学上可接受的盐、水合物、异构体、前药或混合物。
[0020]
本发明中“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定
取代基的化合物与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物的中性形式接触的方式获得碱加成盐。当本发明的化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物的中性形式接触的方式获得酸加成盐,例如本发明式i化合物盐酸盐。
[0021]
这里所采用的术语“药学上可接受的”,是指适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。
[0022]
除非另有说明,本文所用的术语和短语应该按照普通的含义去理解,而不应该被认为是不确定的或不清楚的。当本文中出现商品名时,意在指代其对应的商品或其活性成分。
具体实施方式
[0023]
下面通过举例说明本发明的化合物和中间体的合成方法,下述举例仅作为本发明的示例,而不应作为对本发明范围的限制。除特殊说明外,本发明中所涉及的原料和试剂均可通过商业化渠道获得,具体渠道来源并不影响本发明技术方案的实施。
[0024]
实施例1:(4-(氨基甲基)苯基)膦酸盐酸盐的制备
[0025][0026]
步骤1:(4-(二乙氧基磷酰基)苄基)氨基甲酸叔丁酯的制备
[0027]
称取(4-碘苄基)氨基甲酸叔丁酯(100mg)溶于甲苯(2ml)中,依次加入三(二亚苄基丙酮)二钯(28mg), 1,1'-双(二苯基膦)二茂铁(34mg),三乙胺(84μl),亚磷酸二乙酯(83mg),氩气置换3次,于100℃反应4h,加水淬灭,乙酸乙酯萃取3次,无水硫酸钠干燥,prep-tlc纯化得标题化合物(90mg)。
[0028][0029]
ms(esi)m/z 344.1(m h)
。
[0030]
步骤2:(4-(氨基甲基)苯基)膦酸盐酸盐的制备
[0031]
称取(4-(二乙氧基磷酰基)苄基)氨基甲酸叔丁酯(90mg)溶于12m盐酸水溶液(6ml)中,于110℃下反应6h,减压浓缩,经pre-hplc纯化得标题化合物(10mg)。
[0032][0033]
ms(esi)m/z 188.1(m h) 。
[0034]1h nmr(400mhz,deuterium oxide)δ7.67(dd,j=12.7,7.6hz,2h),7.41(dd,j=
7.6,2.9hz,2h),4.10 (s,2h).
[0035]
实施例2:(5-(氨基甲基)吡啶-2-基)膦酸盐酸盐的制备
[0036][0037]
步骤1:((6-氯吡啶-3-基)甲基)氨基甲酸叔丁酯的制备
[0038]
称取(6-氯吡啶-3-基)甲胺(1.0g)溶于二氯甲烷(20ml)中,依次加入三乙胺(1.9ml)和二碳酸二叔丁酯 (1.8g),室温下反应3h,tlc显示原料消耗完毕,减压浓缩,经柱色谱分离得标题化合物(1.6g)。
[0039][0040]
ms(esi)m/z 243.1(m h)
。
[0041]
步骤2:((6-(二乙氧基磷酰基)吡啶-3-基)甲基)氨基甲酸叔丁酯的制备
[0042]
称取((6-氯吡啶-3-基)甲基)氨基甲酸叔丁酯(200mg)溶于甲苯(5ml)中,依次加入三(二亚苄基丙酮)二钯(76mg),1,1'-双(二苯基膦)二茂铁(92mg),三乙胺(345μl),亚磷酸二乙酯(230mg),氩气置换3次,于 100℃反应4h,加水淬灭,乙酸乙酯萃取3次,无水硫酸钠干燥,prep-tlc纯化得标题化合物(186mg)。
[0043][0044]
ms(esi)m/z 345.2(m h)
。
[0045]
步骤3:(5-(氨基甲基)吡啶-2-基)膦酸盐酸盐的制备
[0046]
称取((6-(二乙氧基磷酰基)吡啶-3-基)甲基)氨基甲酸叔丁酯(186mg)溶于12m盐酸水溶液(10ml)中,于110℃下反应6h,减压浓缩,经pre-hplc纯化得标题化合物(30mg)。
[0047][0048]
ms(esi)m/z 189.0(m h)
。
[0049]1h nmr(400mhz,deuterium oxide)δ8.52(s,2h),8.12(s,1h),4.24(s,2h).
[0050]
实施例3:(6-(氨基甲基)吡啶-3-基)膦酸盐酸盐的制备
[0051][0052]
按照上述合成路线,参考实施例2的操作方法,得标题化合物(6-(氨基甲基)吡啶-3-基)膦酸盐酸盐。
[0053]
ms(esi)m/z 188.9(m h)
。
[0054]1h nmr(400mhz,deuterium oxide)δ8.52(s,2h),8.12(s,1h),4.54(s,2h).
[0055]
实施例4:(5-(氨基甲基)-3-氟吡啶-2-基)膦酸盐酸盐的制备
[0056][0057]
步骤1:(e)-n-((6-氯-5氟吡啶-3-基)亚甲基)-2-甲基丙烷-2-亚磺酰胺的制备
[0058]
称取6-溴吡啶-2-甲醛(300mg),叔丁基亚磺酰胺(345mg),钛酸四乙脂(1ml)溶于2-甲基四氢呋喃中于0℃下反应4h。tlc显示原料消耗完毕,用乙酸乙酯稀释反应液,加水,硅藻土过滤真空减压浓缩,经柱色谱分离得标题化合物(530mg)。
[0059][0060]
ms(esi)m/z 263.0(m h)
[0061]
步骤2:n-((6-氯-5氟吡啶-3-基)甲基)-2-甲基丙烷-2-亚硫酰胺的制备
[0062]
称取(e)-n-((6-氯-5氟吡啶-3-基)亚甲基)-2-甲基丙烷-2-亚磺酰胺(200mg)溶于四氢呋喃(10ml)中,冷却至0℃下,加入三仲丁基硼氢化锂(1.0m四氢呋喃溶液,2.5ml),升至室温反应1小时。tlc显示原料消耗完毕,加水淬灭,乙酸乙酯萃取2次,无水硫酸钠干燥,柱层析纯化得标题化合物(133mg)。
[0063]
[0064]
ms(esi)m/z 265.0(m h)
。
[0065]
步骤3:((6-(二乙氧基磷酰基)-5-氟吡啶-3-基)甲基)氨基甲酸叔丁酯的制备
[0066]
称取n-((6-氯-5氟吡啶-3-基)甲基)-2-甲基丙烷-2-亚硫酰胺(133mg)溶于甲苯(2ml)中,依次加入三(二亚苄基丙酮)二钯(46mg),1,1'-双(二苯基膦)二茂铁(55mg),三乙胺(208μl),亚磷酸二乙酯(138mg),氩气置换3次,于100℃反应4h。tlc显示原料消耗完毕,加水淬灭,乙酸乙酯萃取3次,无水硫酸钠干燥,prep-tlc纯化得标题化合物(61mg)。
[0067][0068]
ms(esi)m/z 367.1(m h)
.
[0069]
步骤4:(5-(氨基甲基)-3-氟吡啶-2-基)膦酸盐酸盐的制备
[0070]
称取((6-(二乙氧基磷酰基)-5-氟吡啶-3-基)甲基)氨基甲酸叔丁酯(61mg)溶于12m盐酸水溶液(6ml) 中,于110℃下反应6h。lcms显示原料消耗完毕,减压浓缩,经pre-hplc纯化得标题化合物(30mg)。
[0071][0072]
ms(esi)m/z 207.0(m h)
[0073]1h nmr(400mhz,deuterium oxide)δ8.68(s,1h),8.42
–
8.25(m,1h),4.37(s,2h).
[0074]
实施例5:(4-(氨基甲基)-3-氟苯基)膦酸盐酸盐的制备
[0075][0076]
按照上述合成路线,参考实施例4的操作方法,得标题化合物(4-(氨基甲基)-3-氟苯基)膦酸盐酸盐。 ms(esi)m/z 206.0(m h)
。
[0077]1h nmr(400mhz,deuterium oxide)δ7.54
–
7.39(m,3h),4.21(s,2h).
[0078]
实施例6:(4-(氨基甲基)-2-氟苯基)膦酸盐酸盐的制备
[0079][0080]
按照上述合成路线,参考实施例4的操作方法,得标题化合物(4-(氨基甲基)-3-氟苯基)膦酸盐酸盐。
[0081]
ms(esi)m/z 205.9(m h)
。
[0082]1h nmr(400mhz,deuterium oxide)δ7.57
–
7.36(m,3h),4.21(s,2h).
[0083]
实施例7:(6-(氨基甲基)吡啶-2-基)膦酸盐酸盐的制备
[0084][0085]
步骤1:(e)-n-((6-溴吡啶-2-基)亚甲基)-2-甲基丙烷-2-亚磺酰胺的制备
[0086]
称取6-溴吡啶-2-甲醛(3g),叔丁基亚磺酰胺(2.34g),无水硫酸铜(7.7g)溶于二氯甲烷(30ml)中于室温下反应12小时。tlc显示原料消耗完毕,硅藻土过滤真空减压浓缩,得标题化合物(5.2g)。
[0087][0088]
ms(esi)m/z 288.8(m h)
。
[0089]
步骤2:n-((6-溴吡啶-2-基)甲基)-2-甲基丙烷-2-亚硫酰胺的制备
[0090]
称取(e)-n-((6-溴吡啶-2-基)亚甲基)-2-甲基丙烷-2-亚磺酰胺(5.2g)溶于四氢呋喃(30ml)中,冷却至0℃下,分批加入硼氢化钠(2.0g),继续反应2小时。tlc显示原料消耗完毕,加水淬灭,乙酸乙酯萃取2次,无水硫酸钠干燥,柱层析纯化得标题化合物(4.8g)。
[0091][0092]
ms(esi)m/z 290.9(m h)
。
[0093]
步骤3:(6-(((叔丁基亚磺酰基)氨基)甲基)吡啶-2-基)膦酸二叔丁酯的制备
[0094]
称取n-((6-溴吡啶-2-基)甲基)-2-甲基丙烷-2-亚硫酰胺(150mg)溶于甲苯(2ml)中,依次加入三(二亚苄基丙酮)二钯(95mg),1,1'-双(二苯基膦)二茂铁(115mg),三乙胺(144μl),二叔丁基亚磷酸盐(202μl),氩气置换3次,于100℃反应过夜。tlc显示原料消耗完毕,加水淬灭,乙酸乙酯萃取3次,无水硫酸钠干燥,prep-hplc纯化得标题化合物(125mg)。
[0095][0096]
ms(esi)m/z 404.9(m h)
。
[0097]
步骤4:(6-(氨基甲基)吡啶-2-基)膦酸盐酸盐的制备
[0098]
称取(6-(((叔丁基亚磺酰基)氨基)甲基)吡啶-2-基)膦酸二叔丁基酯(66mg)溶于6m盐酸水溶液(6ml) 中,于室温下反应过夜。lcms显示原料消耗完毕,减压浓缩,经pre-hplc纯化得标题化合物(64.13mg)。
[0099][0100]
ms(esi)m/z 188.9(m h)
。
[0101]1h nmr(400mhz,deuterium oxide)δ8.09
–
7.97(m,1h),7.83(q,j=6.8hz,1h),7.63
–
7.53(m,1h),4.36 (d,j=3.3hz,2h).
[0102]
前述实施例中例举的制备方法是对本发明化合物制备过程的举例说明,本领域技术人员可参考上述具体方法同时结合本领域一般知识,制备得到本发明范围内的其他化合物,这对本领域技术人员而言是容易实现的。同时,也可以采用本领域常规的实验手段进一步将制备得到的化合物的盐(如前述实施例制备得到的盐酸盐)转化为游离态化合物,例如采用如下方法:取化合物的盐酸盐,用水溶解完全,缓慢加入20%碳酸氢钠溶液,直到有大量固体析出,所得固体过滤干燥可得到游离态化合物。根据本领域的一般常识和发明人的实验研究,可以相信本发明化合物的活性不因化合物为盐型或游离态而受到影响。
[0103]
生物试验
[0104]
测试例1:血浆凝块降解实验
[0105]
1.实验目的
[0106]
测定本发明化合物对人血浆凝块降解的抑制作用。
[0107]
2.实验材料及仪器
[0108][0109]
3.实验步骤
[0110]
3.1采集新鲜健康人血液,用0.109m柠檬酸三钠作为抗凝剂,以1份抗凝剂 9份血液进行混合,室温2000x g离心20分钟,收集上清液(即血浆),分装后于-80℃保存备用。
[0111]
3.2实验当天,将血浆在37℃的水浴中解冻,除tpa外,所有试剂均置于37℃预热。
[0112]
3.3于96孔板中加入12.5μl,80mm的cacl2(hepes buffer,ph 7.4),再分别加入25μl用生理盐水稀释的不同浓度的待测化合物,阴性对照孔加入等体积的生理盐水。
[0113]
3.4将50μl预热的血浆与12.5μl,4nm的tpa混合(hepes buffer,ph 7.4),立即加入96孔板中,于405nm处检测吸收值,每2分钟读值一次,连续测定15小时。
[0114]
3.5吸收值随时间变化,先上升后降低,下降段吸收值中位数对应时间-上升段吸收值中位数对应时间,即为血浆凝块降解时间(clot lysis time)。以阴性对照孔血浆凝块降解时间为参照,计算相对于不同浓度化合物孔中的血浆凝块降解时间,得到抑制率:
[0115]
抑制率%=(1-阴性对照孔
clot lysis time
/化合物孔
clot lysis time
)
×
100%
[0116]
3.6拟合量效曲线
[0117]
以化合物浓度的log值作为x轴,百分比抑制率为y轴,采用分析软件graphpad prism 5的log(抑制剂)vs.响应-可变斜率(variable slope)拟合量效曲线,从而得出各个化合物对细胞活性的ic
50
值。
[0118]
计算公式:y=min (max-min)/(1 10^((logic
50-x)
×
hillslope))。
[0119]
本发明化合物血浆凝块降解的抑制作用通过以上的试验进行测定,并通过计算得到本发明化合物的 ic
50ratio
值(ic
50ratio
=ic
50待测化合物
/ic
50氨甲苯酸
)如表1所示。
[0120]
表1 化合物血浆凝块降解活性测定结果
[0121]
[0122][0123]
注:表1中“/”未计算相关数值。
[0124]
表1的实验数据表明本发明提供的化合物能有效抑制血浆凝块的降解,具有优异的凝血、止血活性,从而具有极佳的成药前景。
[0125]
测试例2:大鼠pk测试
[0126]
1.实验目的
[0127]
通过测定大鼠静脉和灌胃给药后的血浆药物浓度,研究本发明化合物在大鼠体内的药代动力学特性。
[0128]
2.实验动物
[0129]
sd大鼠,sfp级,雄性,n=3,来源:上海西普尔-必凯实验动物有限公司
[0130]
3.药物配制与给药
[0131]
称取化合物加入生理盐水溶解,给药溶液配制后用滤膜过滤,并对过滤前后的给药溶液进行制剂分析。
[0132]
配制:0.2mg/ml的静注给药溶液。
[0133]
实验前一天,大鼠禁食过夜,给药后4小时喂食。
[0134]
实验当天,按表2所示的方案给药。给药后大鼠在各时间点,由颈静脉采血约200μl,置于肝素钠抗凝管中。血液样本采集后置于冰上,并于1小时之内离心分离血浆(离心条件:6800g,6分钟,2-8℃)。血浆样本在分析前存放时放于-80℃冰箱内。
[0135]
表2 给药方案
[0136][0137]
4.生物分析
[0138]
仪器设备:lc-ms/ms-19(tq5500,美国ab sciex公司)。
[0139]
内标:华法林。
[0140]
色谱柱:acquity uplc beh c18,型号1.7um 2.1*50mm,购自深圳市诺亚迪化学科技有限公司;
[0141]
流速:0.60毫升/分钟。
[0142]
柱温:40℃。
[0143]
流动相a:0.1%甲酸水溶液。
[0144]
流动相b:0.1%甲酸的乙腈溶液。
[0145]
洗脱梯度如表3所示。
[0146]
表3 洗脱梯度
[0147]
时间(min)流动相a(%)流动相b(%)09820.6012881.1012881.119821.40982
[0148]
ms检测条件:电喷雾离子源(esi),阳离子模式,mrm扫描。
[0149]
取本实施例第“3”项下制备的血浆样品30μl,用300μl meoh进行蛋白质沉淀,其中含有100ng/ml 内标。将混合物涡旋1分钟并以18000g离心7分钟。将上清液转移至96孔板。取4μl上清液注入 lc-ms/ms进行分析。
[0150]
采用上述lc-ms/ms分析方法测定大鼠血浆中化合物的浓度,通过不同时间点的血药浓度数据,运用 phoenix winnonlin7.0计算药代动力学参数。
[0151]
本发明化合物通过以上的实验进行测定,测得的大鼠药代动力学参数如表4所示。
[0152]
表4 化合物药代动力学参数
[0153]
化合物t
1/2
(h)c
max
(ng/ml)auc
0-t
(h*ng/ml)auc
0-∞
(h*ng/ml)cl(ml/h/kg)氨甲苯酸0.271811.23584.3612.821640.65实施例10.659174.618019.998141.32125.05实施例22.214055.765908.636283.47159.79实施例41.666057.2214112.3514571.8268.67
[0154]
由表4可知,本发明提供的化合物与氨甲苯酸相比,在体内半衰期长,血药浓度及暴露量高,安全性好,具有良好的临床应用前景。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。