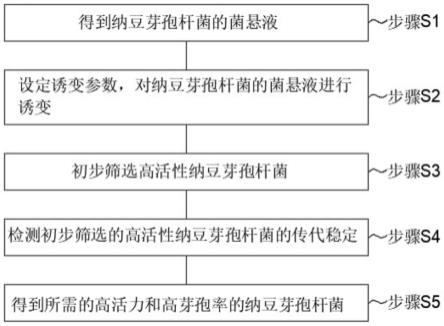
一种高效的利用蓝斑筛选的基因敲除方法
【技术领域】
1.本发明涉及基因工程技术领域,具体涉及一种高效的利用蓝斑筛选的基因敲除方法。
背景技术:
2.基于蓝白斑筛选方法、通过构建自杀载体进行微生物同源重组的方法常用于基因敲除技术。例如中国发明专利申请cn 103789337a公开了一种用于蓝白斑筛选的自杀载体pgmb152,用于构建鸡白痢沙门菌s06004δhsdm基因缺失株,该方法是将laczya基因克隆入自杀载体pgmb151中构建成自杀载体pgmb152;再使用该自杀载体以重组菌蓝白斑筛选法构建鸡白痢沙门菌hsdm基因缺失株。根据其说明书的记载,在自杀载体pgmb151的smai和sali限制性酶切位点之间克隆进入laczya基因(genbank:j01636.1),得到pgmb151-laczya自杀载体(即pgmb152)。后续采用基于蓝白斑筛选方法,经过一次卡那霉素抗性筛选和一次蔗糖平板筛选得到目标菌株。然而,该方法将laczya三个基因克隆入自杀载体pgmb151中,这三个基因长度较大而存在连接不便的缺点,且克隆过程中引入了更多酶切位点,也增加了后续操作的不便。
3.另一方面,这类技术的效率较低,当蔗糖平板的筛选压力迫使菌株中含sacb基因的质粒致死后,无法通过表型鉴定敲除片段是否已经整合到宿主染色体上,需要更多的重复作业才能获得阳性转化子。
技术实现要素:
4.本发明的目的是提高基因工程技术领域中基因敲除的作业效率,提供一种高效的利用蓝斑筛选的基因敲除方法,且具有良好的适用性,可用于构建多种工程菌株。
5.基于上述目的,本发明提供一种高效的利用蓝斑筛选的基因敲除方法,所述方法包括以下步骤:
6.(1)构建插入基因lacz和目的基因上下同源臂的质粒,所得质粒转化入大肠杆菌dh5α,然后涂布于含有生色底物x-gal和50μg/ml卡那霉素的lb抗性平板上,并对平板上的蓝色单菌落提取质粒进行pcr验证;
7.(2)将pcr验证的阳性转化子进行划线纯化,选择紫外分光光度计下a
260
/a
280
》1.8且a
260
/a
230
》2.0的重组质粒;
8.(3)将所得重组质粒转化入目标菌株的感受态细胞中,然后涂布于含有x-gal和25μg/ml卡那霉素的lb抗性平板,进行一次筛选;所述目标菌株的基因中不含完整的β-半乳糖苷酶基因lacz;
9.(4)所述一次筛选中得到的蓝色转化子接种于含25μg/ml卡那霉素的lb抗性液体培养基进行活化,再将菌液划线于含有x-gal和25μg/ml卡那霉素的lb抗性平板,进行纯化;
10.(5)挑取步骤(4)得到的蓝色单菌落于lb无抗液体培养基,将菌液稀释至浓度为10-4
,然后涂布于含有x-gal和质量浓度20%蔗糖的lb平板(不含nacl)进行二次筛选,直至
得到蓝色单菌落;
11.(6)所得蓝色单菌落分别在含有x-gal和质量浓度20%蔗糖的lb平板(不含nacl)与含有25μg/ml卡那霉素的lb抗性平板上进行对点培养;
12.(7)挑选在步骤(6)的含有x-gal和质量浓度20%蔗糖的lb平板上生长且变蓝色,且在含有25μg/ml卡那霉素的lb抗性平板上不生长的单菌落,所得单菌落经pcr验证正确即为成功敲除目的基因的目的菌株。
13.作为一种可选的实施方式,本发明的目的菌株是谷氨酸棒杆菌。
14.基于本发明的思路,满足目标菌株的基因组中不含有完整的β-半乳糖苷酶基因lacz,从而不降解生色底物x-gal条件的其他微生物也可应用本发明的方法,如谷氨酸棒杆菌corynebacterium glutamicum scgg2、大肠埃希氏菌escherichia coli o157:h7 str.sakai等。
15.可选地,以谷氨酸棒杆菌为例,本发明的方法可用于敲除目的基因ncgl1600或ncgl0090。
16.如图1所示,野生型谷氨酸棒杆菌atcc 13032在含有生色底物x-gal的lb琼脂平板上不发生显色反应,全部呈现为黄色菌落。
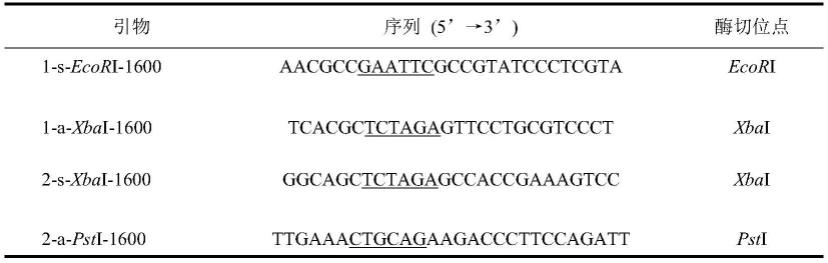
17.根据一种优选的实施方式,当敲除谷氨酸棒杆菌的ncgl1600基因时,步骤(1)中,以谷氨酸棒杆菌atcc 13032为模板,使用引物1-s-ecori-1600和1-a-xbai-1600扩增基因ncgl1600的上同源臂,使用引物2-s-xbai-1600和2-a-psti-1600扩增基因ncgl1600的下同源臂;将所得上下同源臂与自杀质粒pk18mobsacb进行酶切及连接,转入大肠杆菌dh5α;
18.所述pcr验证中使用的引物为1-s-ecori-1600和2-a-psti-1600,其中,引物序列为:
[0019][0020]
根据另一种优选的实施方式,当敲除谷氨酸棒杆菌的ncgl1600基因时,步骤(1)中,以大肠杆菌k12.mg1655基因组为模板,使用引物lacz-s-xbai和lacz-a-xbai进行pcr扩增得到完整的lacz基因序列,所述完整的lacz基因包括基因起始密码子前的sd序列,其中,引物序列为:
[0021][0022]
与现有同源重组技术相比,本发明从敲除菌株的初次筛选到二次筛选通过蓝斑表
型筛选有效提高了筛选概率。发明人发现,当要选择在蔗糖平板上生长而在卡那霉素平板上未生长的单菌落进行验证时,现有的同源重组敲除方法的筛选概率较低,对点90~130个单菌落经常仍未能获得阳性菌株,需要更多的重复作业才能获得阳性转化子,结果如图5所示(全部呈现为黄色单菌落)。而本发明通常在对点50个以内单菌落即可获得阳性菌株,如图6、8所示(部分呈现为黄色单菌落,部分呈现为蓝色单菌落)。
【附图说明】
[0023]
图1为含有x-gal的lb琼脂平板上野生型谷氨酸棒杆菌的表型;
[0024]
图2为实施例1的ncgl1600敲除载体pk18-sa6-l;
[0025]
图3为实施例1和现有技术构建重组质粒pk18-sa6的菌液pcr验证,其中,泳道1和2分别为实施例1和现有技术克隆pk18-sa6的阳性转化子;m为5000bp marker。
[0026]
图4为实施例1中含有敲除载体pk18-sa6-l的菌液pcr验证和敲除株
△
g-6的基因组pcr验证,其中,泳道a~c为蔗糖和卡那霉素平板对点后的阳性单菌落;泳道 为含有pk18-sa6-l的菌液pcr验证;泳道-为阴性对照;泳道g为野生型株;m为5000bp marker。
[0027]
图5为现有敲除技术在蔗糖平板和含有卡那霉素的lb平板上对点单菌落的结果,其中左侧为蔗糖平板,右侧为卡那霉素抗性平板。
[0028]
图6为实施例1在含有x-gal的蔗糖平板和含有卡那霉素的lb平板上对点单菌落的结果,其中左侧为蔗糖平板,右侧为卡那霉素抗性平板。。
[0029]
图7为实施例2和现有技术构建重组质粒pk18-sa9的菌液pcr验证,其中,泳道3和4分别为实施例2和现有技术克隆pk18-sa9的阳性转化子;m为5000bp marker。
[0030]
图8为实施例2在含有x-gal的蔗糖平板和含有卡那霉素的lb平板上对点单菌落的结果,其中左侧为蔗糖平板,右侧为lb平板。
[0031]
图9为实施例2的敲除载体pk18-sa9-l菌液pcr验证和
△
g-9基因组pcr验证,其中,泳道b~c为蔗糖和卡那霉素平板对点后的阳性单菌落;泳道 为含有pk18-sa9-l的菌液pcr验证;泳道-为阴性对照;泳道g为野生型株;m为5000bp marker。
【具体实施方式】
[0032]
以下实施例用于非限制性地解释本发明的技术方案。
[0033]
本发明中涉及以下培养基:
[0034]
lb固体培养基(luria-bertani):nacl 10g/l,胰蛋白胨10g/l,酵母提取粉5g/l,调节ph至7.0,琼脂15g/l。121℃高压湿热灭菌20min。
[0035]
lb液体培养基:nacl 10g/l,胰蛋白胨10g/l,酵母提取粉5g/l,调节ph至7.0。121℃高压湿热灭菌20min。
[0036]
含蔗糖的lb培养基:胰蛋白胨10g/l,酵母提取粉5g/l,调节ph至7.0,琼脂15g/l,121℃高压湿热灭菌20min。20%(w/v)蔗糖,115℃高压湿热灭菌20min。
[0037]
本发明涉及以下微生物:
[0038]
谷氨酸棒杆菌atcc 13032和质粒pk18mobsacb,获赠于内蒙古农业大学食品科学与工程学院食品微生物资源开发与应用创新团队。
[0039]
大肠杆菌k-12mg1655,购买于淼灵质粒平台。
gal和25μg/ml卡那霉素的lb琼脂培养基中以30℃培养48h。将一次筛选得到的蓝色转化子接种于含25μg/ml卡那霉素的lb抗性液体培养基进行活化,再将菌液划线于含有x-gal和25μg/ml卡那霉素的lb抗性平板,进行纯化。然后挑选蓝色单菌落接种于lb液体培养基中30℃200rpm过夜培养。再将菌液进行10-4
梯度稀释并涂布于含有x-gal和质量浓度20%蔗糖的lb平板(不含nacl),30℃培养不超过48h。选择蓝色单菌落在含有x-gal和质量浓度20%蔗糖的lb平板(不含nacl)与含有25μg/ml卡那霉素的lb平板上对点,30℃培养不超过48h。选择在蔗糖平板上生长且表型为蓝色,而在对应的卡那霉素平板上未生长的单菌落,使用引物1-s-ecori-1600与2-a-psti-1600进行基因组pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸6min,共30个循环,16℃降温保藏。
[0049]
结果如图4,6所示,在约50个单菌落之内成功获得阳性转化子。其中,图6中蔗糖平板上圈出的菌落呈现浅黄色,为假阳性转化子,其余菌落呈现蓝色或浅蓝色。
[0050]
对照例1通过现有同源重组技术构建ncgl1600基因敲除的谷氨酸棒杆菌
[0051]
以谷氨酸棒杆菌atcc 13032为模板,分别使用引物1-s-ecori-1600与1-a-xbai-1600、2-s-xbai-1600与2-a-psti-1600扩增基因ncgl1600的上下同源臂。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸1min,共30个循环,16℃降温保藏。所得上同源臂长度为762bp,下同源臂长度为932bp。
[0052]
根据takarabio制造商的说明,采用快切酶(ecori,xbai,psti)和t4连接酶将所得上下同源臂与质粒pk18mobsacb进行酶切及连接,转入大肠杆菌dh5α,在含有50μg/ml卡那霉素的lb琼脂培养基中以37℃培养过夜,筛选阳性转化子并提取重组质粒pk18-sa6。使用引物1-s-ecori-1600和2-a-psti-1600进行菌液pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸2min,共30个循环,16℃降温保藏。验证正确的阳性克隆载体为1676bp,结果如图3所示,表明验证正确。
[0053]
现有技术将敲除基因的上下同源臂与自杀质粒pk18mobsacb连接构建了敲除载体pk18-sa6,电转入谷氨酸棒杆菌中进行第一次卡那霉素抗性筛选和第二次蔗糖平板筛选,在蔗糖平板和卡那霉素平板上对点后进行pcr验证阳性克隆。与本技术不同之处在于,现有技术的敲除载体不插入完整的lacz基因,故而无法通过克隆菌表型提高筛选概率。除此之外,现有技术与本技术对敲除菌株进行pcr验证的扩增条件也不同,因为没有插入lacz基因,现有技术中dna双链延伸的时间较短。
[0054]
构建了敲除ncgl1600基因的载体pk18-sa6,采用电转化方法,将所得载体转入谷氨酸棒杆菌atcc 13032中,在含有25μg/ml卡那霉素的lb琼脂培养基中以30℃培养48h。将一次筛选得到的转化子接种于含25μg/ml卡那霉素的lb抗性液体培养基进行活化,再将菌液划线于25μg/ml卡那霉素的lb抗性平板,进行纯化。然后挑取单菌落接种于lb液体培养基中30℃200rpm过夜培养。再将菌液进行10-4
梯度稀释并涂布于含有质量浓度20%蔗糖的lb平板(不含nacl),30℃培养不超过48h。选择单菌落在含有质量浓度20%蔗糖的lb平板(不含nacl)和含有25μg/ml卡那霉素的lb平板上对点,30℃培养不超过48h。选择在蔗糖平板上生长而在对应的卡那霉素平板上未生长的单菌落,使用引物1-s-ecori-1600与2-a-psti-1600进行基因组pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸2min,共30个循环,16℃降温保藏。
[0055]
结果如图5所示,在约100个单菌落中,各菌落在右侧的卡那霉素抗性平板上均呈
现为黄色,在左侧的蔗糖平板上均呈现为浅黄色,没有得到阳性转化子。
[0056]
实施例2构建ncgl0090基因敲除的谷氨酸棒杆菌
[0057]
以谷氨酸棒杆菌atcc 13032为模板,分别使用引物1-s-bamhi-0090与1-a-xbai-0090、2-s-xbai-0090与2-a-hindiii-0090扩增基因ncgl0090的上下同源臂。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸1min,共30个循环,16℃降温保藏。所得上同源臂长度为789bp,下同源臂长度为843bp。
[0058]
根据takarabio制造商的说明,采用快切酶(bamhi,xbai,hindiii)和t4连接酶将所得上下同源臂与质粒pk18mobsacb进行酶切及连接,转入大肠杆菌dh5a,筛选阳性转化子并提取重组质粒pk18-sa9。使用引物1-s-bamhi-0090和2-a-hindiii-0090进行菌液pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸2min,共30个循环,16℃降温保藏。验证正确的阳性克隆载体为1614bp,结果如图7所示,表明验证正确。
[0059]
与实施例1相同操作,以大肠杆菌k12.mg1655基因组为模板,使用引物lacz-s-xbai和lacz-a-xbai进行pcr,扩增完整的lacz基因序列。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸4min,共30个循环,16℃降温保藏。
[0060]
将扩增的完整lacz基因片段与重组质粒pk18-sa9通过快切酶xbai和t4连接酶进行酶切和连接,从而构建载体pk18-sa9-l,该载体敲除了ncgl0090基因。将敲除载体通过热激法转入大肠杆菌dh5α,在含有x-gal和50μg/ml卡那霉素的lb平板上筛选蓝色转化子,使用引物1-s-bamhi-0090和2-a-hindiii-0090进行菌液pcr验证,结果如图9所示。
[0061]
采用电转化方法,将载体pk18-sa9-l转入谷氨酸棒杆菌atcc13032中,在含有x-gal和25μg/ml卡那霉素的lb琼脂培养基中以30℃培养48h。将一次筛选得到的蓝色转化子接种于含25μg/ml卡那霉素的lb抗性液体培养基进行活化,再将菌液划线于含有x-gal和25μg/ml卡那霉素的lb抗性平板进行纯化。然后挑取蓝色单菌落接种于lb液体培养基中30℃200rpm过夜培养。再将菌液进行10-4
梯度稀释并涂布于含有x-gal和质量浓度20%蔗糖的lb平板(不含nacl),30℃培养不超过48h。选择蓝色单菌落在含有x-gal和质量浓度20%蔗糖的lb平板(不含nacl)与含有25μg/ml卡那霉素的lb平板上对点,30℃培养不超过48h。选择在蔗糖平板上生长且表型为蓝色,而在对应的卡那霉素平板上未生长的单菌落,使用引物1-s-bamhi-0090和2-a-hindiii-0090进行基因组pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸6min,共30个循环,16℃降温保藏。
[0062]
结果如图8所示,各菌落在右侧的卡那霉素抗性平板上均呈现为黄色,在左侧的蔗糖平板上被圈出的菌落呈现浅黄色,为假阳性转化子,其余菌落呈现蓝色或浅蓝色,即在约40个单菌落中成功获得阳性转化子。
[0063]
对照例2通过现有技术构建ncgl0090基因敲除的谷氨酸棒杆菌
[0064]
以谷氨酸棒杆菌atcc13032为模板,分别使用引物1-s-bamhi-0090与1-a-xbai-0090、2-s-xbai-0090与2-a-hindiii-0090扩增基因ncgl0090的上下同源臂。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸1min,共30个循环,16℃降温保藏。所得上同源臂长度为789bp,下同源臂长度为843bp。
[0065]
根据takarabio制造商的说明,采用快切酶(bamhi,xbai,hindiii)和t4连接酶将所得上下同源臂与质粒pk18mobsacb进行酶切及连接,转入大肠杆菌dh5α,在含有50μg/ml卡那霉素的lb琼脂培养基中以37℃培养过夜,筛选阳性转化子并提取重组质粒pk18-sa9。
使用引物1-s-bamhi-0090和2-a-hindiii-0090进行菌液pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸2min,共30个循环,16℃降温保藏。验证正确的阳性克隆载体为1614bp,结果如图7所示,表明验证正确。
[0066]
现有技术将敲除基因的上下同源臂与自杀质粒pk18mobsacb连接构建了敲除载体pk18-sa9,采用电转化方法,将所得载体转入谷氨酸棒杆菌atcc13032中,在含有25μg/ml卡那霉素的lb琼脂培养基中以30℃培养48h。将一次筛选得到的转化子接种于含25μg/ml卡那霉素的lb抗性液体培养基进行活化,再将菌液划线于含有25μg/ml卡那霉素的lb抗性平板,进行纯化。然后挑取单菌落接种于lb液体培养基中30℃200rpm过夜培养。再将菌液进行10-4
梯度稀释并涂布于含有质量浓度20%蔗糖的lb平板(不含nacl),30℃培养不超过48h。选择单菌落在含有质量浓度20%蔗糖的lb平板(不含nacl)与含有25μg/ml卡那霉素的lb平板上对点,30℃培养不超过48h。选择在蔗糖平板上生长而在对应的卡那霉素平板上未生长的单菌落,使用引物1-s-ecori-1600与2-a-psti-1600进行基因组pcr验证。pcr反应程序为:98℃变性10s,55℃退火15s,68℃延伸2min,共30个循环,16℃降温保藏。
[0067]
结果在约100个单菌落中,没有得到阳性转化子。
[0068]
综上所述,本发明的方法对现有技术进行了改进,选择了在质粒内插入敲除基因的上下同源臂,并在上下同源臂之间连接完整的lacz基因,而不是在自杀载体上只插入基因的上下同源臂,或者laczya基因直接连接在载体上再进行上下同源臂的克隆。本技术降低了基因克隆的复杂性,也避免引入过多酶切位点,有效提高了筛选效率。对于基因组中不含有完整的β-半乳糖苷酶基因lacz的目标菌株,由通常需要100-150个单菌落才能获得阳性转化子改进至20-50个单菌落中就能成功获得阳性转化子,提高了筛选效率。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。