1.本发明涉及一种低介电特性和高粘接性优异的环氧树脂组合物、环氧树脂固化物、预浸料、层叠板、印刷布线基板。
背景技术:
2.环氧树脂由于粘接性、挠性、耐热性、耐试剂性、绝缘性、固化反应性优异,所以在涂料、土木粘接、注塑、电气电子材料、膜材料等多领域使用。特别是在作为电气电子材料之一的印刷布线基板用途中,通过对环氧树脂赋予阻燃性而广泛使用。
3.近年来,随着信息设备的小型化、高性能化迅速发展,与此相伴,对半导体、电子部件的领域中使用的材料,要求比以往更高的性能。特别是对于成为电气
·
电子部件的材料的环氧树脂组合物,要求伴随基板的薄型化和高功能化的低介电特性。
4.如下述专利文献1所示,目前,层叠板用途的低介电常数化能够使用导入了脂肪族骨架的双环戊二烯酚醛树脂等,但缺乏改善介电损耗角正切的效果,并且也不满足粘接性。
5.作为用于得到低介电损耗角正切的树脂,如下述专利文献2所示,一直以来使用导入了芳香族骨架的芳香族改性环氧树脂等,其给出了优异的介电损耗角正切,但另一方面又存在粘接力恶化的课题,要求开发出提供低介电损耗角正切且高粘接力的树脂。
6.如上所示,在任一文献中公开的环氧树脂均无法充分满足基于近年来的高功能化的要求性能,不足以确保低介电特性和粘接性。
7.另一方面,专利文献3公开了2,6-二取代酚
·
双环戊二烯型树脂,但并没有公开在酚环中取代了多个双环戊二烯而得到的树脂。
8.现有技术文献
9.专利文献
10.专利文献1:日本特开2001-240654号公报
11.专利文献2:日本特开2015-187190号公报
12.专利文献3:日本特开平5-339341号公报
技术实现要素:
13.因此,本发明要解决的课题是提供一种环氧树脂组合物,其可提供呈现优异的介电损耗角正切,进一步在印刷布线板用途中铜箔剥离强度和层间密合强度的优异的固化物。
14.为了解决上述的课题,本发明人等发现将特定的比率的双环戊二烯与2,6-二取代苯酚类反应而得到的酚醛树脂环氧化时得到的环氧树脂与固化剂固化时,得到的固化物的低介电特性和粘接性均优异,从而完成了本发明。
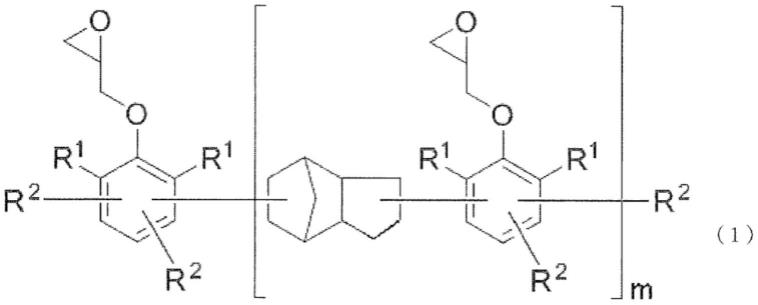
15.即,本发明涉及一种环氧树脂组合物,其特征在于,是含有环氧树脂和固化剂的环氧树脂组合物,环氧树脂的一部分或全部为由下述通式(1)表示的环氧树脂。
[0016][0017]
这里,r1独立地表示碳原子数1~8的烃基,r2独立地表示氢原子或者双环戊烯基,至少一个为双环戊烯基。m表示重复数,其平均值为0~5的数。
[0018]
上述环氧树脂的环氧当量优选为244~3700g/eq.。
[0019]
上述固化剂优选为酚醛树脂类、酸酐类、胺类、氰酸酯类、活性酯类、酰肼类、酸性聚酯类或者芳香族氰酸酯类中的至少一种。
[0020]
另外,本发明是使上述环氧树脂组合物固化而成的固化物。另外,本发明是使用了上述环氧树脂组合物的预浸料、层叠板或者印刷布线基板。
[0021]
本发明的环氧树脂组合物提供一种环氧树脂组合物,该环氧树脂组合物在其固化物中显示优异的介电损耗角正切,进而在印刷布线板用途中铜箔剥离强度和层间密合强度优异。特别是可以优选地用于强烈要求低介电损耗角正切的移动用途、服务器用途等。
附图说明
[0022]
图1是合成例1中得到的多元羟基化合物的gpc谱图。
[0023]
图2是合成例1中得到的多元羟基化合物的ir谱图。
[0024]
图3是合成例4中得到的环氧树脂的gpc谱图。
具体实施方式
[0025]
以下,对本发明的实施方式详细进行说明。
[0026]
本发明的环氧树脂组合物中使用的环氧树脂由上述通式(1)表示。
[0027]
在通式(1)中,r1表示碳原子数1~8的烃基。优选为碳原子数1~8的烷基、碳原子数6~8的芳基、碳原子数7~8的芳烷基、或者烯丙基。作为碳原子数1~8的烷基,可以为直链状、分支状、环状中的任一种,例如可举出甲基、乙基、丙基、异丙基、正丁基、叔丁基、己基、环己基、甲基环己基等,但并不限于此。作为碳原子数6~8的芳基,可举出苯基、甲苯基、二甲苯基、乙基苯基等,但并不限于此。作为碳原子数7~8的芳烷基,可举出苄基、α-甲基苄基等,但并不限于此。从获得容易性和得到固化物时的反应性的观点考虑,优选为苯基、或者甲基,更优选为甲基。
[0028]
r2独立地表示氢原子或者双环戊烯基,分子中的至少一个r2为双环戊烯基。双环戊烯基为来自于双环戊二烯的基团,由下述式(1a)或者式(1b)表示。由于该基团的存在,本发明的环氧树脂组合物的固化物可以降低介电常数、介电损耗角正切。
[0029][0030]
m为重复数,表示0以上的数,该平均值(数均)为0~5,优选为0.5~3,更优选为0.5~2,进一步优选为0.6~1.8。通常的环氧树脂是m为不同成分的混合物,但在这样的情况下,r2平均在1分子中至少一个r2为双环戊烯基即可。此时,可以混合有r2全部为氢原子的反应生成物。
[0031]
作为基于gpc的含量,优选m=0体为10面积%以下,m=1体为50~80面积%,m=2体以上为20~40面积%的范围。
[0032]
由上述通式(1)表示的环氧树脂例如可以通过使下述通式(2)的多元羟基化合物(以下也称为酚醛树脂)与表氯醇等表卤醇反应而得到。该反应根据以往公知的方法进行。
[0033][0034]
在通式(2)中,r1和r2与上述式(1)的定义相同。
[0035]
n为重复数,表示0以上的数,该平均值(数均)为0~5,优选为0.5~3,更优选为0.6~2,进一步优选为0.6~1.8。作为基于gpc的含量,优选n=0体为10面积%以下,n=1体为50~80面积%,n=2体以上为10~40面积%的范围。
[0036]
多元羟基化合物的羟基当量优选为230以上,更优选为240以上,软化点优选为120℃以下,更优选为110℃以下。多元羟基化合物的分子量优选为重均分子量(mw)为400~1000、数均分子量(mn)为350~800的范围。
[0037]
上述多元羟基化合物可以通过在2,6-二取代苯酚类和双环戊二烯在三氟化硼
·
醚催化剂等路易斯酸存在下反应而得到。
[0038]
作为上述2,6-二取代苯酚类,可举出2,6-二甲基苯酚、2,6-二乙基苯酚、2,6-二丙基苯酚、2,6-二异丙基苯酚、2,6-二(正丁基)苯酚、2,6-二(叔丁基)苯酚、2,6-二己基苯酚、2,6-二环己基苯酚、2,6-二苯基苯酚、2,6-二甲苯基苯酚、2,6-二苄基苯酚、2,6-双(α-甲基苄基)苯酚、2-乙基-6-甲基苯酚、2-烯丙基-6-甲基苯酚、2-甲苯基-6-苯基苯酚等,从获得容易性和成为固化物时的反应性的观点考虑,优选为2,6-二苯基苯酚、2,6-二甲基苯酚,特别优选为2,6-二甲基苯酚。
[0039]
用于上述反应的催化剂为路易斯酸,具体而言为三氟化硼、三氟化硼
·
苯酚络合物、三氟化硼
·
醚络合物、氯化铝、氯化锡、氯化锌、氯化铁等,其中,从操作的容易度考虑,优选为三氟化硼
·
醚络合物。催化剂的使用量在三氟化硼
·
醚络合物的情况下,相对于双环戊二烯100重量份,为0.001~20重量份,优选为0.5~10重量份。
[0040]
用于对2,6-二取代苯酚类导入上述式(1a)或者式(1b)的双环戊二烯结构的反应方法是使双环戊二烯以规定的比率与2,6-二取代苯酚反应的方法,也可以使双环戊二烯以多阶段间歇地反应。在一般的反应中,比率是双环戊二烯相对于2,6-二取代苯酚为0.1~0.25倍摩尔,本发明中,为0.28~2倍摩尔。关于使双环戊二烯连续地添加并反应的情况
下的比率,双环戊二烯相对于2,6-二取代苯酚优选为0.28~1倍摩尔,更优选为0.3~0.5倍摩尔。在以多阶段间歇地加入双环戊二烯而反应的情况下,优选为0.8~2倍摩尔,更优选为0.9~1.7倍摩尔。应予说明,各阶段的双环戊二烯的使用量优选为0.28~1倍摩尔。
[0041]
作为确认在由上述通式(2)表示的多元羟基化合物中,导入由式(1a)或者式(1b)表示的双环戊烯基的方法,可以使用质谱法和ft-ir测定。
[0042]
在使用质量分析方法的情况下,可以使用电喷雾质谱法(esi-ms)、场解吸质谱法(fd-ms)等。对利用gpc等将核体数不同的成分分离而成的样品进行质谱法,由此能够确认导入了由式(1a)或者式(1b)表示的取代基。
[0043]
在使用ft-ir测定法的情况下,如果将溶解于thf等有机溶剂的样品涂布于krs-5样品池上,利用ft-ir对使有机溶剂干燥而得到的带样品薄膜的样品池进行测定时,来自于苯酚核中的c-o伸缩振动的峰出现在1210cm
-1
附近,仅在导入有式(1a)或者式(1b)的情况下,来自于双环戊二烯骨架的烯烃部位的c-h伸缩振动的峰出现在3040cm
-1
附近。将作为目的的峰的开始和终点直线地连接而得的线作为基线,将从峰的顶点到基线的长度作为峰高度时,根据3040cm
-1
附近的峰(a
3040
)与1210cm
-1
附近的峰(a
1210
)的比率(a
3040
/a
1210
),能够定量式(1a)或者式(1b)的导入量。可以确认该比率越大,物性值越好,用于满足目的的物性的优选比率(a
3040
/a
1210
)为0.05以上,更优选为0.10以上。
[0044]
本反应可以是将2,6-二取代苯酚类和催化剂投入到反应器中,历经1~10小时滴下双环戊二烯的方式。
[0045]
反应温度优选为50~200℃,更优选为100~180℃,进一步优选为120~160℃。反应时间优选为1~10小时,更优选为3~10小时,进一步优选为4~8小时。
[0046]
反应结束后,加入氢氧化钠、氢氧化钾、氢氧化钙等碱并使催化剂失活。然后可以加入甲苯、二甲苯等芳香族烃类、甲基乙基酮、甲基异丁基酮等酮类等溶剂进行溶解,在水洗后,在减压下回收溶剂,由此能够得到作为目的的酚醛树脂。应予说明,优选尽可能使双环戊二烯全部反应,2,6-二取代苯酚类的一部分未反应、优选10%以下未反应,对其进行减压回收。
[0047]
反应时,可以根据需要使用苯、甲苯、二甲苯等芳香族烃类、氯苯、二氯苯等卤化烃类、乙二醇二甲基醚、二乙二醇二甲基醚等醚类等溶剂。
[0048]
由上述通式(1)表示的环氧树脂例如可以通过对上述酚醛树脂进行环氧化而得到。作为环氧化的方法,例如能够通过如下方式而得到:在酚醛树脂与相对于酚醛树脂的羟基为过量摩尔的表卤醇的混合物中加入氢氧化钠等碱金属氢氧化物作为固体或者浓水溶液,在30~120℃的反应温度中反应0.5~10小时;或者在酚醛树脂与过度摩尔量的表卤醇中加入四乙基氯化铵等季铵盐作为催化剂,在50~150℃的温度下反应1~5小时得到的聚卤代醇醚中加入氢氧化钠等碱金属氢氧化物作为固体或者浓水溶液,在30~120℃的温度下反应1~10小时。
[0049]
在上述反应中,表卤醇的使用量相对于酚醛树脂的羟基为1~20倍摩尔,优选为2~8倍摩尔。并且,碱金属氢氧化物的使用量相对于酚醛树脂的羟基为0.85~1.15倍摩尔。
[0050]
由这些反应得到的环氧树脂由于含有未反应的表卤醇和碱金属的卤化物,所以从反应混合物中蒸发除去未反应的表卤醇,进一步通过基于水的萃取、过滤等方法除去碱金属的卤化物,得到作为目的的环氧树脂。
[0051]
上述环氧树脂的环氧当量(g/eq.)优选为250以上,更优选为300以上,进一步优选为350以上。将双氰胺用作固化剂的情况下,为了防止双氰胺的结晶在预浸料上析出,环氧当量优选为300以上。
[0052]
环氧树脂的软化点优选为100℃以下,更优选为90℃以下。总氯含量优选为1000ppm以下,更优选为700ppm以下。
[0053]
上述环氧树脂的分子量分布能够通过变更环氧化反应时的酚醛树脂和表卤醇的投入比率来进行变更,表卤醇的使用量与酚醛树脂的羟基越靠近等摩尔,越是会成为高分子量分布,越靠近20摩尔倍,则越是会成为低分子量分布。例如能够得到重均分子量(mw)为500~1000的范围,数均分子量(mn)为400~800的范围的环氧树脂。另外,相对于得到的环氧树脂,提供使酚醛树脂再次起作用,由此也能够使其高分子量化。
[0054]
通过使用这样的环氧树脂,由此能够得到本发明的环氧树脂组合物。
[0055]
本发明的环氧树脂组合物是将由上述通式(1)表示的环氧树脂和固化剂作为必须成分。在必须成分的上述环氧树脂之外,根据需要可以同时并用1种或2种以上的各种其他环氧树脂。在并用其它的环氧树脂的情况下,其它的环氧树脂优选为总环氧树脂中的70质量%以下,更优选为50质量%以下。如果其它的环氧树脂过多,则有作为环氧树脂组合物的介电特性变差的担忧。
[0056]
作为上述其它的环氧树脂,可以使用所有在分子中具有2个以上的环氧基的通常的环氧树脂。举例的话,可举出双酚a型环氧树脂、双酚f型环氧树脂、双酚af型环氧树脂、四甲基双酚f型环氧树脂、对苯二酚型环氧树脂、联苯型环氧树脂、二苯乙烯型环氧树脂、双酚芴型环氧树脂、双酚s型环氧树脂、双硫醚型环氧树脂、间苯二酚型环氧树脂、联苯芳烷基苯酚型环氧树脂、萘二酚型环氧树脂、酚醛清漆型环氧树脂、芳香族改性苯酚酚醛清漆型环氧树脂、甲酚酚醛清漆型环氧树脂、烷基酚醛清漆型环氧树脂、双酚酚醛清漆型环氧树脂、联萘型环氧树脂、萘酚酚醛清漆型环氧树脂、β-萘酚芳烷基型环氧树脂、二萘酚芳烷基型环氧树脂、α-萘酚芳烷基型环氧树脂、三苯基甲烷型环氧树脂等三官能环氧树脂、四苯基乙烷型环氧树脂等四官能环氧树脂、双环戊二烯型环氧树脂(不包括通式(1)中包含的树脂)、1,4-丁二醇二缩水甘油基醚、1,6-己二醇二缩水甘油基醚、甘油聚缩水甘油基醚、三羟甲基丙烷聚缩水甘油基醚、三羟甲基乙烷聚缩水甘油基醚、季戊四醇聚缩水甘油基醚等多元醇聚缩水甘油基醚、丙二醇二缩水甘油基醚等亚烷基二醇型环氧树脂、环己烷二甲醇二缩水甘油基醚等脂肪族环状环氧树脂、二聚酸聚缩水甘油基酯等缩水甘油基酯类、苯基二缩水甘油基胺、甲苯基二缩水甘油基胺、二氨基二苯基甲烷四缩水甘油基胺、氨基苯酚型环氧树脂等缩水甘油基胺型环氧树脂、celloxide 2021p(株式会社大赛璐制)等脂环式环氧树脂、含磷的环氧树脂、含溴的环氧树脂、氨基甲酸酯改性环氧树脂、含唑烷酮环的环氧树脂等,但并不限于这些树脂。从获得容易度的观点考虑,更优选使用由下述通式(3)表示的环氧树脂、双环戊二烯型环氧树脂(不包括通式(1)的树脂)、萘二酚型环氧树脂、苯酚酚醛清漆型环氧树脂、芳香族改性苯酚酚醛清漆型环氧树脂、甲酚酚醛清漆型环氧树脂、α-萘酚芳烷基型环氧树脂、双环戊二烯型环氧树脂、含磷的环氧树脂、含唑烷酮环的环氧树脂。
[0057][0058][0059]
这里,r3独立地表示碳原子数1~8的烃基,例如为甲基、乙基、正丙基、异丙基、正丁基、叔丁基、正己基、环己基等烷基,可以相互相同也可以相互不同。
[0060]
x表示2价的基团,例如表示亚甲基、亚乙基、异亚丙基、异亚丁基、六氟异亚丙基等亚烷基、-co-、-o-、-s-、-so2-、-s-s-、或者由式(4)表示的亚芳烷基。
[0061]
r4独立地表示氢原子或者碳原子数1以上的烃基,例如为甲基,可以相互相同也可以相互不同。
[0062]
ar为苯环或者萘环,这些苯环或者萘环可以具有碳原子数1~10的烷基、碳原子数1~10的烷氧基、碳原子数6~11的芳基、碳原子数7~12的芳烷基、碳原子数6~11的芳氧基、或者碳原子数7~12的芳烷基氧基作为取代基。
[0063]
作为固化剂,根据需要可以并用1种或者2种以上的各种酚醛树脂类、酸酐类、胺类、氰酸酯类、活性酯类、酰肼类、酸性聚酯类、芳香族氰酸酯类等通常使用的固化剂。
[0064]
在本发明的环氧树脂组合物中,相对于总环氧树脂的环氧基1摩尔,固化剂的活性氢基的摩尔比优选为0.2~1.5摩尔,更优选为0.3~1.4摩尔,进一步优选为0.5~1.3摩尔,特别优选为0.8~1.2摩尔。在超出该范围的情况下,有可能固化会不完全,无法得到良好的固化物性。例如在使用酚醛树脂系固化剂、胺系固化剂的情况下,相对于环氧基几乎等摩尔地配合活性氢基。在使用酸酐系固化剂的情况下,相对于环氧基1摩尔,配合酸酐基团0.5~1.2摩尔,优选为配合0.6~1.0摩尔。在将本发明的酚醛树脂单独用作固化剂的情况下,优选相对于环氧树脂1摩尔以0.9~1.1摩尔的范围使用。
[0065]
本发明中的活性氢基是指具有环氧基和反应性的活性氢的官能团(包含具有通过水解等产生活性氢的潜在性活性氢的官能团、表示同等的固化作用的官能团),具体而言可举出酸酐基、羧基、氨基、酚羟基等。应予说明,涉及到活性氢基,1摩尔的羧基、酚羟基按1摩尔计算,氨基(nh2)按2摩尔计算。另外,在活性氢基不明确的情况下,可以根据测定求出活性氢当量。例如通过使环氧当量已知的苯基缩水甘油基醚等单环氧树脂与活性氢当量未知的固化剂反应,测定消耗的单环氧树脂的量,从而能够求出使用的固化剂的活性氢当量。
[0066]
作为能够在本发明的环氧树脂组合物中使用的酚醛树脂系固化剂,其具体例可举出双酚a、双酚f、双酚c、双酚k、双酚z、双酚s、四甲基双酚a、四甲基双酚f、四甲基双酚s、四甲基双酚z、四溴双酚a、二羟基二苯基硫醚、4,4’-硫代双(3-甲基-6-叔丁基苯酚)等双酚类、儿茶酚、间苯二酚、甲基间苯二酚、对苯二酚、单甲基对苯二酚、二甲基对苯二酚、三甲
基对苯二酚、单-叔丁基对苯二酚、二-叔丁基对苯二酚等二羟基苯类、二羟基萘、二羟基甲基萘、二羟基甲基萘、三羟基萘等羟基萘类、lc-950pm60(shin-at&c公司制)等含磷的苯酚固化剂、shonol brg-555(aica工业株式会社制)等苯酚酚醛清漆树脂、dc-5(日铁化学&材料株式会社制)等甲酚酚醛清漆树脂、含有三嗪骨架的酚醛树脂、芳香族改性苯酚酚醛清漆树脂、双酚a酚醛清漆型树脂、resitop tpm-100(群荣化学工业株式会社制)等三羟基苯基甲烷型酚醛清漆型树脂、萘酚酚醛清漆型树脂等苯酚类、萘酚类和/或双酚类与醛类的缩合物、sn-160、sn-395、sn-485(日铁化学&材料株式会社制)等苯酚类、苯酚类和/或萘酚类和/或双酚类与二甲苯二醇的缩合物、苯酚类和/或萘酚类与异丙烯基苯乙酮的缩合物、苯酚类和/或萘酚类和/或双酚类与双环戊二烯的反应物、苯酚类和/或萘酚类和/或双酚类与二乙烯基苯的反应物、苯酚类和/或萘酚类和/或双酚类与萜烯类的反应物、苯酚类和/或萘酚类和/或双酚类与联苯系交联剂的缩合物等所谓的清漆型酚醛树脂所谓的苯酚化合物、聚丁二烯改性酚醛树脂、具有螺环的酚醛树脂等。从得到容易度的观点考虑,优选为苯酚酚醛清漆树脂、双环戊二烯型酚醛树脂、三羟基苯基甲烷型酚醛清漆树脂、芳香族改性苯酚酚醛清漆树脂等。
[0067]
苯酚酚醛清漆树脂可以由苯酚类和交联剂得到。作为苯酚类,可举出苯酚、甲酚、二甲酚、丁基苯酚、戊基苯酚、壬基苯酚、丁基甲基苯酚、三甲基苯酚、苯基苯酚、1-萘酚、2-萘酚等,另外,可举出作为上述酚醛树脂系固化剂举出的双酚类。作为交联剂的醛类,可例示甲醛、乙醛、丙醛、丁醛、戊醛、己醛、苯甲醛、氯醛、溴醛、乙二醛、丙二醛、丁二醛、戊二醛、己二醛、庚二醛、癸二醛、丙烯醛、巴豆醛、水杨醛、苯二醛、羟基苯甲醛等。作为联苯系交联剂,可举出双(羟甲基)联苯、双(甲氧基甲基)联苯、双(乙氧基甲基)联苯、双(氯甲基)联苯等。
[0068]
作为酸酐系固化剂,具体而言可举出马来酸酐、甲基四氢邻苯二甲酸酐、六氢邻苯二甲酸酐、4-甲基六氢邻苯二甲酸酐、甲基双环[2.2.1]庚烷-2,3-二羧酸酐、双环[2.2.1]庚烷-2,3-二羧酸酐、1,2,3,6-四氢邻苯二甲酸酐、均苯四酸酐、邻苯二甲酸酐、偏苯三酸酐、甲基纳迪克酸、苯乙烯单体与马来酸酐的共聚物、茚类与马来酸酐的共聚物等。
[0069]
作为胺系固化剂,具体而言可举出作为二乙烯三胺、三乙烯四胺、间苯二甲胺、异佛尔酮二胺、二氨基二苯基甲烷、二氨基二苯基砜、二氨基二苯基醚、苄基二甲基胺、2,4,6-三(二甲基氨基甲基)苯酚、聚醚胺、双胍化合物、双氰胺、茴香胺等芳香族胺类、作为二聚酸等酸类与多胺类的缩合物的聚酰胺胺等胺系化合物等。
[0070]
作为氰酸酯化合物,如果是在1分子中具有2个以上的氰基(氰酸酯基)的化合物,则没有特别限定。例如可举出苯酚酚醛清漆型、烷基苯酚酚醛清漆型等酚醛清漆型氰酸酯系固化剂、萘酚芳烷基型氰酸酯系固化剂、联苯烷基型氰酸酯系固化剂、双环戊二烯型氰酸酯系固化剂、双酚a型、双酚f型、双酚e型、四甲基双酚f型、双酚s型等双酚型氰酸酯系固化剂以及它们的一部分三嗪化而成的预聚物等。作为氰酸酯系固化剂的具体例,例如可以使用双酚a二氰酸酯、聚苯酚氰酸酯(低聚(3-亚甲基-1,5-亚苯基氰酸酯)、双(3-甲基-4-氰酸酯苯基)甲烷、双(3-乙基-4-氰酸酯苯基)甲烷、双(4-氰酸酯苯基)-1,1-乙烷、4,4-二氰酸酯-二苯基、2,2-双(4-氰酸酯苯基)-1,1,1,3,3,3-六氟丙烷、4,4’-亚甲基双(2,6-二甲基苯基氰酸酯)、4,4’-亚乙基二苯基二氰酸酯、六氟双酚a二氰酸酯、
2,2-双(4-氰酸酯)苯基丙烷、1,1-双(4-氰酸酯苯基甲烷)、双(4-氰酸酯-3,5-二甲基苯基)甲烷、1,3-双(4-氰酸酯苯基-1-(甲基亚乙基))苯、双(4-氰酸酯苯基)硫基醚、双(4-氰酸酯苯基)醚等二官能氰酸酯树脂、三(4-氰酸酯苯基)-1,1,1-乙烷、双(3,5-二甲基-4-氰酸酯苯基)-4-氰酸酯苯基-1,1,1-乙烷等3元苯酚的氰酸酯、由含有苯酚酚醛清漆、甲酚酚醛清漆、双环戊二烯结构的酚醛树脂等衍生出的多官能氰酸酯树脂、这些氰酸酯树脂一部分三嗪化而成的预聚物等。这些可以使用1种或者2种以上。
[0071]
作为活性酯系固化剂,没有特别限制,一般而言,优选使用苯酚酯类、硫基苯酚酯类、n-羟基胺酯类、杂环羟基化合物的酯类等在1分子中具有2个以上的反应活性高的酯基的化合物。该活性酯系固化剂优选为通过羧酸化合物和/或硫基羧酸化合物与羟基化合物和/或硫醇化合物的缩合反应而得到。特别是从耐热性提高的观点考虑,优选为由羧酸化合物和羟基化合物得到的活性酯系固化剂,更优选为由羧酸化合物与苯酚化合物和/或萘酚化合物得到的活性酯系固化剂。作为羧酸化合物,例如可举出安息香酸、乙酸、琥珀酸、马来酸、衣康酸、富马酸、异富马酸、对苯二甲酸、均苯四酸等。作为苯酚化合物或者萘酚化合物,可举出对苯二酚、间苯二酚、双酚a、双酚f、双酚s、酸式酚酞、甲基化双酚a、甲基化双酚f、甲基化双酚s、苯酚、邻甲酚、间甲酚、对甲酚、儿茶酚、α-萘酚、β-萘酚、1,5-二羟基萘、1,6-二羟基萘、2,6-二羟基萘、二羟基二苯甲酮、三羟基二苯甲酮、四羟基二苯甲酮、间苯三酚、苯三醇、双环戊二烯基二苯酚、苯酚酚醛清漆、上述通式(2)的多元羟基化合物等。活性酯系固化剂可以使用1种或者2种以上。作为活性酯系固化剂,具体而言,优选为包含双环戊二烯基二苯酚结构的活性酯系固化剂、包含萘结构的活性酯系固化剂、作为苯酚清漆的乙酰化物的活性酯系固化剂、作为苯酚清漆的苯甲酰化物的活性酯系固化剂等,其中,从剥离强度的提高优异的观点考虑,更优选为包含上述通式(2)的多元羟基化合物等双环戊二烯基二苯酚结构的活性酯系固化剂。
[0072]
作为其它的固化剂,具体而言,可举出三苯基膦等膦化合物、四苯基溴化等盐、2-甲基咪唑、2-苯基咪唑、2-乙基-4-甲基咪唑、2-十一烷基咪唑、1-氰基乙基-2-甲基咪唑等咪唑类、作为咪唑类与三苯膦或异氰脲酸或硼等的盐的咪唑盐类、三甲基氯化铵等季铵盐类、二氮杂双环化合物、二氮杂双环化合物与苯酚类或苯酚酚醛清漆树脂类等的盐类、三氟化硼与胺类、醚化合物等的配位化合物、芳香族或者碘盐等。
[0073]
环氧树脂组合物中可以根据需要使用固化促进剂。作为可使用的固化促进剂的例子,可举出2-甲基咪唑、2-乙基咪唑、2-乙基-4-甲基咪唑等咪唑类、4-二甲基氨基吡啶、2-(二甲基氨基甲基)苯酚、1,8-二氮杂双环(5,4,0)十一碳烯-7等叔胺类、三苯基膦、三环己基膦、三苯基膦三苯基硼烷等膦类、辛酸锡等金属化合物。在使用固化促进剂的情况下,该使用量优选相对于本发明的环氧树脂组合物中的环氧树脂成分100重量份为0.02~5重量份。通过使用固化促进剂,由此能够降低固化温度,或者缩短固化时间。
[0074]
环氧树脂组合物中可以使用有机溶剂或者反应性稀释剂用于粘度调整。
[0075]
作为有机溶剂,例如可举出n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等酰胺类、乙二醇单甲基醚、二甲氧基二乙二醇、乙二醇二乙基醚、二乙二醇二乙基醚、三乙二醇二甲基醚等醚类、丙酮、甲基乙基酮、甲基异丁基酮、环己酮等酮类、甲醇、乙醇、1-甲氧基-2-丙醇、2-乙基-1-己醇、苄基醇、乙二醇、丙二醇、丁基二乙二醇、松油等醇类、乙酸丁基酯、
乙酸甲氧基丁基酯、甲基乙酸溶纤剂、乙基乙酸溶纤剂、乙基乙酸二乙二醇酯、丙二醇单甲基醚乙酸酯、卡必醇乙酸酯、苄基醇乙酸酯等乙酸酯类、安息香酸甲酯、安息香酸乙酯等安息香酸酯类、甲基溶纤剂、溶纤剂、丁基溶纤剂等溶纤剂类、甲基卡必醇、卡必醇、丁基卡必醇等卡必醇类、苯、甲苯、二甲苯等芳香族烃类、二甲基亚砜、乙腈、n-甲基吡咯烷酮等,但并不限于此。
[0076]
作为反应性稀释剂,例如可举出烯丙基缩水甘油基醚、丁基缩水甘油基醚、2-乙基己基缩水甘油基醚、苯基缩水甘油基醚、甲苯基缩水甘油基醚等单官能缩水甘油基醚类、新癸酸缩水甘油基酯等单官能缩水甘油基酯类等,但并不限于此。
[0077]
这些有机溶剂或者反应性稀释剂优选为以不挥发成分为90质量%以下而单独或者混合多个种类使用的有机溶剂或者反应性稀释剂,其适当的种类、使用量可以根据用途适当地选择。例如在印刷布线板用途中,优选为甲基乙基酮、丙酮、1-甲氧基-2-丙醇等沸点为160℃以下的极性溶剂,该使用量以不挥发成分计优选为40~80质量%。另外,粘接膜用途中,例如优选使用酮类、乙酸酯类、卡必醇类、芳香族烃类、二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮等,该使用量以不挥发成分计优选为30~60质量%。
[0078]
环氧树脂组合物可以在不损害特性的范围内配合其它的热固化性树脂、热塑性树脂。例如可举出酚醛树脂、苯并嗪树脂、双马来酰亚胺树脂、双马来酰亚胺三嗪树脂、丙烯酸树脂、石油树脂、茚树脂、香豆酮茚树脂、苯氧基树脂、聚氨基甲酸酯树脂、聚酯树脂、聚酰胺树脂、聚酰亚胺树脂、聚酰胺酰亚胺树脂、聚醚酰亚胺树脂、聚苯醚树脂、改性聚苯醚树脂、聚醚砜树脂、聚砜树脂、聚醚醚酮树脂、聚苯硫醚树脂、聚乙烯醇缩甲醛树脂、聚硅氧烷化合物、含有羟基的聚丁二烯等含有反应性官能团的亚烷基树脂类,但并不限于此。
[0079]
出于提高得到的固化物的阻燃性的目的,环氧树脂组合物中可以使用公知的各种阻燃剂。作为可使用的阻燃剂,例如可举出卤素系阻燃剂、磷系阻燃剂、氮系阻燃剂、有机硅系阻燃剂、无机系阻燃剂、有机金属盐系阻燃剂等。从相对于环境的观点考虑,优选为不包含卤素的阻燃剂,特别是优选为磷系阻燃剂。这些阻燃剂可以单独使用,也可以同时采用两种以上。
[0080]
磷系阻燃剂可以使用无机磷系化合物、有机磷系化合物中的任一种。作为无机磷系化合物,例如可使用红磷、磷酸一铵、磷酸二铵、磷酸三铵、聚磷酸铵等磷酸铵类、磷酸酰胺等无机系含氮磷化合物。作为有机磷系化合物,例如可举出脂肪族磷酸酯、磷酸酯化合物、例如px-200(大八化学工业株式会社制)等缩合磷酸酯类、聚磷腈、膦酸化合物、次膦酸化合物、膦氧化物、膦烷化合物、有机系含氮磷化合物等通用有机磷系化合物、次膦酸的金属盐,除此之外,还可举出9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物、10-(2,5-二羟基苯基)-10h-9-氧杂-10-磷杂菲-10-氧化物、10-(2,7-二羟基萘)-10h-9-氧杂-10-磷杂菲-10-氧化物等环状有机磷化合物、作为使这些与环氧树脂、酚醛树脂等化合物反应的衍生物的含磷的环氧树脂、含磷的固化剂等。
[0081]
作为阻燃剂的配合量,根据磷系阻燃剂的种类、环氧树脂组合物的成分、所希望的阻燃性的程度可适当地选择。例如环氧树脂组合物中的有机成分(不包括有机溶剂)中的磷含量优选为0.2~4质量%,更优选为0.4~3.5质量%,进一步优选为0.6~3质量%。如果磷含量少,则可能难以确保阻燃性,如果过多,则可能给耐热性带来负面影响。并且,使用磷系阻燃剂的情况下,可以同时采用氢氧化镁等阻燃助剂。
[0082]
环氧树脂组合物中可以根据需要使用填充材料。具体而言,可举出熔融二氧化硅、结晶二氧化硅、氧化铝、氮化硅、氢氧化铝、勃姆石、氢氧化镁、滑石、云母、碳酸钙、硅酸钙、氢氧化钙、碳酸镁、碳酸钡、硫酸钡、氮化硼、碳、碳纤维、玻璃纤维、氧化铝纤维、二氧化硅氧化铝纤维、碳化硅纤维、聚酯纤维、纤维素纤维、芳族聚酰胺纤维、陶瓷纤维、微粒橡胶、硅酮橡胶、热塑性弹性体、炭黑、颜料等。一般而言,作为使用填充材料的理由,可举出耐冲击性的提高效果。另外,在使用氢氧化铝、勃姆石、氢氧化镁等金属氢氧化物的情况下,有作为阻燃助剂发挥作用而使阻燃性提高的效果。这些填充材料的配合量相对于环氧树脂组合物整体,优选为1~150质量%,更优选为10~70质量%。如果配合量多,则可能存在需要作为层叠板用途的粘接性降低,进而固化物变脆,可能得不到足够的机械物性的担忧。另外,如果配合量少,则有无法发挥固化物的耐冲击性提高等填充剂的配合效果的担忧。
[0083]
在将环氧树脂组合物形成为板状基板等的情况下,从其尺寸稳定性、弯曲强度等观点出发,可举出纤维状的环氧树脂组合物作为优选的填充材料。更优选举出将玻璃纤维编织成网眼状的玻璃纤维基板。
[0084]
环氧树脂组合物可以进一步根据需要配合硅烷偶联剂、抗氧化剂、脱模剂、消泡剂、乳化剂、触变性赋予剂、平滑剂、阻燃剂、颜料等各种添加剂。这些添加剂的配合量相对于环氧树脂组合物优选为0.01~20质量%的范围。
[0085]
环氧树脂组合物可以通过含浸于纤维状基材而制成印刷布线板等中使用的预浸料。作为纤维状基材,可使用玻璃等无机纤维、聚酯树脂等、多胺树脂、聚丙烯酸树脂、聚酰亚胺树脂、芳香族聚酰胺树脂等有机质纤维的编织物或者无纺布,但并不限于此。作为由环氧树脂组合物制造预浸料的方法,没有特别限定,例如是将环氧树脂组合物浸渍并含浸于利用有机溶剂进行粘度调整而制成的树脂清漆后,进行加热干燥,将树脂成分半固化(b阶段化)而得到,例如能够在100~200℃下加热干燥1~40分钟。这里,预浸料中的树脂量优选为树脂成分为30~80质量%。
[0086]
另外,为了将预浸料固化,一般而言能够使用制造印刷布线板时使用的层叠板的固化方法,并不限于此。例如在使用预浸料形成层叠板的情况下,将预浸料层叠一张或多张,在单侧或者双侧配置金属箔而构成层叠物,对该层叠物进行加热
·
加压而层叠成为一体。这里,作为金属箔,可单独使用铜、铝、黄铜、镍等,也可使用合金、复合的金属箔。并且,通过对制成的层叠物进行加压加热而使预浸料固化,能够得到层叠板。此时,优选将加热温度设为160~220℃,将加压压力设为50~500n/cm2,将加热加压时间设为40~240分钟,能够得到作为目的的固化物。加热温度低时,无法充分地进行固化反应,加热温度高时,环氧树脂组合物的分解可能会开始。另外,如果加压压力低,则存在在得到的层叠板的内部残留有气泡,电特性降低的情况,如果加压压力高,则存在树脂在固化前可能会流动,无法得到所希望的厚度的固化物的担忧。另外,如果加热加压时间短,则有可能无法充分地进行固化反应,如果加热加压时间长,则有可能导致预浸料中的环氧树脂组合物的热分解,因而不优选。
[0087]
环氧树脂组合物能够通过利用与公知的环氧树脂组合物同样的方法进行固化而得到环氧树脂固化物。作为用于得到固化物的方法,能够采取与公知的环氧树脂组合物相同的方法,优选使用通过注塑、注入、灌注、浸渍、滴涂、转印成型、压缩成型等或通过制成树脂片、附有树脂的铜箔、预浸料等形态层叠并进行加热加压固化而制成层叠板等的方法。此
时的固化温度通常为100~300℃,固化时间通常为1小时~5小时左右。
[0088]
本发明的环氧树脂固化物可以采用层叠物、成型物、粘接物、涂膜、膜等形态。
[0089]
制备环氧树脂组合物并通过加热固化来评价层叠板和固化物的结果是,能够提供一种在固化物中显示出优异的低介电特性,进而在印刷布线板用途中铜箔剥离强度和层间密合强度优异的环氧固化性树脂组合物。
[0090]
实施例
[0091]
举出实施例和比较例来具体地说明本发明,但本发明并不限于此。只要没有特别说明,“份”表示重量份,“%”表示质量%,“ppm”表示质量ppm。另外,测定方法分别通过以下的方法测定。
[0092]
·
羟基当量:
[0093]
基于jis k 0070规格进行测定,单位表示为“g/eq.”。应予说明,只要没有特别说明,酚醛树脂的羟基当量是指酚羟基当量。
[0094]
·
软化点:
[0095]
基于jis k 7234规格、环球法进行了测定。具体而言,使用自动软化点装置(株式会社meitec制,asp-mg4)。
[0096]
·
环氧当量:
[0097]
基于jis k 7236规格进行测定,单位表示为“g/eq.”。具体而言,使用自动电位差滴定装置(平沼工业株式会社制,com-1600st),作为溶剂使用氯仿,加入溴化四乙基铵乙酸溶液,以0.1mol/l过氯酸-乙酸溶液进行了滴定。
[0098]
·
总氯含量:
[0099]
基于jis k 7243-3规格进行测定,单位由“ppm”表示。具体而言,作为溶剂使用二乙二醇单丁基醚,加入1mol/l氢氧化钾1,2-丙二醇溶液进行加热处理后,使用自动电位差滴定装置(平沼工业株式会社制,com-1700),以0.01mol/l的硝酸银溶液进行了滴定。
[0100]
·
铜箔剥离强度以及层间粘接力:
[0101]
基于jis c 6481进行了测定,层间粘接力是在第7层和第8层之间进行剥离测定。
[0102]
·
阻燃性:
[0103]
基于ul94,利用垂直法进行了评价。评价记为v-0、v-1、v-2。
[0104]
·
玻璃化转变温度(tg):
[0105]
基于ipc-tm-6502.4.25.c,由利用示差扫描热量测定装置(株式会社日立高科技制,exstar6000dsc6200),在20℃/分钟的升温条件下进行了测定,以此时的dsc
·
tgm(相对于玻璃状态与橡胶状态的切线为变异曲线的中间温度)的温度表示。
[0106]
·
相对介电常数和介电损耗角正切:
[0107]
基于ipc-tm-650 2.5.5.9使用材料分析仪(agilent technologies公司制),利用容量法,求出频率1ghz中的相对介电常数和介电损耗角正切,由此进行了评价。
[0108]
·
gpc(凝胶渗透色谱)测定:
[0109]
使用在本体(东曹株式会社制,hlc-8220gpc)中串联地具备柱(东曹株式会社制,tskgelg4000h
xl
,tskgelg3000h
xl
,tskgelg2000h
xl
)而成的装置,柱温为40℃。另外,洗脱液使用四氢呋喃(thf),设为1ml/分钟的流速,检测器使用示差折射率检测器。测定试样使用50μl的将样品0.1g溶解于10ml的thf中并利用微滤器过滤得到的试样。数据处理使用东曹
株式会社制gpc-8020model ii version 6.00。
[0110]
·
ir:
[0111]
使用傅立叶变换型红外分光光度计(perkin elmer precisely制,spectrum one ft-ir spectrometer 1760x),样品池使用krs-5,将溶解于thf的样品涂布在样品池上,干燥后,测定波数650~4000cm
-1
的吸光度。
[0112]
·
esi-ms:
[0113]
使用质谱仪(岛津制作所制,lcms-2020),使用乙腈和水作为流动相,测定溶解于乙腈的样品,由此进行了质量分析。
[0114]
在实施例、比较例中使用的缩写如下所示。
[0115]
[环氧树脂]
[0116]
e1:合成例1中得到的环氧树脂
[0117]
e2:合成例2中得到的环氧树脂
[0118]
e3:合成例3中得到的环氧树脂
[0119]
e4:联苯芳烷基型环氧树脂(日本化药株式会社制,nc-3000,环氧当量274,软化点60℃)
[0120]
e5:三苯酚甲烷型环氧树脂(日本化药株式会社制,eppn-501h,环氧当量166)
[0121]
e6:含磷环氧树脂(日铁化学&材料株式会社制,fx-1225,环氧当量317,磷含有率)
[0122]
e7:萘型环氧树脂(日铁化学&材料株式会社制,esn-475v,环氧当量325)
[0123]
e8:联苯型环氧树脂(三菱化学株式会社制,yx-4000h,环氧当量195,熔点105℃)
[0124]
e9:含硫原子的环氧树脂(日铁化学&材料株式会社制,yslv-120te,环氧当量250,熔点121℃)
[0125]
e10:对苯二酚型环氧树脂(日铁化学&材料社制,ydc-1312,环氧当量176,熔点142℃)
[0126]
e11:双环戊二烯型环氧树脂(dic株式会社制,hp-7200h,环氧当量280,软化点82℃)
[0127]
[固化剂]
[0128]
p1:苯酚酚醛清漆树脂(aicasdk phenol株式会社制,brg-557,羟基当量105,软化点85℃)
[0129]
p2:双环戊二烯型酚醛树脂(群荣化学工业株式会社制,gdp-6140,羟基当量196,软化点130℃)
[0130]
p3:三羟基苯基甲烷型酚醛清漆型树脂(群荣化学工业株式会社制,resitop tpm-100,羟基当量98,软化点108℃)
[0131]
p4:联苯芳烷基型酚醛树脂(明和化成株式会社制,meh-7851,羟基当量223,软化点75℃)
[0132]
p5:萘酚型固化剂(日铁化学&材料株式会社制,sn-485,羟基当量215,软化点85℃)
[0133]
p6:合成例4中得到的双环戊二烯型活性酯树脂
[0134]
p7:双氰胺(nippon carbide industry co.,inc.制,dihard,活性氢当量21)
[0135]
[苯并嗪树脂]
[0136]
b1:bpf型苯并嗪树脂(四国化成工业株式会社制,f-a型苯并嗪树脂)
[0137]
[固化促进剂]
[0138]
c1:2e4mz:2-乙基-4-甲基咪唑(四国化成工业株式会社制,curezol2e4mz)
[0139]
c2:三苯基膦(北兴化学工业株式会社制,hokko tpp)
[0140]
c3:2-苯基咪唑(四国化成工业株式会社制,curezol2pz)
[0141]
c4:4-二甲基氨基吡啶(岸田化学株式会社制)
[0142]
[填充剂]
[0143]
f1:中空玻璃填料(3m japan株式会社制,glass bubbles im30k,平均粒径(d50)16μm)
[0144]
合成例1
[0145]
在具备搅拌机、温度计、氮吹入管、滴液漏斗以及冷却管的反应装置中加入2,6-二甲酚140份、47%bf3醚络合物9.3份(相对于最初添加的双环戊二烯为0.1倍摩尔),一边进行搅拌一边加热到110℃。一边保持为相同温度一边历经1小时滴下双环戊二烯86.6份(相对于2,6-二甲酚为0.57倍摩尔)。进而在110℃的温度下反应3小时后,一边保持为相同温度一边历经1小时滴下双环戊二烯68份(相对于2,6-二甲酚为0.44倍摩尔)。进而在120℃下反应2小时。加入氢氧化钙14.6份。另外,添加10%的草酸水溶液45份。然后,加温到160℃脱水后,在5mmhg的减压下加温到200℃,蒸发除去未反应的原料。加入mibk700份,溶解生成物,加入80℃的温水200份进行水洗,分离除去下层的水层。然后,在5mmhg的减压下,加温到160℃,蒸发除去mibk,得到红褐色的多元羟基化合物274份。是羟基当量299、软化点97℃的树脂,吸收比(a
3040
/a
1210
)为0.17。测定基于esi-ms(负极)的质谱,其结果确认为m-=253、375、507、629。将得到的多元羟基化合物的gpc示于图1,将ft-ir示于图2。gpc中的mw为690,mn为510,n=0体含量为6.5面积%,n=1体含量为61.5%,n=2体以上的含量为32.0%。图1的a表示式(2)的n=1体和式(8)的没有r2附加体的n=1体的混合体,b表示式(2)的n=0体。图2的c表示来自于双环戊二烯骨架的烯烃部位的c-h伸缩振动的峰,d表示基于苯酚核的c-o伸缩振动的吸收。
[0146]
在具备搅拌机、温度计、氮吹入管、滴液漏斗以及冷却管的反应装置中,加入该多元羟基化合物200份、表氯醇309份和二乙二醇二甲基醚93部,加温到65℃。在125mmhg的减压下,一边保持在63~67℃的温度一边历经4小时滴下49%氢氧化钠水溶液60份。此时,表氯醇与水共沸,流出来的水依次向体系外除去。反应结束后,在成为5mmhg、180℃的条件下回收表氯醇,加入mibk550份,溶解生成物。然后,加入150份的水来溶解副生成的食盐,静置而分离除去下层的食盐水。在磷酸水溶液中和后,将树脂溶液水洗过滤,直到水洗液为中性。在5mmhg的减压下,加热到180℃,馏去mibk,得到红褐色透明的2,6-二甲酚
·
双环戊二烯型环氧树脂(e1)226份。是环氧当量358、总氯含量520ppm、软化点80℃的树脂。将得到的环氧树脂(e1)的gpc示于图3。gpc中的mw为870,mn为570,m=0体含量为5.5面积%,m=1体含量为61.8%,m=2体以上的含量为32.6%。
[0147]
合成例2
[0148]
在与合成例1同样的反应装置中,投入2,6-二甲酚140份、47%bf3醚络合物9.3份(相对于最初添加的双环戊二烯为0.1倍摩尔),一边进行搅拌一边加热到110℃。一边保持
在相同温度一边历经1小时滴下双环戊二烯86.6份(相对于2,6-二甲酚为0.57倍摩尔)。进而在110℃的温度下反应3小时后,一边保持在相同温度一边历经1小时滴下双环戊二烯90.6份(相对于2,6-二甲酚为0.60倍摩尔)。进而在120℃下反应2小时,加入氢氧化钙14.6份。进一步添加10%的草酸水溶液45份。然后,加热到160℃进行脱水后,在5mmhg的减压下,加热到200℃而蒸发除去未反应的原料。加入mibk740份,溶解生成物,加入80℃的温水200份进行水洗,将下层的水层分离除去。然后,在5mmhg的减压下,加热到160℃,蒸发除去mibk,得到红褐色的多元羟基化合物310份。是羟基当量341、软化点104℃的树脂,吸收比(a
3040
/a
1210
)为0.27。测定基于esi-ms(负极)的质谱,其结果确认了m-=253、375、507、629。gpc中的mw为830,mn为530,n=0体含量为5.9面积%,n=1体含量为60.1%,n=2体以上的含量为34.0%。
[0149]
在反应装置中加入该多元羟基化合物200份、表氯醇271份和二乙二醇二甲基醚81份,加热到65℃。在125mmhg的减压下,一边保持在63~67℃的温度一边历经4小时滴加49%氢氧化钠水溶液53份。此时,表氯醇与水共沸,流出来的水依次向体系外除去。反应结束后,在5mmhg、180℃的条件下回收表氯醇,加入mibk540份溶解生成物。然后,加入150份的水来溶解副生成的食盐,静置并分离除去下层的食盐水。利用磷酸水溶液中和后,将树脂溶液水洗过滤,直到水洗液变为中性。在5mmhg的减压下,加热到180℃,馏去mibk,得到红褐色透明的2,6-二甲酚
·
双环戊二烯型环氧树脂(e2)221份。是环氧当量421、总氯含量530ppm、软化点84℃的树脂。gpc中的mw为880,mn为570,m=0体含量为5.5面积%,m=1体含量为58.8%,m=2体以上的含量为35.7%。
[0150]
合成例3
[0151]
在与合成例1同样的反应装置中,投入2,6-二甲酚140份、47%bf3醚络合物9.3份(相对于最初添加的双环戊二烯为0.1倍摩尔),一边进行搅拌一边加热到110℃。一边保持为相同温度一边历经1小时滴加双环戊二烯86.6份(相对于2,6-二甲酚为0.57倍摩尔)。进而在110℃的温度下反应3小时后,一边保持相同温度一边历经1小时滴加双环戊二烯56.7份(相对于2,6-二甲酚为0.37倍摩尔)。进而在120℃下反应2小时。加入氢氧化钙14.6份。另外,添加10%的草酸水溶液45份。然后,加热到160℃进行脱水后,在5mmhg的减压下,加热到200℃而蒸发除去未反应的原料。加入mibk660份,溶解生成物,加入80℃的温水200份,进行水洗,分离除去下层的水层。然后,在5mmhg的减压下,加热到160℃,蒸发除去mibk,得到红褐色的多元羟基化合物280份。是羟基当量为272,软化点91℃的树脂,吸收比(a
3040
/a
1210
)为0.14。测定基于esi-ms(负极)的质谱,其结果确认了m-=253、375、507、629。gpc中的mw为680,mn为530,n=0体含量为5.9面积%,n=1体含量为75.1%,n=2体以上的含量为19.0%。
[0152]
在反应装置中加入该多元羟基化合物200份、表氯醇170份和二乙二醇二甲基醚51份,加热到65℃。在125mmhg的减压下,一边保持在63~67℃的温度一边历经4小时滴加49%氢氧化钠水溶液66份。此时,表氯醇与水共沸,流出来的水依次向体系外除去。反应结束后,在成为5mmhg、180℃的条件下回收表氯醇,加入mibk560份,溶解生成物。然后,加入150份的水来溶解副生成的食盐,静置而分离除去下层的食盐水。利用磷酸水溶液中和后,将树脂溶液水洗过滤,直到水洗液为中性。在5mmhg的减压下,加热到180℃,馏去mibk,得到红褐色透明的2,6-二甲酚
·
双环戊二烯型环氧树脂(e3)229份。是环氧当量358、总氯含量570ppm、
软化点76℃的树脂。gpc中的mw为800,mn为470,m=0体含量为4.6面积%,m=1体含量为63.2%,m=2体以上的含量为32.2%。
[0153]
合成例4
[0154]
在与合成例1同样的反应装置中投入苯酚400份、47%bf3醚络合物7.5份,一边进行搅拌一边加热到70℃。一边保持在相同温度一边历经2小时滴加双环戊二烯70.2份。另外,在125~135℃的温度下反应4小时,加入氢氧化钙11.7份。另外,添加10%的草酸水溶液35份。然后,加热到160℃并脱水后,在5mmhg的减压下加热到200℃,蒸发除去未反应的原料。加入mibk1097份,溶解生成物,加入80℃的温水108份,进行水洗,分离除去下层的水层。然后,在5mmhg的减压下加热到160℃,蒸发除去mibk,得到红褐色的多元羟基化合物158份。羟基当量为177,软化点为92℃。
[0155]
在反应装置中投入该多元羟基化合物64.8份、1-萘酚17.4份、四正丁基铵溴化物0.01份、异富马酸溴化物49.4份以及甲苯329份,升温到50℃并使其溶解。一边将体系内控制到60℃以下一边历经3小时滴加20%氢氧化钠水溶液97.3份,然后,在相同温度下持续搅拌1小时。将反应混合物静置分液,除去水层。重复该操作直到水层的ph为7。然后,通过回流脱水,除去水分,得到不挥发成分65%的处于甲苯溶液状态的活性酯树脂(p6)161份。由原料的投入量计算的活性酯当量为235。
[0156]
实施例1
[0157]
配合100份作为环氧树脂的e1,配合37份作为固化剂的p1,配合0.22份作为固化促进剂的c1,溶解于利用mek、丙二醇单甲基醚、n,n-二甲基甲酰胺调整而得的混合溶剂中,得到环氧树脂组合物清漆。将得到的环氧树脂组合物清漆浸入到玻璃布(日东纺织株式会社制,wea7628xs13,0.18mm厚)。将浸入的玻璃布在150℃的热风循环烘箱中干燥9分钟,得到预浸料。将得到的预浸料8片与铜箔(三井金属矿业株式会社制3ec-iii,厚度35μm)在上下方向重叠,在130℃
×
15分钟 190℃
×
80分钟的温度条件下进行2mpa的真空压制,得到1.6mm厚度的层叠板。将层叠板的铜箔剥离强度和层间粘接力的结果示于表1。
[0158]
将得到的预浸料拆开,制成通过100目的筛粉状的预浸料粉末。将得到的预浸料粉末加入到氟树脂制的模具中,在130℃
×
15分钟 190℃
×
80分钟的温度条件下进行2mpa的真空加压,得到50mm见方
×
2mm厚的试验片。将试验片的相对介电常数和介电损耗角正切的结果示于表1。
[0159]
实施例2~11、比较例1~12
[0160]
按照表1~3的配合量(份)配合,进行与实施例1同样的操作,得到层叠板和试验片。固化促进剂的使用是能够将清漆凝胶时间调整为300秒左右的量。进行与实施例1同样的试验,将其结果示于表1~3。
[0161]
[表1]
[0162][0163]
[表2]
[0164][0165]
[表3]
[0166][0167]
实施例12和比较例13~15
[0168]
以表4的配合量(份)进行配合,进行与实施例1同样的操作,得到层叠板和试验片。将层叠板的阻燃性、铜箔剥离强度、层间粘接力以及tg的测定结果、以及试验片的相对介电常数和介电损耗角正切的测定结果示于表4。
[0169]
[表4]
[0170][0171]
实施例13
[0172]
为了进行作为注塑树脂的评价,将50份作为环氧树脂的e2和50份e8、32份作为固化剂的p1、以及1.0份作为固化促进剂的c2,而得到树脂组合物。使用得到的环氧树脂组合物,在175℃下成型,另外在175℃下进行12小时的二次固化,得到固化物。将固化物的相对
介电常数、介电损耗角正切以及tg的测定结果示于表5。
[0173]
实施例14~15以及比较例16~18
[0174]
以表5的配合量(份)进行配合,进行与实施例13同样的操作,得到固化物。将进行了与实施例13同样的试验的结果示于表5。
[0175]
[表5]
[0176] 实施例13实施例14实施例15比较例16比较例17比较例18e2505050
ꢀꢀꢀ
e850
ꢀꢀ
100
ꢀꢀ
e9 50
ꢀꢀ
100 e10
ꢀꢀ
50
ꢀꢀ
100p1323342384260c21.01.01.01.01.01.0f1404045454550相对介电常数2.582.532.622.792.672.82介电损耗角正切0.0140.0130.0150.0190.0180.020tg(℃)158157163149147158
[0177]
根据这些结果可知,本发明的环氧树脂组合物可以提供显示出非常良好的低介电特性、且粘接力优异的树脂固化物。
[0178]
产业上的可利用性
[0179]
本发明的环氧树脂组合物的介电性、耐热性、粘接性优异,能够利用于层叠、成型、粘接等各种用途,特别是作为高速通信设备的电子材料有用。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。