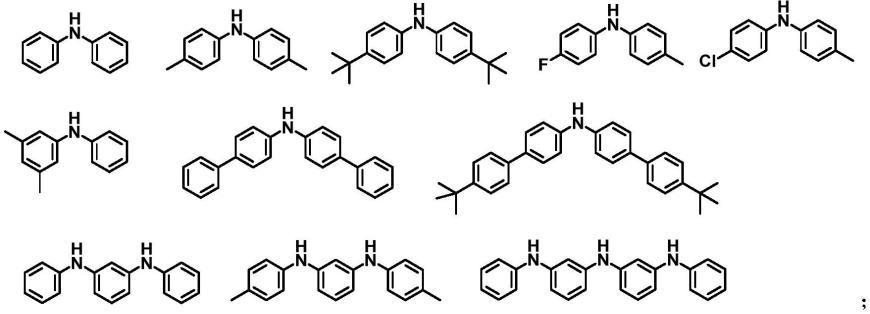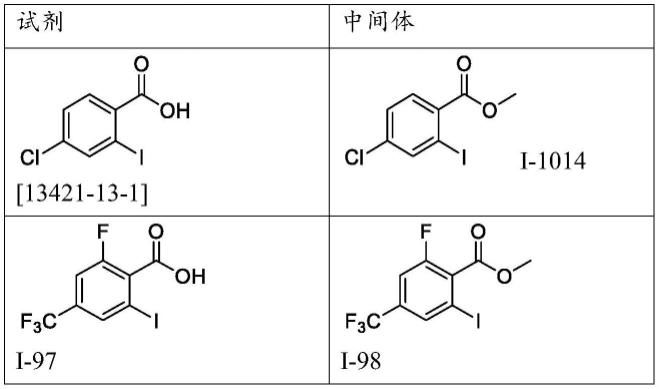
1.本发明涉及可用作nod样受体蛋白3(nlrp3)炎症小体途径抑制剂的新型三嗪酮。本发明还涉及制备所述化合物的方法、包含所述化合物的药物组合物、使用所述化合物治疗各种疾病和障碍的方法和含有它们的药剂,以及它们在由nlrp3介导的疾病和障碍中的用途。
背景技术:
2.炎症小体被认为是先天免疫系统的中枢信号传导枢纽,是多蛋白复合物,这些多蛋白复合物在多种病原体相关或危险相关分子模式(pamp或damp)激活一组特定的细胞内模式识别受体(prr)后组装。迄今为止,已经表明炎症小体可以由核苷酸结合寡聚化结构域(nod)样受体(nlr)以及含有脓素和hin200结构域的蛋白质形成(van opdenbosch n和lamkanfi m.immunity[免疫力],2019年6月18日;50(6):1352-1364)。nlrp3炎症小体在检测到环境晶体、污染物、宿主衍生的damp和蛋白质聚集体后组装(tartey s和kanneganti td.immunology[免疫学],2019年4月;156(4):329-338)。与nlrp3接合的临床相关damp包括导致痛风和动脉粥样硬化的尿酸和胆固醇晶体、阿尔茨海默病中的具有神经毒性的淀粉样蛋白β原纤维和导致间皮瘤的石棉颗粒(kelley等人,int j mol sci[国际分子科学杂志],2019年7月6日;20(13))。此外,nlrp3被以下激活:感染因子,例如霍乱弧菌;真菌病原体,例如烟曲霉和白色念珠菌;腺病毒、甲型流感病毒和sars-cov-2(tartey和kanneganti,2019(参见上文);fung等人emerg microbes infect[新现微生物感染],2020年3月14日;9(1):558-570)。
[0003]
虽然确切的nlrp3激活机制仍不清楚,但对于人类单核细胞,有人建议一步激活就足够了,而在小鼠中则是两步机制。鉴于触发因素众多,nlrp3炎症小体需要在转录和转录后水平进行附加调节(yang y等人,cell death dis[细胞死亡与疾病],2019年2月12日;10(2):128)。
[0004]
nlrp3蛋白由以下组成:n末端的脓素结构域,然后是核苷酸结合位点结构域(nbd)和位于c末端的富含亮氨酸的重复(lrr)基序(sharif等人,nature[自然],2019年6月;570(7761):338-343)。在识别pamp或damp后,nlrp3与衔接蛋白、凋亡相关斑点样蛋白(asc)和蛋白酶半胱天冬酶-1聚集形成功能性炎症小体。激活后,半胱天冬酶-1酶原进行自体蛋白水解,从而切割焦孔素(gasdermin)d(gsdmd)以产生n末端gsdmd分子,其最终将导致质膜中的孔形成和细胞死亡的裂解形式,称为细胞焦亡。可替代地,半胱天冬酶-1切割促炎细胞因子pro-il-1β和pro-il-18,以允许通过细胞焦亡释放其生物活性形式(kelley等人,2019-参见上文)。
[0005]
nlrp3炎症小体或其下游介质的失调与以下范围内的多种病状相关:从免疫性/炎性疾病、自身免疫性/自身炎性疾病(冷吡啉相关周期性综合征(miyamae t.paediatr drugs[儿科药物],2012年4月1日;14(2):109-17);镰状细胞病;系统性红斑狼疮(sle))到肝脏障碍(例如非酒精性脂肪性肝炎(nash)、慢性肝病、病毒性肝炎、酒精性脂肪性肝炎和
酒精性肝病)(szabo g和petrasek j.nat rev gastroenterol hepatol[自然综述胃肠病学和肝病学],2015年7月;12(7):387-400)和炎性肠病(例如克罗恩病、溃疡性结肠炎)(zhen y和zhang h.front immunol[免疫学前沿],2019年2月28日;10:276)。此外,炎性关节障碍(例如痛风、假痛风(软骨钙质沉着病)、关节病、骨关节炎和类风湿性关节炎(vande walle l等人,nature[自然],2014年8月7日;512(7512):69-73)与nlrp3相关联。此外,肾脏相关疾病(高草酸尿症(knauf等人,kidney int[国际肾脏学],2013年11月;84(5):895-901)、狼疮性肾炎、高血压肾病(krishnan等人,br j pharmacol[英国药理学会杂志],2016年2月;173(4):752-65)、血液透析相关炎症和糖尿病肾病(其是糖尿病(1型、2型和糖尿病)的肾脏相关并发症,也称为糖尿病肾疾病(shahzad等人,kidney int[国际肾脏学],2015年1月;87(1):74-84))与nlrp3炎症小体激活相关。报告将神经炎症相关障碍(例如脑感染、急性损伤、多发性硬化、阿尔茨海默病)和神经退行性疾病(帕金森病)的发生和进展与nlrp3炎症小体激活联系起来(sarkar等人,npj parkinsons dis[npj帕金森病],2017年10月17日;3:30)。此外,心血管或代谢障碍(例如心血管风险降低(cvrr)、动脉粥样硬化、i型和ii型糖尿病及相关并发症(例如肾病、视网膜病变)、外周动脉疾病(pad)、急性心力衰竭和高血压(ridker等人,cantos试验组n engl j med[新英格兰医学杂志],2017年9月21日;377(12):1119-1131;以及toldo s和abbate a.nat rev cardiol[自然综述心脏病学],2018年4月;15(4):203-214)最近与nlrp3相关联。此外,还描述了皮肤相关疾病(例如伤口愈合和疤痕形成;炎性皮肤病,例如痤疮、化脓性汗腺炎(kelly等人,br j dermatol[英国皮肤病学杂志],2015年12月;173(6))。此外,呼吸系统病症与nlrp3炎症小体活性(例如哮喘、结节病、严重急性呼吸系统综合征(sars)(nieto-torres等人,virology[病毒学],2015年11月;485:330-9))以及年龄相关黄斑变性(doyle等人,nat med[自然医学],2012年5月;18(5):791-8)相关。描述了几种与nlrp3相关的癌症相关疾病/障碍(例如骨髓增生性肿瘤、白血病、骨髓增生异常综合征(mos)、骨髓纤维化、肺癌、结肠癌(ridker等人,lancet[柳叶刀],2017年10月21日;390(10105):1833-1842;derangere等人,cell death differ[细胞死亡与分化].2014年12月;21(12):1914-24;basiorka等人,lancet haematol[柳叶刀血液学],2018年9月;5(9):e393-e402,zhang等人,hum immunol[人免疫学],2018年1月;79(1):57-62))。
[0006]
一些专利申请描述了nlrp3抑制剂,其中最近的专利包括例如国际专利申请wo 2020/018975、wo 2020/037116、wo 2020/021447、wo 2020/010143、wo 2019/079119、wo 2019/0166621和wo 2019/121691,它们公开了一系列特异性化合物。
[0007]
需要nlrp3炎症小体途径抑制剂来为本文提及的疾病/障碍提供新的和/或可替代的治疗。
技术实现要素:
[0008]
本发明提供抑制nlrp3炎症小体途径的化合物。
[0009]
因此,在本发明的一个方面,现在提供具有式(i)的化合物,
[0010][0011]
或其药学上可接受的盐,其中:
[0012]
r1代表:
[0013]
(i)c
3-6
环烷基,其任选地被一个或多个独立地选自-oh和-c
1-3
烷基的取代基取代;
[0014]
(ii)芳基或杂芳基,其各自任选地被1至3个独立地选自卤代、-oh、-o-c
1-3
烷基、-c
1-3
烷基、卤代c
1-3
烷基、羟基c
1-3
烷基、c
1-3
烷氧基、卤代c
1-3
烷氧基的取代基取代;或
[0015]
(iii)杂环基,其任选地被1至3个独立地选自c
1-3
烷基和c
3-6
环烷基的取代基取代;
[0016]
r2代表:
[0017]
(i)c
1-3
烷基,其任选地被一个或多个独立地选自卤代、-oh和-oc
1-3
烷基的取代基取代;
[0018]
(ii)c
3-6
环烷基;
[0019]
(iii)c
2-4
烯基,其任选地被-oc
1-3
烷基取代;或
[0020]
(iv)-n(r
2a
)r
2b
;
[0021]r2a
和r
2b
各自代表氢或c
1-4
烷基,或者r
2a
和r
2b
可以连接在一起形成3元至4元任选地被一个或多个氟原子取代的环;
[0022]
r3代表:
[0023]
(i)氢;
[0024]
(ii)卤代;
[0025]
(iii)c
1-4
烷基,其任选地被一个或多个独立地选自卤代、-oh和-oc
1-3
烷基的取代基取代;
[0026]
(iv)c
2-4
烯基,其任选地被-oc
1-3
烷基取代;
[0027]
(v)c
3-6
环烷基;或
[0028]
(vi)-oc
1-3
烷基,
[0029]
这些化合物在本文可以被称为“本发明的化合物”。
[0030]
在一个实施例中,可以提及的本发明的化合物包括以下那些,其中:
[0031]
(i)当r3代表氢,r2代表甲基时,则r1不代表4-甲基苯基;
[0032]
(ii)当r3代表氢,r2代表环己基时,则r1不代表2-茚满基(在2位置处连接的2,3-二氢-1h-茚),
[0033]
这些在本文可以被称为“所述条件”。
[0034]
例如,提供了如上文所定义的具有式(i)的化合物或其药学上可接受的盐,用作nlrp3抑制剂(例如,用于治疗与nlrp3炎症小体活性相关的疾病或障碍),前提是其不是所述条件中的化合物。还提供了如上文所定义的具有式(i)的化合物或其药学上可接受的盐,用作nlrp3抑制剂,用于治疗癌症,前提是其不是所述条件中的化合物(i)。还提供了如上文所定义的具有式(i)的化合物或其药学上可接受的盐,用作nlrp3抑制剂,用于治疗阿尔茨海默病,前提是其不是所述条件中的化合物(ii)。
[0035]
在本发明的一个方面,提供了如上文所定义的具有式(i)的化合物或其药学上可
接受的盐,其中:
[0036]
r1代表:
[0037]
(i)c
3-6
环烷基,其任选地被一个或多个独立地选自卤代、-oh、-c
1-3
烷基(它本身任选地被一个或多个选自氟和-oh的取代基取代)和-oc
1-3
烷基的取代基取代;
[0038]
(ii)芳基或杂芳基,其各自任选地被1至3个独立地选自卤代、-cn、-oh、-o-c
1-3
烷基、-c
1-6
烷基(例如-c
1-3
烷基)、卤代c
1-3
烷基、羟基c
1-3
烷基、c
1-3
烷氧基c
1-3
烷基、卤代c
1-3
烷氧基、氨基c
1-3
烷基(例如h2n-c
1-3
烷基或(ch3)2n-c
1-3
烷基)、c
3-6
环烷基或芳基/杂芳基(其中除前三个基团以外这些靠后的基团本身任选地被一个或多个选自卤代、c
1-3
烷基和-oc
1-3
烷基的取代基取代)的取代基取代;或
[0039]
(iii)杂环基,其任选地被1至3个独立地选自卤代、=o、-oh、-c
1-4
烷基(它本身任选地被一个或多个选自氟、=o和-oh的取代基取代)、-oc
1-3
烷基、c
3-6
环烷基和3-6元杂环基环的取代基取代;
[0040]
r2代表:
[0041]
(i)c
1-6
烷基(例如c
1-4
烷基或c
1-3
烷基),其任选地被一个或多个独立地选自卤代、=o、-oh和-oc
1-3
烷基的取代基取代;
[0042]
(ii)c
3-6
环烷基,其任选地被一个或多个选自卤代(例如氟)、c
1-3
烷基和-oc
1-3
烷基的取代基取代;
[0043]
(iii)c
2-4
烯基,其任选地被-oc
1-3
烷基取代;或
[0044]
(iv)-n(r
2a
)r
2b
;
[0045]r2a
和r
2b
各自代表氢或c
1-4
烷基,或者r
2a
和r
2b
可以连接在一起形成3元至4元任选地被一个或多个氟原子取代的环;
[0046]
r3代表:
[0047]
(i)氢;
[0048]
(ii)卤代或-cn;
[0049]
(iii)c
1-6
烷基(例如c
1-4
烷基),其任选地被一个或多个独立地选自卤代、-oh和-oc
1-3
烷基的取代基取代;
[0050]
(iv)c
2-4
烯基,其任选地被-oc
1-3
烷基取代;
[0051]
(v)c
3-6
环烷基,其任选地被一个或多个氟原子取代;
[0052]
(vi)-nh2、-n(h)(c
1-3
烷基)或n(c
1-3
烷基)2;或
[0053]
(vii)-oc
1-3
烷基,其任选地被一个或多个氟原子取代;
[0054]
并且其中该含有r3的苯环还可以任选地被一个选自卤代(例如氟)、-oh和-cn的取代基取代(在三个相关位置处),
[0055]
这些化合物在本文也可以被称为“本发明的化合物”。
[0056]
在一个实施例中,提供了如上文所定义的具有式(i)的化合物或其药学上可接受的盐,其中r3不代表氢。
[0057]
在另一方面,提供了用作药剂的本发明化合物。在另一方面,提供了包含治疗有效量的本发明化合物的药物组合物。
[0058]
在另外的方面,提供了用于以下用途的本发明化合物(和/或包含这样的化合物的药物组合物):用于治疗与nlrp3活性(包括炎症小体活性)相关的疾病或障碍;用于治疗其
中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍;用于抑制nlrp3炎症小体活性(包括在有需要的受试者中);和/或用作nlrp3抑制剂。特定的疾病或障碍可在本文中提及,并且可例如选自炎症小体相关疾病或障碍、免疫性疾病、炎性疾病、自身免疫性疾病或自身炎性疾病。
[0059]
在另一方面,提供了本发明化合物(和/或包含这样的化合物的药物组合物)在以下中的用途:用于治疗与nlrp3活性(包括炎症小体活性)相关的疾病或障碍;用于治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍;用于抑制nlrp3炎症小体活性(包括在有需要的受试者中);和/或用作nlrp3抑制剂。
[0060]
在另一方面,提供了本发明化合物(和/或包含这样的化合物的药物组合物)在制造药剂中的用途,该药剂用于:治疗与nlrp3活性(包括炎症小体活性)相关的疾病或障碍;治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍;和/或抑制nlrp3炎症小体活性(包括在有需要的受试者中)。
[0061]
在另一方面,提供了治疗疾病或障碍的方法(其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展),该方法包括例如向受试者(有需要)施用治疗有效量的本发明化合物。在另外的方面,提供了抑制受试者(有需要)的nlrp3炎症小体活性的方法,该方法包括向有需要的受试者施用治疗有效量的本发明化合物。
[0062]
在另外的方面,提供了与一种或多种治疗剂(例如如本文所述)组合(包括药物组合)的本发明化合物。这样的组合也可提供用于如本文关于本发明化合物所述的用途,或如本文关于本发明化合物所述的这样的组合的用途。还可以提供如本文关于本发明化合物所述的方法,但其中该方法包括施用治疗有效量的这样的组合。
具体实施方式
[0063]
本发明提供了具有式(i)的化合物,
[0064][0065]
或其药学上可接受的盐,其中:
[0066]
r1代表:
[0067]
(i)c
3-6
环烷基,其任选地被一个或多个独立地选自-oh和-c
1-3
烷基的取代基取代;
[0068]
(ii)芳基或杂芳基,其各自任选地被1至3个独立地选自卤代、-oh、-o-c
1-3
烷基、-c
1-3
烷基、卤代c
1-3
烷基、羟基c
1-3
烷基、c
1-3
烷氧基、卤代c
1-3
烷氧基的取代基取代;或
[0069]
(iii)杂环基,其任选地被1至3个独立地选自c
1-3
烷基和c
3-6
环烷基的取代基取代;
[0070]
r2代表:
[0071]
(i)c
1-3
烷基,其任选地被一个或多个独立地选自卤代、-oh和-oc
1-3
烷基的取代基取代;
[0072]
(ii)c
3-6
环烷基;
[0073]
(iii)c
2-4
烯基,其任选地被-oc
1-3
烷基取代;或
[0074]
(iv)-n(r
2a
)r
2b
;
酰基衍生物和n-曼尼希碱。有关前药的一般信息可以例如在bundegaard,h.“design of prodrugs[前药的设计]”第l-92页,elesevier[爱思唯尔出版社],new york-oxford[纽约牛津](1985)中找到。
[0095]
本发明的化合物可以包含双键并且因此可以作为关于每个单独双键的e(异侧)和z(同侧)几何异构体存在。位置异构体也可以被包括在本发明的这些化合物中。所有这样的异构体(例如,如果本发明的化合物包含双键或稠环,则包括顺式和反式形式)及其混合物都包括在本发明的范围之内(例如,单一的位置异构体和位置异构体的混合物都可以包括在本发明的范围之内)。
[0096]
本发明的化合物还可以展示出互变异构现象。所有的互变异构形式(或互变异构体)及其混合物都包括在本发明的范围之内。术语“互变异构体”或“互变异构形式”指的是具有不同能量的结构异构体,这些异构体可经由低能量势垒相互转换。例如,质子互变异构体(也称作质子移变互变异构体)包括经由质子移变产生的相互转换,例如酮-烯醇和亚胺-烯胺异构化。价键互变异构体包括由一些成键电子的重组产生的相互转换。
[0097]
本发明的化合物还可以包含一个或多个不对称碳原子并且因此可以展示出旋光和/或非对映异构现象。可以使用常规技术,例如,色谱法或分步结晶来分离非对映异构体。可以通过使用常规技术,例如分步结晶或hplc,对这些化合物的外消旋混合物或其他混合物进行分离来分选不同的立体异构体。可替代地,所希望的旋光异构体可以通过适当的旋光起始材料在不会引起外消旋作用或差向异构作用(epimerisation)的条件(即
‘
手性池’(
‘
chiral pool’)方法)下的反应;通过衍生化作用(即,拆分,包括动态拆分)适当的起始材料与可以在合适的阶段被去除的
‘
手性助剂’(例如与纯手性酸)反应,随后通过常规手段(例如色谱法)分离非对映异构体衍生物;或者通过与适当的手性试剂或手性催化剂反应来制备,所有都在技术人员已知的条件下。
[0098]
所有的立体异构体(包括但不限于非对映异构体、对映异构体和阻转异构体)及其混合物(例如,外消旋混合物)都包含在本发明的范围之内。
[0099]
本文示出的这些结构中,在任何具体的手性原子的立体化学都未指明的情况下,那么所有的立体异构体都被认为是本发明的化合物并且包括在本发明的化合物中。在立体化学通过表示具体构型的实楔形线或虚线被指明的情况下,那么该立体异构体是所指明和定义的。
[0100]
当指定绝对构型时,它是根据卡恩-英戈尔德-普雷洛格(cahn-ingold-prelog)系统。不对称原子处的构型由r或s指定。绝对构型未知的已拆分的化合物可以根据它们旋转平面偏振光的方向而由( )或(-)指定。
[0101]
当鉴定特定的立体异构体时,这意指所述立体异构体基本上不含其他异构体,即与小于50%、优选地小于20%、更优选地小于10%、甚至更优选地小于5%,特别是小于2%并且最优选地小于1%的其他立体异构体相关联。因此,当具有式(i)的化合物例如被指定为(r)时,这意指该化合物基本上不含(s)异构体。
[0102]
本发明的化合物能以非溶剂化的形式连同与药学上可接受的溶剂(例如水、乙醇等)的溶剂化的形式存在,并且意在表明本发明包括溶剂化的以及非溶剂化的形式两者。
[0103]
本发明还涵盖本发明的同位素标记的化合物,这些同位素标记的化合物与本文列举的那些相同,但是事实上一个或多个原子被原子质量或质量数不同于自然中通常发现
(或自然中发现的最多的那一个)的原子质量或质量数的原子所替换。如在本文中指明的任何具体的原子或元素的所有同位素都被认为是在本发明的这些化合物的范围之内。可掺入本发明的化合物的示例性同位素包括氢、碳、氮、氧、磷、硫、氟、氯和碘的同位素,例如2h、3h、
11
c、
13
c、
14
c、
13
n、
15
o、
17
o、
18
o、
32
p、
33
p、
35
s、
18
f、
36
cl、
123
i、和
125
i。本发明的某些同位素标记的化合物(例如,用3h和
14
c标记的那些)在化合物中是有用的并且用于底物组织分布测定。氚化(3h)和碳-l4(
14
c)同位素因其制备容易和可检测性而是有用的。此外,用更重同位素(例如氘)(即,2h)取代可以提供由于更大的代谢稳定性而产生的某些治疗优点(例如,增加的体内半衰期或降低的剂量需求)并且因此在一些环境下可以是优选的。正电子发射同位素(例如
15
o、
13
n、
11
c和
18
f)可用于正电子发射断层成像术(pet)研究以检查底物受体占用率。一般可以通过与在说明书/或在下文的实例中披露的那些类似的以下程序、通过用同位素标记的试剂取代非同位素标记的试剂来制备本发明的同位素标记的化合物。
[0104]
除非另外指明,本文定义的c
1-q
烷基基团(其中q是该范围的上限)可以是直链的,或者,当存在足够数目(即,如果适当的话,最少两个或三个)的碳原子时,可以是支链的。这样的基团通过单键连接到分子的其余部分。
[0105]c2-q
烯基当在本文中使用时(同样,其中q是范围的上限)是指烷基基团,该烷基基团包含不饱和度,即至少一个双键。
[0106]c3-q
环烷基(其中q是范围的上限)是指环状烷基基团,例如环烷基基团可以是单环的,或者如果有足够的原子,可以是二环的。在一个实施例中,这样的环烷基基团是单环的。这样的环烷基基团是不饱和的。多个取代基可以附接在环烷基基团上的任何位点处。
[0107]
当在本文中使用时,术语“卤代”优选包含氟、氯、溴和碘。
[0108]c1-q
烷氧基(其中q是范围的上限)是指式-ora的基团,其中ra是如本文所定义的c
1-q
烷基。
[0109]
卤代c
1-q
烷基(其中q是范围的上限)基团是指c
1-q
烷基基团,如本文所定义,其中该基团被一个或多卤代取代。羟基c
1-q
烷基(其中q是范围的上限)是指c
1-q
烷基基团,如本文所定义,其中该基团被一个或多个(例如一个)羟基(-oh)基团取代(或一个或多个,例如一个氢原子被-oh取代)。类似地,卤代c
1-q
烷氧基和羟基c
1-q
烷氧基代表分别被一个或多个卤代取代的或被一个或多个(例如一个)羟基取代的相应-oc
1-q
烷基基团。
[0110]
可以被提及的杂环基基团包括非芳香族单环和二环的杂环基基团,其中在该环系统中的这些原子中的至少一个(例如,一至四个)不是碳(即杂原子),并且其中在该环系统中的原子的总数在3与20之间(例如,在三和十之间,例如,在3和8之间,例如5至8)。这样的杂环基基团还可以是桥接的。这样的杂环基基团是饱和的。可以被提及的c
2-q
杂环基基团包括7-氮杂二环[2.2.1]庚基、6-氮杂二环[3.1.1]庚基、6-氮杂二环[3.2.1]-辛基、8-氮杂二环-[3.2.1]辛基、吖丙啶基、氮杂环丁烷基、二氢吡喃基、二氢吡啶基、二氢吡咯基(包括2,5-二氢吡咯基)、二氧戊环基(包括1,3-二氧戊环基)、二噁烷基(包括1,3-二噁烷基和1,4-二噁烷基)、二噻烷基(包括1,4-二噻烷基)、二硫戊环基(包括1,3-二硫戊环基)、咪唑烷基、咪唑啉基、吗啉基、7-氧杂二环[2.2.1]庚基、6-氧杂二环-[3.2.1]辛基、氧杂环丁烷基、环氧乙烷基、哌嗪基、哌啶基、非芳香族的吡喃基、吡唑烷基、吡咯烷酮基、吡咯烷基、吡咯啉基、奎宁环基、环丁砜基、3-丁二烯砜基、四氢吡喃基、四氢呋喃基、四氢吡啶基(例如1,2,3,4-四氢吡啶基和1,2,3,6-四氢吡啶基)、硫杂环丁烷基、硫杂环丙烷基、硫杂环戊烷基、硫代
吗啉基、三噻烷基(包括1,3,5-三噻烷基)、托烷基等。在适当的情况下,杂环基基团上的取代基位于该环系统中的任何原子(包括杂原子)上。杂环基基团的附接点可以是经由该环系统中的任何原子(在适当的情况下),包括杂原子(例如氮原子),或在可以作为该环系统的一部分存在的任何稠合的碳环上的原子。杂环基基团还可以处于n-或s-氧化的形式。在一个实施例中,本文提及的杂环基基团是单环的。
[0111]
可以被提及的芳基基团包括c
6-20
,例如c
6-12
(例如,c
6-10
)芳基基团。这样的基团可以是单环的、二环的或三环的并且具有6与12(例如,6与10)个之间的环碳原子,其中至少一个环是芳香族的。c
6-10
芳基基团包括苯基、萘基等基团,例如1,2,3,4-四氢萘基。芳基基团的附接点可以是经由该环系统的任何原子。例如,当该芳基基团是多环的时候,该附接点可以是经由原子,包括非芳香族环的原子。然而,当芳基基团是多环(例如,二环或三环)的时候,它们优选地是经由一个芳香族环连接到该分子的其余部分上。当芳基基团是多环时,在一个实施例中,每个环是芳香族的。在一个实施例中,本文提及的芳基基团是单环的或二环的。在另外的实施例中,本文提及的芳基基团是单环的。
[0112]“杂芳基”当在本文中使用时指的是包含一个或多个杂原子(例如一个至四个杂原子)的芳香族基团,该一个或多个杂原子优选地选自n、o和s。杂芳基基团包括具有5元和20元之间(例如,5元和10元之间)的那些,并且可以是单环的、二环的或三环的,前提是这些环中至少一个是芳香族的(这样形成,例如,单环、二环或三环的杂芳香族基团)。当该杂芳基基团是多环的时,该附接点可以是经由任何原子,包括非芳香族环的原子。然而,当杂芳基基团是多环(例如,二环或三环)的时,它们优选地是经由一个芳香族环连接到该分子的其余部分上。在一个实施例中,当杂芳基基团是多环时,则每个环是芳香族的。可以被提及的杂芳基基团包括3,4-二氢-1h-异喹啉基、1,3-二氢异吲哚基、1,3-二氢异吲哚基(例如,3,4-二氢-1h-异喹啉-2-基、1,3-二氢异吲哚-2-基、1,3-二氢异吲哚-2-基;即,经由一个非芳香族环连接的杂芳基基团),或者优选地包括吖啶基、苯并咪唑基、苯并二噁烷基、苯并二氧杂环庚基、苯并二氧杂环戊烯基(包括1,3-苯并二氧杂环戊烯基)、苯并呋喃基、苯并呋咱基、苯并噻二唑基(包括2,1,3-苯并噻二唑基)、苯并噻唑基、苯并噁二唑基(包括2,1,3-苯并噁二唑基)、苯并噁嗪基(包括3,4-二氢-2h-1,4-苯并噁嗪基)、苯并噁唑基、苯并吗啉基、苯并硒杂二唑基(包括2,1,3-苯并硒杂二唑基)、苯并噻吩基、咔唑基、色满基、噌啉基、呋喃基、咪唑基、咪唑并[1,2-a]吡啶基、吲唑基、二氢吲哚基、吲哚基、异苯并呋喃基、异色满基、异二氢吲哚基、异吲哚基、异喹啉基、异噻唑基、异硫代色满基(isothiochromanyl)、异噁唑基、萘啶基(包括1,6-萘啶基,或者优选地是1,5-萘啶基和1,8-萘啶基)、噁二唑基(包括1,2,3-噁二唑基、1,2,4-噁二唑基和1,3,4-噁二唑基)、噁唑基、吩嗪基、吩噻嗪基、酞嗪基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑基、哒嗪基、吡啶基、嘧啶基、吡咯基、喹唑啉基、喹啉基、喹嗪基、喹喔啉基、四氢异喹啉基(包括1,2,3,4-四氢异喹啉基和5,6,7,8-四氢异喹啉基)、四氢喹啉基(包括1,2,3,4-四氢喹啉基和5,6,7,8-四氢喹啉基)、四唑基、噻二唑基(包括1,2,3-噻二唑基、1,2,4-噻二唑基和1,3,4-噻二唑基)、噻唑基、硫代色满基、硫代乙氧苯基、噻吩基、三唑基(包括1,2,3-三唑基、1,2,4-三唑基和1,3,4-三唑基)等基团。在适当的情况下,杂芳基基团上的取代基位于该环系统中的任何原子(包括杂原子)上。杂芳基基团的附接点可以是经由该环系统中的任何原子(在适当的情况下),包括杂原子(例如氮原子),或在可以作为该环系统的部分存在的任何稠合的碳环上的原子。杂芳基基团还可以处于n-或
s-氧化的形式。当杂芳基基团是其中存在非芳香族环的多环时,该非芳香族环可以被一个或多个=o基团取代。在一个实施例中,本文提及的杂芳基基团可以是单环或二环的。在另外的实施例中,本文提及的杂芳基基团是单环的。
[0113]
可以被提及的杂原子包括磷、硅、硼,并且优选地是氧、氮和硫。
[0114]
为避免疑义,在本文指出一个基团可以被一个或多个取代基(例如,选自c
1-6
烷基)取代的情况下,这些取代基(例如烷基基团)是彼此独立的。即,这样的基团可以被相同的取代基(例如相同的烷基取代基)或不同的(例如烷基)取代基取代。
[0115]
本文中提及的所有个体特征(例如,优选特征)可以独立地或与本文中提及的任何其他特征(包括优选特征)组合地采用(因此,优选特征可以与其他优选特征结合或独立于它们地采用)。
[0116]
技术人员将理解为作为本发明主题的本发明的化合物包括稳定的那些。即,本发明的化合物包括足够稳固以承受从例如反应混合物分离至有用纯度的那些。
[0117]
现在将描述本发明的各种实施例,包括本发明的化合物的实施例。
[0118]
在一个实施例中,提供了如上文所定义的具有式(i)的化合物或其药学上可接受的盐,其中r3不代表氢。
[0119]
在一个实施例中,提供了如上文所定义的具有式(i)的化合物或其药学上可接受的盐,其中r3代表:
[0120]
(i)卤代;
[0121]
(ii)c
1-4
烷基,其任选地被一个或多个独立地选自卤代、-oh和-oc
1-3
烷基的取代基取代;
[0122]
(iii)c
2-4
烯基,其任选地被-oc
1-3
烷基取代;
[0123]
(iv)c
3-6
环烷基;或
[0124]
(v)-oc
1-3
烷基。
[0125]
在一个实施例中,本发明的化合物包括以下那些,其中r1代表:(i)c
3-6
环烷基;(ii)芳基或杂芳基;或(iii)或杂环基,所有这些都如本文所定义任选地被取代。在一个特定实施例中,r1代表:(i)c
3-6
环烷基;或(ii)芳基或杂芳基,所有这些都如本文所定义任选地被取代。
[0126]
在一个实施例中,当r1代表任选地被取代的c
3-6
环烷基时,则其代表任选地被一个或两个选自c
1-3
烷基(例如甲基)和-oh的取代基取代的c
3-6
环烷基(或者在一个实施例中,c
3-4
环烷基)。在另外的实施例中,r1代表环丙基(例如未取代的)或环丁基。在另外的实施例中,r1代表环己基。在又另外的实施例中,r1代表未取代的环丙基或被-oh和甲基(例如在相同碳原子处)取代的环丁基。在又另外的实施例中,r1代表环己基,例如被-oh(例如被一个-oh基团)取代。因此,在一个实施例中,r1代表:
[0127][0128]
其中每个r
1a
代表一个或两个选自-oh和c
1-3
烷基(例如甲基)的任选取代基。在该方面的一个特定实施例中,r1代表c
3-6
环烷基,例如任选地被取代的环己基、任选地被取代的环丁基或未取代的(或任选地被取代的)环丙基,例如:
[0129][0130]
其中每个r
1ab
代表一个或两个任选的选自由r
1a
定义的那些的取代基,并且在一个实施例中,代表一个任选的选自-oh的取代基;
[0131][0132]
其中每个r
1aa
代表一个或两个选自由r
1a
定义的那些的任选取代基,并且在一个实施例中,代表两个取代基甲基和-oh;或者
[0133][0134]
其中r
1a
如以上所定义,但在一个特定实施例中,它不存在。
[0135]
在一个实施例中,在r1代表如本文所定义任选地被取代的芳基或杂芳基时,则其可以代表:(i)苯基;(ii)5元或6元单环杂芳基基团;或(iii)9元或10元二环杂芳基基团,所有这些基团任选地被一到三个如本文定义的取代基取代。在一个实施例中,上述芳基和杂芳基基团任选地被一个或两个(例如一个)选自以下的取代基取代:卤代(例如氟)、-oh、c
1-3
烷基和-oc
1-3
烷基。在一个实施例中,r1代表苯基或单环6元杂芳基基团并且在另一个实施例中它可以代表9元或10元(例如9元)二环杂芳基基团。因此,在一个实施例中,r1可以代表:
[0136][0137]
其中r
1b
代表一个或两个选自卤代、-ch3、-oh和-och3的任选取代基(并且在另外的实施例中,这样的任选取代基选自氟和甲氧基),并且rb、rc、rd、re和rf中的至少一个代表氮杂原子(并且其他代表ch)。在一个实施例中,rb、rc、rd、re和rf中的一个或两个代表氮杂原子,例如,rd代表氮,并且任选地,rb代表氮,或者,rc代表氮。在一个方面:(i)rb和rd代表氮;(ii)rd代表氮;或(iii)rc代表氮。因此,r1可以代表3-吡啶基或4-嘧啶基,这两个基团都如本文所定义任选地被取代;然而,在一个实施例中,这样的基团是未取代的。
[0138]
在另一个实施例中,r1可以代表:
[0139][0140]
其中r
1b
如以上所定义(即,代表一个或两个任选的取代基),但在一个方面,其优选地不存在(因此,在一个实施例中,代表未取代的5元杂芳基基团),并且rk、r
l
、rm和rn中的至少一个代表杂原子,并且在一个实施例中,这些中的至少一个代表n,其他独立地选自ch、n、o和s(前提是,遵守化合价规则);例如,在一个实施例中,rk和rn中的一个代表n,另一个代表
oc
1-2
烷基取代。在另外的实施例中,r2代表任选地被一个或多个独立地选自卤代、-oh和-oc
1-2
烷基的取代基取代的c
1-3
烷基。在又另外的实施例中,r2代表未取代的c
1-3
烷基。
[0156]
在一个特定实施例中,r2代表未取代的异丙基或未取代的乙基。
[0157]
在一个实施例中,r3代表(i)氢;(ii)卤代(例如溴);(iii)c
1-4
烷基,其任选地被一个或多个独立地选自卤代、-oh和-oc
1-2
烷基的取代基取代;(iv)c
3-6
环烷基(例如环丙基);或(v)-oc
1-3
烷基。在一个实施例中,当r3代表任选地被取代的c
1-4
烷基时,则其代表任选地被一个或多个氟原子取代的c
1-3
烷基。在一个实施例中,当r3代表c
3-6
环烷基时,则其代表环丙基。在一个实施例中,当r3代表-oc
1-3
烷基时,则其代表-oc
1-2
烷基(例如-och3)。
[0158]
在一个特定实施例中,r3代表氢、溴、甲基、乙基、异丙基、-cf3、-chf2、环丙基或甲氧基。
[0159]
本发明的化合物的名称是根据由化学文摘服务社(cas)认同的命名法法则、使用先进化学开发公司(advanced chemical development,inc.)软件(acd/产物命名版本(name product version)10.01,build15494,2006年12月1日)或根据由国际理论和应用化学联合会(international union of pure and applied chemistry(iupac))认同的命名法法则、使用先进化学开发公司软件(acd/产物命名版本10.01.0.14105,2006年10月)而产生的。在互变异构形式的情况下,产生该结构的描绘的互变异构形式的名称。其他未描绘的互变异构形式也被包括在本发明的范围内。
[0160]
化合物的制备
[0161]
在本发明的一个方面,提供了制备本发明化合物的方法,其中在此提及如本文定义的具有式(i)的化合物。
[0162]
具有式(i)的化合物可以通过以下来制备:
[0163]
(i)将具有式(ii)的化合物,
[0164][0165]
或其衍生物(例如盐),其中r2和r3如上文所定义,与具有式(iii)的化合物
[0166]
h2n-r1ꢀꢀ
(iii)
[0167]
或其衍生物,其中r1如上文所定义,在酰胺形成反应条件(也称为酰胺化)下进行反应,例如在合适的偶联剂(例如,丙基膦酸酐、1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐(o-(7-氮杂苯并三唑-1-基)-n,n,n’,n
’‑
四甲基脲鎓六氟磷酸盐)、1,1'-羰基二咪唑、n,n
’‑
二环己基碳二亚胺、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(或其盐酸盐)、n,n
’‑
二琥珀酰亚胺碳酸酯、苯并三唑-1-基氧基三(二甲基氨基)鏻六氟磷酸盐、2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐(即o-(1h-苯并三唑-1-基)-n,n,n
′
,n
′‑
四甲基脲鎓六氟磷酸盐)、苯并三唑-1-基氧基三-吡咯烷鏻六氟磷酸盐、溴-三-吡咯烷鏻六氟磷酸盐、2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓四氟碳酸酯、1-环己基碳二亚胺-3-丙基氧基甲基聚苯乙烯、o-苯并三唑-1-基-n,n,n’,n
’‑
四甲基脲鎓四氟硼酸盐)存在下,任选地在合适的碱(例如氢化钠、碳酸氢钠、碳酸钾、吡啶、三乙
胺、二甲基氨基吡啶、二异丙胺、氢氧化钠、叔丁醇钾和/或二异丙基氨基锂(或其变体)和适当的溶剂(例如四氢呋喃、吡啶、甲苯、二氯甲烷、氯仿、乙腈、二甲基甲酰胺、三氟甲基苯、二噁烷或三乙胺)存在下。这样的反应可以在另外的添加剂(例如1-羟基苯并三唑水合物)存在下进行。可替代地,羧酸基团可以在标准条件下被转化为相应的酰氯(例如在socl2或草酰氯存在下),然后例如在与上述那些相似的条件下,该酰氯与具有式(ii)的化合物反应;
[0168]
(ii)将具有式(iv)的化合物,
[0169][0170]
其中r2和r3如上文所定义,与具有式(v)的化合物,
[0171]
lg
a-ch
2-c(o)-n(h)r1ꢀꢀ
(v)
[0172]
其中lga代表合适的离去基团(例如卤代,例如氯)并且r1如本文所定义,在合适的反应条件下,例如在合适的碱(例如cs2co3、k2co3或lihmds等)存在下,或在替代性烷基化反应条件下进行反应;
[0173]
(iii)通过将某个具有式(i)的化合物转化(这样的转化步骤也可以对中间体进行)为另一个,例如:
[0174]-对于其中r2代表-n(r
2a
)r
2b
的具有式(i)的化合物,将其中r2代表卤代的具有式(i)的相应化合物与适当的胺hn(r
2a
)r
2b
(其中r
2a
和r
2b
如本文所定义)在胺化反应中进行反应,在适当的条件下,例如使用标准偶联条件,在催化剂(例如cui)、配体(例如d/l-脯氨酸)和碱(例如k2co3)存在下;可以对其中另一基团代表卤代的化合物进行类似的转化,并且在另一位置处的胺是需要的;
[0175]-对于含有烯烃的具有式(i)的化合物,在还原条件下还原为相应的含有烷烃的具有式(i)的化合物,例如在合适的催化剂(例如钯碳)存在下,在合适的反应惰性溶剂(例如乙醇或甲醇)中用氢气还原;
[0176]-偶联以将卤代或三氟甲磺酸酯基团转化为例如烷基、烯基或环烷基基团,例如在合适的偶联剂存在下,例如其中该偶联剂包含附接到合适的基团(例如-b(oh)2、-b(or
wx
)2、锌酸盐(例如包括-zn(r
wx
)2、-znbrr
wx
)或-sn(r
wx
)3,其中每个r
wx
独立地代表c
1-6
烷基基团,或在-b(or
wx
)2的情况下,各个r
wx
基团可以连接在一起以形成4元至6元环状基团(例如4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基基团)上的适当烷基、烯基或芳基/杂芳基基团,从而形成例如频哪醇硼酸酯基团。该反应可以在以下存在下进行:合适的催化剂系统,例如金属(或盐或其络合物)(例如pd、cui、pd/c、pdcl2、pd(oac)2、pd(ph3p)2cl2、pd(ph3p)4(即四(三苯基膦)钯)、pd2(dba)3和/或nicl2(优选的催化剂包括ruphos pd g3、xphos pd和双(三-叔-丁基膦)钯(0)),以及任选的配体(例如pdcl2(dppf).dcm、t-bu3p、(c6h
11
)3p、ph3p、asph3、p(o-tol)3、1,2-双(二苯基膦基)乙烷、2,2'双(二-叔丁基膦基)-1,1'-联苯基、2,2'-双(二苯基膦基)-1,1'-二-萘基、1,1
’‑
双(二苯基-膦基-二茂铁)、1,3-双(二苯基膦基)丙烷、xantphos或其混合物),与合适的碱(例如na2co3、k3po4、cs2co3、naoh、koh、k2co3、csf、et3n、(i-pr)2net、t-buona或t-buok(或其混合物;优选的碱包括na2co3和k2co3)一起,在合适的溶剂(例如二噁烷、甲苯、乙醇、二甲基甲酰胺、二甲氧基乙烷、乙二醇二甲醚、水、二甲
基亚砜、乙腈、二甲基乙酰胺、n-甲基吡咯烷酮、四氢呋喃或其混合物(优选的溶剂包括二甲基甲酰胺和二甲氧基乙烷)中;
[0177]-在合适的还原条件(例如nabh4或类似物)存在下,将酮还原为醇;
[0178]-通过适当的格氏试剂(例如烷基mgbr)的反应,将-c(o)烷基部分转化为-c(oh)(烷基)(烷基)部分;
[0179]-将烯烃=ch2部分转化为羰基=o部分,例如,在ad-mix-α和甲磺酰胺存在下,例如可以将-ch=ch2部分转化为-c(o)h部分(例如通过与四氧化锇反应),该-c(o)h部分又可以通过与dast反应转化为-chf2基团;
[0180]-将酮转化为醇-oh部分;
[0181]-在适当的反应条件下,将-oh部分烷基化(烷基化成-o-烷基)。
[0182]
具有式(ii)的化合物可以通过相应羧酸酯的水解(例如在标准水解条件下,例如在碱金属氢氧化物(例如氢氧化锂)存在下的碱水解)来制备,该羧酸酯又通过具有式(iv)的化合物,
[0183][0184]
其中r2和r3如上文所定义,与具有式(vi)的化合物,
[0185]
lg-ch
2-c(o)o-r
aa
ꢀꢀ
(vi)
[0186]
的反应来制备,其中r
aa
代表c
1-6
烷基(例如乙基)并且lg代表合适的离去基团,例如卤代(例如氯),例如在反应条件下并使用例如本文所述的那些试剂。
[0187]
因此,一般而言,本发明的化合物可以参考以上程序制备。然而,出于通用性的考虑,下面提供了另外的方案以提供本发明的中间体和最终化合物。在以下方案中(以及在下文所述的实验的具体细节中)提供了进一步的细节。
[0188]
在这方面,方案1概述了一个典型的合成:
[0189]
方案1
[0190][0191]
如本文所述的本发明化合物可以通过方案1(以上)所示的反应顺序来制备,其中将适当的酰氯(m1)(其中r3如本文所定义)与2-氨基-2-甲基-1-丙醇反应,以获得相应的噁
唑基化合物(m2),将其与有机金属(例如有机锂)反应,以提供具有邻位-间位取代基的相应化合物(例如邻位锂化中间体),将其用适当的化合物(例如适当的醛)淬灭,以提供化合物(m3)。再将(m3)氧化,例如用戴斯-马丁试剂氧化,以提供相应的酮(m4)。(m4)的噁唑基部分可以例如在相应的酸(例如h2so4)存在下被水解以得到相应的酯(m5),然而(m4)或(m5)可以在适当的条件下与肼(例如呈水合物的形式)反应,以提供化合物(m6)(在本文中也称为具有式(iv)的化合物)。然后在碱(例如k2co3)、亲核催化剂(例如ki)和冠醚(例如18-冠醚-6)存在下,将该化合物用适当的卤代乙酸烷基酯(其中r是c
1-4
烷基)烷基化,以提供酯(m7),通常将其例如在碱性条件(例如在thf中的lioh水溶液或在meoh中的naoh水溶液)切割,以产生酸中间体(m8)(在本文中也称为具有式(ii)的化合物),随后使用标准偶联条件(例如1-丙烷膦酸酐)和碱(例如三乙胺)用r
1-nh2(其中如果r1具有官能团例如oh、nh2、co2h,则此类基团被任选地保护)酰胺化,随后任选地再进行脱保护步骤,以提供具有式(i)的化合物或其药学上可接受的盐。
[0192]
此外,以下方案2和3中描绘的以下转化也显示了在允许在此类中间体(以及最终化合物)的r2位置引入其他取代基方面的通用性。
[0193]
方案2
[0194][0195]
在方案2(以上)中,提供了获得化合物(m6)的替代方法。从(m9)开始,与苯胺反应以提供(m10),其可以进行格氏(grignard)反应以提供(m11)-在这种情况下,格氏试剂可以代表其中r2代表适当的烷基基团的格氏试剂-然后,该中间体可以与肼(例如呈水合物的形式)反应以提供(m6)。此后,例如,可以按照方案1所概述的程序进行转化。
[0196]
方案3
[0197][0198]
可替代地,方案3提供了获得具有式(iv)的化合物(上文也称为化合物(m6))的其
他途径。例如,按照该方案,具有式(m6a)的化合物可以进行溴化反应,以提供具有式(iv)但其中r2代表溴的化合物(m6b)。此后,可以在下游产品中获得r2基团的其他变型。例如,从(m6b),布赫瓦尔德偶联可以提供其他化合物,例如其中r2代表氨基(例如-n(r
2a
)(r
2b
)基团,或可转化为这种基团的另一个胺基团)的那些具有式(iv)的化合物,例如通过在胺(例如hn(r
2a
)r
2b
)和适当的催化剂(例如pd基催化剂或如本文所述的另一种催化剂)存在下,任选地与合适的碱和配体(例如,如本文关于具有式(i)的化合物的制备所述的一种碱和配体)反应。可替代地,可以将该化合物(m6b)转化为(m6d),例如在适当的锡基试剂存在下。然后,该化合物(m6d)可以通过还原或格氏反应进一步转化为(m6e)或(m6f),这提供替代的r2基团,例如任选地被取代的烷基基团(如所描绘的)。
[0199]
某些中间体化合物可以是可商购的,可以在文献中是已知的,或可以通过与本文所述的方法类似或通过常规的合成程序、根据标准的技术、使用适当的试剂和反应条件从可获得的起始材料中来获得。
[0200]
在本发明的最终化合物或相关的中间体上/中的某些取代基可以在以上所述的方法之后或过程中,通过本领域技术人员所熟知的方法被修饰一次或多次。这样的方法的实例包含取代、还原、氧化、烷化、酰化、水解、酯化、醚化、卤化、硝化或偶联。
[0201]
本发明的化合物可以使用常规技术(例如重结晶,在可能的情况下在标准条件下)从它们的反应混合物中分离。
[0202]
本领域技术人员将理解,在上述和在下文方法中,可能需要通过保护基团对中间体化合物的官能团进行保护。
[0203]
这种保护的需求将取决于远端官能度的性质和制备方法的条件而变化(并且该需求可以由本领域普通技术人员容易地确定)。合适的氨基保护基团包括乙酰基、三氟乙酰基、叔丁氧基羰基(boc)、苯甲氧基羰基(cbz)、9-芴基亚甲基氧基羰基(fmoc)和2,4,4-三甲基戊烷-2-基(其可以在酸(例如在水/醇(例如meoh)中的hcl)存在下,通过反应而脱保护)等。对这种保护的需求易于由本领域技术人员确定。例如,-c(o)o-叔丁酯基团可以用作对于-c(o)oh基团的保护基团,并且因此前者可以例如通过在弱酸(例如tfa或类似物)存在下反应而被转化为后者。
[0204]
官能团的保护和脱保护可以在上述方案中的反应之前或之后进行。
[0205]
保护基团可以根据本领域技术人员所熟知的和如在下文中所述的技术而去除。例如,本文所述的经保护的化合物/中间体可以使用标准的脱保护技术被化学转化为未保护的化合物。
[0206]
所涉及的化学类型将指示保护基团的需求和类型以及用于完成合成的顺序。
[0207]
保护基团的使用被完全描述于“protective groups in organic synthesis[有机合成中的保护基团]”,第3版,t.w.greene和p.g.m.wutz,wiley-interscience[威力跨学科出版社](1999)中。
[0208]
如上文所述的方法制备的本发明的化合物可合成为对映异构体的外消旋混合物形式,这些对映异构体可依照本领域已知的拆分程序彼此分离。那些以外消旋形式获得的本发明的化合物可以通过与合适的手性酸发生反应而转化成相应的非对映异构体盐形式。所述非对映异构盐形式随后例如通过选择性或分步结晶而分离,并且对映异构体通过碱由其释放。分离本发明的化合物的对映异构形式的替代方式涉及使用手性固定相的液相色谱
法。所述纯立体化学异构形式还可以衍生自适当起始材料的相应的纯立体化学异构形式,前提是反应立体定向地发生。优选地,如果一种具体的立体异构体是所希望的,那么所述化合物将通过立体专一的制备方法被合成。这些方法将有利地采用对映异构体纯的起始材料。
[0209]
药理学
[0210]
有证据表明nlrp3诱导的il-1和il-18在与多种不同障碍相关发生或作为其结果的炎性应答中发挥作用(menu等人,clinical and experimental immunology[临床和实验免疫学],2011,166,1-15;strowig等人,nature[自然],2012,481,278-286)。已发现nlrp3突变导致一组称为caps的罕见自身炎性疾病(ozaki等人,j.inflammation research[炎症研究杂志],2015,8,15-27;schroder等人,cell[细胞],2010,140:821-832;menu等人,clinical and experimental immunology[临床和实验免疫学],2011,166,1-15)。caps是一种以反复发热和炎症为特征的遗传性疾病,由形成临床连续体的三种自身炎性障碍构成。这些疾病按严重程度依次为家族性感冒自身炎性综合征(fcas)、穆-韦(muckle-wells)综合征(mws)和慢性婴儿皮肤神经关节综合征(cinca;也称为新生儿多系统炎性疾病,nomid),并且都已被证明是由nlrp3基因中的功能获得性突变引起的,这导致il-1β的分泌增加。nlrp3还与许多自身炎性疾病有关,包括化脓性关节炎、坏疽性脓皮病和痤疮(papa)、斯威特综合征(sweet's syndrome)、慢性非细菌性骨髓炎(cno)和寻常痤疮(cook等人,eur.j.lmmunol.[欧洲免疫学杂志],2010,40,595-653)。
[0211]
许多自身免疫性疾病已被证明涉及nlrp3,尤其包括多发性硬化、1型糖尿病(t1d)、牛皮癣、类风湿性关节炎(ra)、白塞氏病(behcet's disease)、施尼茨勒综合征(schnitzler syndrome)、巨噬细胞活化综合征(braddock等人,nat.rev.drug disc.[自然综述药物发现]2004,3,1-10;inoue等人,immunology[免疫学],2013,139,11-18;coll等人,nat.med.[自然医学]2015,21(3),248-55;scott等人,clin.exp.rheumatol.[临床和实验风湿病学]2016,34(1),88-93)、系统性红斑狼疮及其并发症例如狼疮性肾炎(lu等人,j.lmmunol.[免疫学杂志],2017,198(3),1119-29)、以及系统性硬化(artlett等人,arthritis rheum.[关节炎与风湿病]2011,63(11),3563-74)。nlrp3也已被证明在许多肺部疾病中发挥作用,包括慢性阻塞性肺疾病(copd)、哮喘(包括类固醇抵抗性哮喘)、石棉肺和矽肺病(de nardo等人,am.j.pathol.[美国病理学杂志],2014,184:42-54;kim等人,am.j.respir.crit.care med[美国呼吸与危重症医学杂志],2017,196(3),283-97)。nlrp3还被认为在许多中枢神经系统病症中发挥作用,包括多发性硬化(ms)、帕金森病(pd)、阿尔茨海默病(ad)、痴呆症、亨廷顿病、脑型疟疾、肺炎球菌脑膜炎引起的脑损伤(walsh等人,nature reviews[自然综述],2014,15,84-97;和dempsey等人,brain.behav.lmmun.[脑行为与免疫学]2017,61,306-16)、颅内动脉瘤(zhang等人,j.stroke and cerebrovascular dis.[中风与脑血管病杂志],2015,24,5,972-9)和创伤性脑损伤(ismael等人,j.neurotrauma.[神经创伤杂志],2018,35(11),1294-1303)。nlrp3活性也已被证明涉及各种代谢疾病,包括2型糖尿病(t2d)及其器官特异性并发症、动脉粥样硬化、肥胖、痛风、假痛风、代谢综合征(wen等人,nature immunology[自然免疫学],2012,13,352-357;duewell等人,nature[自然],2010,464,1357-1361;strowig等人,nature[自然],2014,481,278-286)和非酒精性脂肪性肝炎(mridha等人,j.hepatol.[肝病学杂志]2017,66(5),1037-46)。也
56;hu等人,pnas.[美国国家科学院院刊],2010,107(50),21635-40)、多发性骨髓瘤(li等人,hematology[血液学],2016 21(3),144-51)和头颈部鳞状细胞癌(huang等人,j.exp.clin.cancer res.[实验与临床癌症研究杂志],2017,36(1),116)。nlrp3炎症小体的激活也已显示介导肿瘤细胞对5-氟尿嘧啶的化学抗性(feng等人,j.exp.clin.cancer res.[实验与临床癌症研究杂志],2017,36(1),81),并且周围神经中nlrp3炎症小体的激活促成化学疗法诱导的神经性疼痛(jia等人,mol.pain.[分子疼痛],2017,13,1-11)。nlrp3也显示是有效控制病毒、细菌和真菌所必需的。
[0215]
nlrp3的激活导致细胞焦亡,这一特征在临床疾病的表现中起着重要作用(yan-gang等人,cell death and disease[细胞死亡和疾病],2017,8(2),2579;alexander等人,hepatology[肝脏病学],2014,59(3),898-910;baldwin等人,j.med.chem.[药物化学杂志],2016,59(5),1691-1710;ozaki等人,j.inflammation research[炎症研究杂志],2015,8,15-27;zhen等人,neuroimmunology neuroinflammation[神经免疫学神经炎症],2014,1(2),60-65;mattia等人,j.med.chem.[药物化学杂志],2014,57(24),10366-82;satoh等人,cell death and disease[细胞死亡与疾病],2013,4,644)。因此,预计nlrp3抑制剂将阻断细胞焦亡以及促炎细胞因子(例如il-1β)从细胞中释放。
[0216]
因此,如本文所述(例如在本文所述的任何实施例中,包括通过实例和/或以本文所述的任何形式,例如盐形式或游离形式等)的本发明的化合物表现出有价值的药理特性,例如nlrp3对nlrp3炎症小体途径的抑制特性,例如如本文提供的体外测试所示,因此适用于疗法或用作研究化学品,例如作为工具化合物。本发明的化合物可用于治疗选自以下的适应症:炎症小体相关疾病/障碍、免疫性疾病、炎性疾病、自身免疫性疾病或自身炎性疾病,例如nlrp3信号传导促成病理学和/或症状和/或进展的疾病、障碍或病症,和/或根据本文所述的任何方法/用途(例如通过使用或施用本发明的化合物)可能对nlrp3抑制有应答并且可能被治疗或预防的疾病、障碍或病症,因此,在一个实施例中,这样的适应症可以包括:
[0217]
i.炎症,包括由炎性障碍引起的炎症(例如自身炎性疾病),作为非炎性障碍的症状发生的炎症,作为感染结果发生的炎症,或继发于外伤、损伤或自身免疫的炎症。可以治疗或预防的炎症的实例包括与以下相关地发生、或作为以下的结果发生的炎性应答:
[0218]
a.皮肤病症,例如接触性超敏反应、大疱性类天疱疮、晒伤、银屑病、特应性皮炎、接触性皮炎、过敏性接触性皮炎、脂溢性皮炎、扁平苔藓、硬皮病、天疱疮、大疱性表皮松解症、荨麻疹、红斑、或脱发;
[0219]
b.关节病症,例如骨关节炎、全身型幼年特发性关节炎、成人斯蒂尔病(still's disease)、复发性多软骨炎、类风湿性关节炎、幼年慢性关节炎、晶体诱导关节病(例如假痛风、痛风)、或血清阴性脊柱关节病(例如强直性脊柱炎、银屑病关节炎或莱特尔氏病(reiter's disease));
[0220]
c.肌肉病症,例如多肌炎或重症肌无力;
[0221]
d.胃肠道病症,例如炎性肠病(包括克罗恩病和溃疡性结肠炎)、胃溃疡、乳糜泻、直肠炎、胰腺炎、嗜酸性胃肠炎、肥大细胞增多症、抗磷脂综合征、或食物相关过敏,所述食物相关过敏的影响可能远离肠道(例如、偏头痛、鼻炎或湿疹);
[0222]
e.呼吸系统病症,例如慢性阻塞性肺病(copd)、哮喘(包括支气管、过敏性、内源
性、外源性或粉尘性哮喘、尤其是慢性或顽固性哮喘、例如晚期哮喘和气道高反应性)、支气管炎、鼻炎(包括急性鼻炎、过敏性鼻炎、萎缩性鼻炎、慢性鼻炎、干酪性鼻炎、肥厚性鼻炎、脓性鼻炎、干燥性鼻炎、药物性鼻炎、膜性鼻炎、季节性鼻炎(例如花粉热)、和血管运动性鼻炎)、鼻窦炎、特发性肺纤维化(ipf)、结节病、农民肺、矽肺病、石棉肺、成人呼吸窘迫综合征、过敏性肺炎、或特发性间质性肺炎;
[0223]
f.血管病症,例如动脉粥样硬化、白塞氏病、血管炎、或韦格纳肉芽肿病;
[0224]
g.免疫性病症,例如自身免疫性病症,例如系统性红斑狼疮(sle)、干燥综合征、系统性硬化、桥本甲状腺炎、i型糖尿病、特发性血小板减少性紫癜、或格雷夫斯病;
[0225]
h.眼部病症,例如葡萄膜炎、过敏性结膜炎、或春季卡他性结膜炎;
[0226]
i.神经系统病症,例如多发性硬化或脑脊髓炎;
[0227]
j.感染或感染相关病症,例如获得性免疫缺陷综合症(aids)、急性或慢性细菌感染、急性或慢性寄生虫感染、急性或慢性病毒感染、急性或慢性真菌感染、脑膜炎、肝炎(a、b或c、或其他病毒性肝炎)、腹膜炎、肺炎、会厌炎、疟疾、登革出血热、利什曼病、链球菌肌炎、结核分枝杆菌、胞内分枝杆菌、卡氏肺囊虫肺炎、睾丸炎/附睾炎、军团菌、莱姆病、甲型流感、艾普斯登-巴尔病毒(epstein-barr virus)、病毒性脑膜炎/无菌性脑膜炎、或盆腔炎性疾病;
[0228]
k.肾病症,例如系膜增生性肾小球肾炎、肾病综合征、肾炎、肾小球肾炎、急性肾衰竭、尿毒症、或肾炎综合征;
[0229]
l.淋巴系统病症,例如卡斯尔曼病(castleman's disease);
[0230]
m.免疫系统的病症或涉及免疫系统的病症,例如高lge综合征、瘤型麻风、家族性噬血细胞综合征、或移植物抗宿主病;
[0231]
n.肝脏病症,例如慢性活动性肝炎、非酒精性脂肪性肝炎(nash)、酒精性肝炎、非酒精性脂肪肝病(nafld)、酒精性脂肪性肝病(afld)、酒精性脂肪性肝炎(ash)或原发性胆汁性肝硬化;
[0232]
o.癌症,包括下文列出的那些;
[0233]
p.烧伤、伤口、外伤、出血或中风;
[0234]
q.辐射暴露;
[0235]
r.肥胖;和/或
[0236]
s.疼痛,例如炎性痛觉过敏;
[0237]
ii.炎性疾病,包括由炎性障碍引起的炎症,例如自身炎性疾病,例如冷吡啉相关周期性综合征(caps)、穆-韦综合征(muckle-wells syndrome,mws)、家族性感冒自身炎性综合征(fcas)、家族性地中海热(fmf)、新生儿多系统炎性疾病(nomid)、马吉德综合征(majeed syndrome)、化脓性关节炎、坏疽性脓皮病和痤疮综合征(papa)、成人斯蒂尔病(aosd)、a20单倍型不足(ha20)、小儿肉芽肿性关节炎(pga)、plcg2相关抗体缺乏和免疫失调(plaid)、plcg2相关自身炎性、抗体缺乏和免疫失调(aplaid)、或铁粒细胞性贫血伴b细胞免疫缺陷、周期性发热和发育迟缓(sifd);
[0238]
iii.免疫性疾病,例如自身免疫性疾病,例如急性播散性脑炎、艾迪生氏病(addison's disease)、强直性脊椎炎、抗磷脂抗体综合征(aps)、抗合成酶抗体综合征、再生障碍性贫血、自身免疫性肾上腺炎、自身免疫性肝炎、自身免疫性卵巢炎、自身免疫性多
腺体衰竭、自身免疫性甲状腺炎、乳糜泻、克罗恩病、1型糖尿病(t1d)、古德帕斯丘综合征(goodpasture's syndrome)、格雷夫斯病、格林-巴利综合征(guillain-barre syndrome,gbs)、桥本甲状腺炎、原发性血小板减少性紫癜、川崎病、红斑狼疮(包括系统性红斑狼疮(sle))、多发性硬化(ms)(包括原发性进行性多发性硬化(ppms))、继发性进行性多发性硬化(spms)和复发性缓解性多发性硬化(rrms)、重症肌无力、肌阵挛综合征(oms)、视神经炎、奥德氏甲状腺炎(ord's thyroiditis)、天疱疮、恶性贫血、多关节炎、原发性胆汁性肝硬化、类风湿性关节炎(ra)、银屑病关节炎、青少年特发性关节炎或斯蒂尔病、难治性痛风性关节炎、莱特尔氏综合征(reiter's syndrome)、干燥综合征(sjogren's syndrome)、系统性硬化(一种系统性结缔组织疾病)、多发性大动脉炎、颞动脉炎、温热自身免疫性溶血性贫血、韦格纳肉芽肿病、全身脱毛、布里尔氏病(beliefs disease)、恰加斯氏病(chagas'disease)、家族性自主神经异常、子宫内膜异位症、化脓性汗腺炎(hs)、间质性膀胱炎、神经性肌强直、银屑病、结节病、硬皮病、溃疡性结肠炎、施尼茨勒综合征(schnitzler syndrome)、巨噬细胞活化综合征、blau综合征、巨细胞颞动脉炎、白癜风或外阴痛;
[0239]
iv.癌症,包括肺癌、肾细胞癌、非小细胞肺癌(nsclc)、朗格汉斯细胞组织细胞增生症(lch)、骨髓增生性肿瘤(mpn)、胰腺癌、胃癌、骨髓增生异常综合征(mos)、白血病(包括急性淋巴细胞性白血病(all)和急性髓细胞白血病(aml)、早幼粒细胞白血病(apml、或apl))、肾上腺癌、肛门癌、基底和鳞状细胞皮肤癌、胆管癌、膀胱癌、骨癌、脑和脊髓肿瘤、乳腺癌、宫颈癌、慢性淋巴细胞白血病(cll)、慢性粒细胞白血病(cml)、慢性粒单核细胞白血病(cmml)、结直肠癌、子宫内膜癌、食管癌、尤因肉瘤家族(ewing family of tumours)、眼癌、胆囊癌、胃肠道类癌肿瘤、胃肠道间质瘤(gist)、妊娠滋养细胞疾病、神经胶质瘤、霍奇金淋巴瘤、卡波西肉瘤、肾癌、喉癌和下咽癌、肝癌、肺类癌肿瘤、淋巴瘤(包括皮肤t细胞淋巴瘤)、恶性间皮瘤、黑色素瘤皮肤癌、默克尔细胞皮肤癌、多发性骨髓瘤、鼻腔和鼻窦癌、鼻咽癌、神经母细胞瘤、非霍奇金淋巴瘤、非小细胞肺癌、口腔癌和口咽癌、骨肉瘤、卵巢癌、阴茎癌、垂体瘤、前列腺癌、视网膜母细胞瘤、横纹肌肉瘤、唾液腺癌、皮肤癌、小细胞肺癌、小肠癌、软组织肉瘤、胃癌、睾丸癌、胸腺癌、甲状腺癌(包括未分化甲状腺癌)、子宫肉瘤、阴道癌、外阴癌、华氏巨球蛋白血症(waldenstrom macroglobulinemia)、和肾母细胞瘤(wilms tumour);
[0240]
v.感染,包括病毒感染(例如来自流感病毒、人类免疫缺陷病毒(hiv)、甲病毒属(如基孔肯雅热病毒(chikungunya)和罗斯河病毒(ross river virus))、黄病毒属(如登革热病毒和寨卡病毒)、疱疹病毒(如艾普斯登-巴尔病毒、巨细胞病毒、水痘带状疱疹病毒、和kshv)、痘病毒(例如痘苗病毒(改良型痘苗病毒安卡拉(ankara))和粘液瘤病毒)、腺病毒(例如腺病毒5)、乳头瘤病毒、或sars-cov-2),细菌感染(例如来自金黄色葡萄球菌、幽门螺杆菌、炭疽杆菌、百日咳博代氏杆菌(bordatella pertussis)、类鼻疽伯克氏菌、白喉棒状杆菌、破伤风芽孢梭菌、肉毒杆菌、肺炎链球菌、酿脓链球菌、单核细胞增生李斯特菌、流感嗜血杆菌、多杀性巴氏杆菌、痢疾杆菌、结核分枝杆菌、麻风分枝杆菌、肺炎支原体、人支原体、脑膜炎奈瑟菌、淋病奈瑟菌、立氏立克次体、嗜肺军团菌、肺炎克雷伯菌、铜绿假单胞菌、痤疮丙酸杆菌、梅毒螺旋体、沙眼衣原体、霍乱弧菌、减毒沙门氏菌、伤寒沙门氏菌、伯氏疏螺旋体或鼠疫耶尔森菌),真菌感染(例如来自念珠菌属或曲霉菌属物种),原生动物感染(例如来自疟原虫属、巴贝虫属、贾弟虫属、内变形虫属、利什曼原虫属或锥虫),蠕虫感染
(例如来自血吸虫、蛔虫、绦虫或吸虫),和朊病毒感染;
[0241]
vi.中枢神经系统疾病,例如帕金森病、阿尔茨海默病、痴呆、运动神经元病、亨廷顿氏舞蹈病、脑型疟疾、肺炎球菌脑膜炎引起的脑损伤、颅内动脉瘤、创伤性脑损伤、多发性硬化、和肌萎缩侧索硬化;
[0242]
vii.代谢疾病,例如2型糖尿病(t2d)、动脉粥样硬化、肥胖、痛风、和假痛风;
[0243]
viii.心血管疾病,例如高血压、局部贫血、再灌注损伤(包括mi后缺血再灌注损伤)、中风(包括缺血性中风)、短暂性脑缺血发作、心肌梗塞(包括复发性心肌梗塞)、心力衰竭(包括充血性心力衰竭和射血分数保留的心力衰竭)、栓塞、动脉瘤(包括腹主动脉瘤)、心血管风险降低(cvrr)、和心包炎(包括德雷斯勒综合征(dressler's syndrome));
[0244]
ix.呼吸系统疾病,包括慢性阻塞性肺疾病(copd)、哮喘(例如过敏性哮喘和类固醇抵抗性哮喘)、石棉肺、矽肺病、纳米颗粒诱导的炎症、囊性纤维化、和特发性肺纤维化;
[0245]
x.肝脏疾病,包括非酒精性脂肪性肝病(nafld)和非酒精性脂肪性肝炎(nash)(包括晚期纤维化期f3和f4)、酒精性脂肪性肝病(afld)、和酒精性脂肪性肝炎(ash);
[0246]
xi.肾病,包括急性肾病、高草酸尿症、慢性肾病、草酸盐肾病、肾钙质沉着症、肾小球肾炎、和糖尿病肾病;
[0247]
xii.眼部疾病,包括眼上皮疾病、年龄相关黄斑变性(amo)(干性和湿性)、葡萄膜炎、角膜感染、糖尿病性视网膜病变、视神经损伤、干眼症、和青光眼;
[0248]
xiii.皮肤病,包括皮炎(例如接触性皮炎和特应性皮炎)、接触性超敏反应、晒伤、皮肤损伤、化脓性汗腺炎(hs)、其他引起囊肿的皮肤病、和聚合性痤疮;
[0249]
xiv.淋巴系统病症,例如淋巴管炎和卡斯尔曼病;
[0250]
xv.心理障碍,例如抑郁症和心理压力;
[0251]
xvi.移植物抗宿主病;
[0252]
xvii.骨骼病,包括骨质疏松症、骨硬化症;
[0253]
xviii.血液病,包括镰状细胞病;
[0254]
xix.异常性疼痛,包括机械性异常性疼痛;以及
[0255]
xx.被确定携带nlrp3生殖细胞或体细胞非沉默突变的任何疾病。
[0256]
更具体地,本发明的化合物可用于治疗选自以下的适应症:炎症小体相关疾病/障碍、免疫性疾病、炎性疾病、自身免疫性疾病、或自身炎性疾病、(例如自身炎性发热综合征(例如冷吡啉相关周期性综合征)、镰状细胞病、系统性红斑狼疮(sle))、肝脏相关疾病/障碍(例如慢性肝病、病毒性肝炎、非酒精性脂肪性肝炎(nash)、酒精性脂肪性肝炎、和酒精性肝病)、炎性关节炎相关障碍(例如痛风、假痛风(软骨钙质沉着症)、骨关节炎、类风湿性关节炎、关节病(例如急性、慢性))、肾脏相关疾病(例如高草酸尿症、狼疮性肾炎、i/ii型糖尿病和相关并发症(例如肾病、视网膜病变)、高血压肾病、血液透析相关炎症)、神经炎症相关疾病(例如多发性硬化、脑感染、急性损伤、神经退行性疾病、阿尔茨海默病)、心血管/代谢疾病/障碍(例如心血管风险降低(cvrr)、高血压、动脉粥样硬化、i型和ii型糖尿病及相关并发症、外周动脉疾病(pad)、急性心力衰竭)、炎性皮肤病(例如化脓性汗腺炎、痤疮)、伤口愈合和疤痕形成、哮喘、结节病、年龄相关黄斑变性、和癌症相关疾病/障碍(例如结肠癌、肺癌、骨髓增生性肿瘤、白血病、骨髓增生异常综合征(mos)、骨髓纤维化)。特别地,自身炎性发热综合征(例如caps)、镰状细胞病、i型/ii型糖尿病和相关并发症(例如肾病、视网膜病
变)、高草酸尿症、痛风、假痛风(软骨钙质沉着症)、慢性肝病、nash、神经炎症相关障碍(例如多发性硬化、脑感染、急性损伤、神经退行性疾病、阿尔茨海默病)、动脉粥样硬化和心血管风险(例如心血管风险降低(cvrr)、高血压)、化脓性汗腺炎、伤口愈合和疤痕形成、以及癌症(例如结肠癌、肺癌、骨髓增生性肿瘤、白血病、骨髓增生异常综合征(mos)、骨髓纤维化)。
[0257]
特别地,本发明的化合物可用于治疗选自以下的疾病或障碍:自身炎性发热综合征(例如caps)、镰状细胞病、i型/ii型糖尿病和相关并发症(例如肾病、视网膜病变)、高草酸尿症、痛风、假痛风(软骨钙质沉着症)、慢性肝病、nash、神经炎症相关疾病(例如多发性硬化、脑感染、急性损伤、神经退行性疾病、阿尔茨海默病)、动脉粥样硬化和心血管风险(例如心血管风险降低(cvrr)、高血压)、化脓性汗腺炎、伤口愈合和疤痕形成、以及癌症(例如结肠癌、肺癌、骨髓增生性肿瘤、白血病、骨髓增生异常综合征(mos)、骨髓纤维化)。因此,作为另外的方面,本发明提供了本发明的化合物(因此,包括通过本文的任何实施例/形式/实例所定义的化合物)在疗法中的用途。在另外的实施例中,疗法选自可通过抑制nlrp3炎症小体来治疗的疾病。在另一个实施例中,疾病如本文任何列表中所定义。因此,提供了本文(包括任何实施例/形式/实例)所述的本发明的化合物的任一种,用于治疗本文所述的任何疾病或障碍(例如,如上述列表中所述)。
[0258]
药物组合物和组合
[0259]
在一个实施例中,本发明还涉及一种组合物,该组合物包含药学上可接受的载体以及作为活性成分的、治疗有效量的本发明化合物。本发明的化合物可以被配制为用于施用目的的不同药物形式。可引用通常用于全身性施用药物的所有组合物作为适当的组合物。为制备本发明的药物组合物,将有效量的任选地呈盐形式的具体化合物作为活性成分与药学上可接受的载体组合于紧密混合物中,该载体可采用众多种形式,这取决于施用所希望的制剂形式。所希望的是,这些药物组合物处于单位剂型,特别地是适用于口服施用或通过肠胃外注射施用的单位剂型。例如,在制备呈口服剂型的组合物时,可采用任何常见药物介质,在口服液体制剂(例如悬浮液、糖浆剂、酏剂、乳液以及溶液)的情况下,例如采用水、二醇、油、醇等;或者在粉末、丸剂、胶囊剂和片剂的情况中,固体载体例如淀粉、糖、高岭土、稀释剂、润滑剂、粘合剂、崩解剂等。因为其容易施用,片剂和胶囊剂代表了最有利的口服单位剂型,在该情况下显然使用固体药物载体。对于肠胃外组合物而言,载体通常将至少在很大程度上包含无菌水,但也可以包括其他成分例如以辅助溶解性。例如可制备可注射溶液,其中载体包含盐溶液、葡萄糖溶液或盐水和葡萄糖溶液的混合物。也可以制备可注射悬浮液,在这种情况下可以采用适当的液体载体、助悬剂等。还包括旨在使用前不久转化为液体形式制剂的固体形式制剂。
[0260]
在一个实施例中,并且取决于施用模式,该药物组合物将优选地包含按重量计从0.05%至99%、更优选地按重量计从0.1%至70%、甚至更优选地按重量计从0.1%至50%的这种或这些活性成分,以及按重量计从1%至99.95%、更优选地按重量计从30%至99.9%、甚至更优选地按重量计从50%至99.9%的药学上可接受的载体,所有的百分数都基于组合物的总重量。
[0261]
药物组合物另外可以包含本领域已知的不同其他成分,例如,润滑剂、稳定剂、缓冲剂、乳化剂、粘度调节剂、表面活性剂、防腐剂、调味剂或着色剂。
[0262]
为了易于施用和剂量的均一性,将上述药物组合物配制成单位剂型是特别有利的。如本文所使用的单位剂型是指适合作为单位剂量的物理离散单位,每个单位含有预定量的活性成分,该预定量的活性成分经计算会与所希望的药物载体相结合而产生所需治疗效果。这样的单位剂型的实例是片剂(包括刻痕或包衣的片剂)、胶囊剂、丸剂、粉末包(powder packet)、糯米纸囊剂(wafer)、栓剂、可注射溶液或悬浮液等剂型,及其分离多倍剂(segregated multiple)。当然,根据本发明的化合物的每日剂量将随着所采用的化合物、施用模式、所希望的治疗以及所针对的分枝杆菌疾病而变化。然而,一般而言,当根据本发明的化合物以不超过1克(例如,在从10mg/kg至50mg/kg体重的范围内)的每日剂量施用时,将获得令人满意的结果。
[0263]
在一个实施例中,提供了一种组合,其包含治疗有效量的根据本文所述的任一实施例的本发明化合物和另一治疗剂(包括一种或多种治疗剂)。在另外的实施例中,提供了这样的组合,其中另一治疗剂选自(并且当存在多于一种治疗剂时,各自独立地选自):法尼醇x受体(fxr)激动剂;抗脂变剂(anti-steatotics);抗纤维化剂;jak抑制剂;检查点抑制剂,包括抗pd1抑制剂、抗lag-3抑制剂、抗tim-3抑制剂或抗pol 1抑制剂;化学疗法、放射疗法和外科手术;降尿酸疗法;合成代谢和软骨再生疗法;il-17的阻断剂;补体抑制剂;布鲁顿酪氨酸激酶抑制剂(btk抑制剂);toll样受体抑制剂(tlr7/8抑制剂);car-t疗法;抗高血压剂;胆固醇降低剂;白三烯a4水解酶(ltah4)抑制剂;sglt2抑制剂;132-激动剂;抗炎剂;非甾体类抗炎药(“nsaid”);乙酰水杨酸药(asa),包括阿司匹林;扑热息痛;再生疗法治疗;囊性纤维化治疗;或动脉粥样硬化治疗。在另外的实施例中,还提供了用于如本文所述的关于本发明化合物使用的一个或多个组合,例如用于治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍,或与nlrp3活性(包括nlrp3炎症小体活性)相关(包括抑制nlrp3炎症小体活性)的疾病或障碍,并且在这方面,本文提及的特定疾病/障碍在此同样适用。还可以提供如本文所述的关于本发明化合物的方法,但其中该方法包括施用治疗有效量的这样的组合(并且,在一个实施例中,这样的方法可以是在抑制nlrp3炎症小体活性的背景下治疗本文提及的疾病或障碍)。本文提及的组合可以在单一制剂中或者它们可以被配置为分开的制剂,这样使得它们可以同时地、分开地或顺序地施用。因此,在一个实施例中,本发明还涉及一种组合产品,其包含(a)根据本文所述的任一实施例的本发明的化合物,和(b)一种或多种其他治疗剂(其中这样的治疗剂如本文所述),作为组合制剂在治疗与抑制nlrp3炎症小体活性相关的疾病或障碍中同时、单独或顺序使用(并且其中该疾病或障碍可以是本文所述的那些中的任一种),例如,在一个实施例中,该组合可以是成套试剂盒。这样的组合可称为“药物组合”。作为组合的组分的本发明化合物的施用途径可以与跟其组合的一种或多种其他治疗剂相同或不同。其他治疗剂是例如化学化合物、肽、抗体、抗体片段或核酸,当将该治疗剂与本发明的化合物组合施用至患者时,该治疗剂具有治疗活性或增强治疗活性。
[0264]
当作为组合给出时,本领域技术人员可以确定(a)根据本发明的化合物以及(b)一种或多种其他治疗剂的重量比。如本领域技术人员所熟知的,所述比率以及精确的剂量以及施用的频率取决于根据本发明的具体化合物以及所使用的一种或多种其他抗细菌剂,正治疗的具体病症,正治疗的病症的严重程度,具体患者的年龄、体重、性别、饮食、施用时间以及总体身体状况,施用模式连同个体可以服用的其他药物。此外,显而易见的是,有效日
用量可以降低或提高,这取决于所治疗的受试者的应答和/或取决于给出本发明的化合物处方的医生的评估。本发明的化合物与另一种抗细菌剂的具体重量比可以在从1/10到10/1、更特别从1/5到5/1、甚至更特别从1/3到3/1的范围内。
[0265]
对于约50-70kg的受试者,本发明的药物组合物或组合可以处于约1-1000mg的一种或多种活性成分的单位剂量,或约1-500mg、或约1-250mg、或约1-150mg、或约1-100mg、或约1-50mg的活性成分的单位剂量。化合物、药物组合物或其组合的治疗有效剂量取决于受试者的种类、体重、年龄和个体状况、所治疗的障碍或疾病或其严重程度。具有普通技能的内科医师、临床医师或兽医可以容易地确定预防、治疗或抑制障碍或疾病进展所需的每种活性成分的有效量。
[0266]
以上引用的剂量特性可通过有利地使用哺乳动物例如小鼠、大鼠、狗、猴或其分离的器官、组织和制品的体外和体内试验证明。本发明的化合物可以以溶液的形式(例如,水溶液)体外施用,以及肠内、肠胃外、有利地静脉内,例如,作为悬浮液或在水溶液中体内施用。体外剂量可以在约10-3
摩尔和10-9
摩尔的浓度之间的范围内。体内治疗有效量的范围取决于施用途径,在约0.1-500mg/kg之间,或在约1-100mg/kg之间。
[0267]
如本文所用,术语“药物组合物”是指本发明的化合物或其药学上可接受的盐与至少一种药学上可接受的载体一起,呈适合口服或肠胃外施用的形式。
[0268]
如本文所用,术语“药学上可接受的载体”是指在制备或使用药物组合物中有用的物质并且包括例如合适的稀释剂、溶剂、分散介质、表面活性剂、抗氧化剂、防腐剂、等渗剂、缓冲剂、乳化剂、吸收延迟剂、盐、药物稳定剂、粘合剂、赋形剂、崩解剂、润滑剂、润湿剂、甜味剂、调味剂、染料及其组合,如本领域技术人员已知的(参见例如,remington the science and practice of pharmacy[雷明顿:药学科学与实践],第22版pharmaceutical press[药学出版社],2013,第1049-1070页)。
[0269]
如本文所用,术语“受试者”是指动物,优选地是哺乳动物,最优选地是人类,例如该受试者是或已经成为治疗、观察或实验的对象。
[0270]
如本文所用,术语“治疗有效量”是指本发明的化合物(包括,在适用的情况下,包含本发明的这样的化合物的形式、组合物、组合)引起受试者的生物学或医学应答(例如,酶或蛋白活性的减小或抑制,或改善症状、缓解病症、减慢或延迟疾病进展或预防疾病等)的量。在一个非限制性实施例中,术语“治疗有效量”是指当施用至受试者时在下列方面起效的本发明的化合物的量:(1)至少部分缓解、抑制、防止和/或改善(i)由nlrp3介导的,(ii)与nlrp3活性相关的,或(iii)以nlrp3的活性(正常或异常)为特征的病症或障碍或疾病;或(2)降低或抑制nlrp3的活性;或(3)降低或抑制nlrp3的表达。在另一个非限制性实施例中,术语“治疗有效量”是指当被施用至细胞、或组织、或非细胞生物材料、或介质时,有效地至少部分降低或抑制nlrp3的活性;或至少部分地降低或抑制nlrp3的表达。
[0271]
如本文所用,术语“抑制(inhibit、inhibition或inhibiting)”是指减少或抑制给定的病症、症状、或障碍、或疾病,或生物活性或过程的基础活性的显著降低。具体地,抑制nlrp3或抑制nlrp3炎症小体途径包括降低nlrp3或nlrp3炎症小体途径诱导il-1和/或il-18产生的能力。这可以通过包括但不限于灭活、去稳定和/或改变nlrp3分布的机制来实现。
[0272]
如本文所用,术语“nlrp3”意指包括但不限于核酸、多核苷酸、寡核苷酸、有义和反义多核苷酸链、互补序列、肽、多肽、蛋白质、同源和/或直系同源nlrp分子、同种型、前体、突
变体、变体、衍生物、剪接变体、等位基因、不同物种、及其活性片段。
[0273]
如本文所用,术语任何疾病或障碍的“治疗(treat、treating或treatment)”是指缓解或改善疾病或障碍(即,减慢或阻止疾病或其至少一种临床症状的发展);或缓解或改善至少一种与疾病或障碍相关的物理参数或生物标志物,包括患者可能无法辨别的那些。
[0274]
如本文所用,术语任何疾病或障碍的“预防(prevent、preventing或prevention)”是指疾病或障碍的预防性治疗;或延迟疾病或障碍的发作或进展。
[0275]
如本文所用,如果受试者将在生物学、医学或生活质量方面受益于治疗,则这样的受试者“需要”这样的治疗。
[0276]“组合”是指一种剂量单位形式的固定组合,或组合施用(其中本发明的化合物与组合配伍体(partner)(例如下文所解释的另一种药物,也称为“治疗剂”或“共同药剂(co-agent)”)可以同时地独立地施用或在时间间隔内分开施用。单个组分可以包装在试剂盒中或分开包装。可在施用之前将一种或两种组分(例如粉末或液体)重构或稀释至所希望的剂量。如本文所用,术语“共同施用”或“组合施用”等意在涵盖将所选择的组合配伍体施用至对其有需要的单个受试者(例如患者),并且旨在包括其中药剂不一定通过相同的施用途径施用或同时施用的治疗方案。
[0277]
如本文所用,术语“药物组合”意指由多于一种治疗剂的混合或组合所产生的产品,并且包括治疗剂的固定和非固定组合两者。如本文所用,术语“药物组合”是指在一个剂量单位形式中的固定组合、或用于组合施用的非固定组合或成套试剂盒,其中两种或多种治疗剂可以同时地独立地施用或在时间间隔内分开施用。术语“固定组合”意指治疗剂(例如本发明的化合物和组合配伍体)以单一实体或剂量的形式同时地施用至患者。术语“非固定组合”意指治疗剂(例如本发明的化合物和组合配伍体)作为分开的实体同时地、并行地或依序地(没有特定的时间限制)施用至患者,其中这样的施用在患者体内提供治疗有效水平的两种化合物。后者也适用于鸡尾酒疗法,例如三种或更多种治疗剂的施用。
[0278]
术语“组合疗法”是指施用两种或更多种治疗剂以治疗本公开中所述的治疗病症或障碍。这样的施用涵盖以基本上同时的方式共同施用这些治疗剂,如以具有固定比率的活性成分的单个胶囊施用。可替代地,这样的施用也涵盖在每种活性成分的多个容器中或分开的容器(例如片剂、胶囊剂、粉末和液体)中共同施用。可以将粉末和/或液体在施用之前重构或稀释到所期望的剂量。另外,这样施用也涵盖在大致相同的时间或在不同的时间顺序使用每种类型的治疗剂。在任何一种情况下,治疗方案将在治疗本文所述的病症或障碍方面提供药物组合的有益作用。
[0279]
药理学、用途、组合物和组合的总结
[0280]
在一个实施例中,提供了一种药物组合物,其包含治疗有效量的根据本文所述的任一实施例的本发明化合物和药学上可接受的载体(包括一种或多种药学上可接受的载体)。
[0281]
在一个实施例中,提供了根据本文所述的任一实施例的本发明化合物,用作药剂。
[0282]
在一个实施例中,提供了根据本文所述的任一实施例的本发明的化合物(和/或包含根据本文所述的任一实施例的本发明的这样的化合物的药物组合物),用于在以下中使用:用于治疗与nlrp3活性(包括炎症小体活性)相关的疾病或障碍;用于治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍;用于抑制nlrp3炎
症小体活性(包括在有需要的受试者中);和/或用作nlrp3抑制剂。
[0283]
在一个实施例中,提供了根据本文所述的任一实施例的本发明的化合物(和/或包含根据本文所述的任一实施例的本发明的这样的化合物的药物组合物)在以下中的用途:用于治疗与nlrp3活性(包括炎症小体活性)相关的疾病或障碍;用于治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍;用于抑制nlrp3炎症小体活性(包括在有需要的受试者中);和/或用作nlrp3抑制剂。
[0284]
在一个实施例中,提供了根据本文所述的任一实施例的本发明的化合物(和/或包含根据本文所述的任一实施例的本发明的这样的化合物的药物组合物)在制造药剂中的用途,该药剂用于:治疗与nlrp3活性(包括炎症小体活性)相关的疾病或障碍;治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍;和/或抑制nlrp3炎症小体活性(包括在有需要的受试者中)。
[0285]
在一个实施例中,提供了治疗疾病或障碍的方法(其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展),该方法包括例如向受试者(有需要)施用治疗有效量的根据本文所述的任一实施例的本发明的化合物(和/或包含根据本文所述的任一实施例的本发明的这样的化合物的药物组合物)。在另外的实施例中,提供了一种抑制受试者(有需要)的nlrp3炎症小体活性的方法,该方法包括向有需要的受试者施用治疗有效量的根据本文所述的任一实施例的本发明的化合物(和/或包含根据本文所述的任一实施例的本发明的这样的化合物的药物组合物)。
[0286]
在本发明的所有相关实施例中,在提及疾病或障碍(例如上文)(例如其中nlrp3信号传导促成疾病/障碍的病理学、和/或症状、和/或进展的疾病或障碍,或与nlrp3活性(包括nlrp3炎症小体活性)相关(包括抑制nlrp3炎症小体活性)的疾病或障碍)的情况下,这样的疾病可包括炎症小体相关疾病或障碍、免疫性疾病、炎性疾病、自身免疫性疾病或自身炎性疾病。在另外的实施例中,这样的疾病或障碍可包括自身炎性发热综合征(例如冷吡啉相关周期性综合征)、肝脏相关疾病/障碍(例如慢性肝病、病毒性肝炎、非酒精性脂肪性肝炎(nash)、酒精性脂肪性肝炎、和酒精性脂肪性肝病)、炎性关节炎相关障碍(例如痛风、假痛风(软骨钙质沉着病)、骨关节炎、类风湿性关节炎、关节病例如急性、慢性)、肾脏相关疾病(例如高草酸尿症、狼疮性肾炎、i型/ii型糖尿病及相关并发症(例如肾病、视网膜病变)、高血压肾病、血液透析相关炎症)、神经炎症相关疾病(例如多发性硬化、脑感染、急性损伤、神经退行性疾病、阿尔茨海默病)、心血管/代谢疾病/障碍(例如心血管风险降低(cvrr)、高血压、动脉粥样硬化、i型和ii型糖尿病及相关并发症、外周动脉疾病(pad)、急性心力衰竭)、炎性皮肤病(例如化脓性汗腺炎、痤疮)、伤口愈合和疤痕形成、哮喘、结节病、年龄相关黄斑变性、和癌症相关疾病/障碍(例如结肠癌、肺癌、骨髓增生性肿瘤、白血病、骨髓增生异常综合征(mos)、骨髓纤维化)。在一个特定方面,这样的疾病或障碍选自自身炎性发热综合征(例如caps)、镰状细胞病、i型/ii型糖尿病和相关并发症(例如肾病、视网膜病变)、高草酸尿症、痛风、假痛风(软骨钙质沉着病)、慢性肝病、nash、神经炎症相关障碍(例如多发性硬化、脑感染、急性损伤、神经退行性疾病、阿尔茨海默病)、动脉粥样硬化和心血管风险(例如心血管风险降低(cvrr)、高血压)、化脓性汗腺炎、伤口愈合和疤痕形成、和癌症(例如结肠癌、肺癌、骨髓增生性肿瘤、白血病、骨髓增生异常综合征(mos)、骨髓纤维化)。在一个特定实施例中,与抑制nlrp3炎症小体活性相关的疾病或障碍选自炎症小体相关疾病和障碍、免
疫性疾病、炎性疾病、自身免疫性疾病、自身炎性发热综合征、冷吡啉相关周期性综合征、慢性肝病、病毒性肝炎、非酒精性脂肪性肝炎、酒精性脂肪性肝炎、酒精性肝病、炎性关节炎相关障碍、痛风、软骨钙质沉着病、骨关节炎、类风湿性关节炎、慢性关节病、急性关节病、肾脏相关疾病、高草酸尿症、狼疮性肾炎、i型和ii型糖尿病、肾病、视网膜病变、高血压肾病、血液透析相关炎症、神经炎症相关疾病、多发性硬化、脑感染、急性损伤、神经退行性疾病、阿尔茨海默病、心血管疾病、代谢疾病、心血管风险降低、高血压、动脉粥样硬化、外周动脉疾病、急性心力衰竭、炎性皮肤病、痤疮、伤口愈合和疤痕形成、哮喘、结节病、年龄相关黄斑变性、结肠癌、肺癌、骨髓增生性肿瘤、白血病、骨髓增生异常综合征和骨髓纤维化。
[0287]
在一个实施例中,提供了一种组合,其包含治疗有效量的根据本文所述的任一实施例的本发明化合物和另一治疗剂(包括一种或多种治疗剂)。在另外的实施例中,提供了这样的组合,其中另一治疗剂选自(并且当存在多于一种治疗剂时,各自独立地选自):法尼醇x受体(fxr)激动剂;抗脂变剂;抗纤维化剂;jak抑制剂;检查点抑制剂,包括抗pd1抑制剂、抗lag-3抑制剂、抗tim-3抑制剂或抗pol 1抑制剂;化学疗法、放射疗法和外科手术;降尿酸疗法;合成代谢和软骨再生疗法;il-17的阻断剂;补体抑制剂;布鲁顿酪氨酸激酶抑制剂(btk抑制剂);toll样受体抑制剂(tlr7/8抑制剂);car-t疗法;抗高血压剂;胆固醇降低剂;白三烯a4水解酶(ltah4)抑制剂;sglt2抑制剂;132-激动剂;抗炎剂;非甾体类抗炎药(“nsaid”);乙酰水杨酸药(asa),包括阿司匹林;扑热息痛;再生疗法治疗;囊性纤维化治疗;或动脉粥样硬化治疗。在另外的实施例中,还提供了用于如本文所述的关于本发明化合物使用的一个或多个组合,例如用于治疗其中nlrp3信号传导促成所述疾病/障碍的病理学和/或症状和/或进展的疾病或障碍,或与nlrp3活性(包括nlrp3炎症小体活性)相关(包括抑制nlrp3炎症小体活性)的疾病或障碍,并且在这方面,本文提及的特定疾病/障碍在此同样适用。还可以提供如本文所述的关于本发明化合物的方法,但其中该方法包括施用治疗有效量的这样的组合(并且,在一个实施例中,这样的方法可以是在抑制nlrp3炎症小体活性的背景下治疗本文提及的疾病或障碍)。本文提及的组合可以在单一制剂中或者它们可以被配置为分开的制剂,这样使得它们可以同时地、分开地或顺序地施用。因此,在一个实施例中,本发明还涉及一种组合产品,其包含(a)根据本文所述的任一实施例的本发明的化合物,和(b)一种或多种其他治疗剂(其中这样的治疗剂如本文所述),作为组合制剂在治疗与抑制nlrp3炎症小体活性相关的疾病或障碍中同时、单独或顺序使用(并且其中该疾病或障碍可以是本文所述的那些中的任一种)。
[0288]
本发明的化合物(包括包含本发明的化合物的形式和组合物/组合),无论是用于上述适应症或其他适应症可以具有比现有技术已知化合物更有效、毒性更低、作用时间更长、更强效、产生更低副作用、更容易吸收,和/或具有更佳的药代动力学特性(例如更高的口服生物利用度和/或更低的清除率)的优点,和/或具有优于现有技术已知化合物的其他有用的药理、生理或化学性质。
[0289]
例如,本发明的化合物可具有这样的优点,即它们具有良好或改良的热动力学溶解度(例如相较于现有技术中已知的化合物;以及例如,如通过已知的方法和/或本文所述的方法所测定的)。本发明的化合物可具有这样的优点,即它们将阻断细胞焦亡以及促炎细胞因子(例如il-1β)从细胞中释放。本发明的化合物还可具有避免副作用的优点,例如与现有技术的化合物相比,这可能是由于nlrp3抑制的选择性。本发明的化合物也可具有这样的
优点,即它们具有良好或改善的体内药物动力学和口服生物利用度。它们还可具有这样的优点,即它们具有良好或改良的体内疗效。具体而言,当在下文概述的测试中(例如在实例c和d中)进行比较时,本发明的化合物也可以具有优于现有技术化合物的优点。
[0290]
通用制备和分析方法
[0291]
根据本发明的化合物通常可以通过一系列步骤进行制备,其中的每个步骤可以是技术人员已知的或在本文中所述的。
[0292]
显然在前述和以下反应中,反应产物可以从反应介质中分离出,并且,如果必要的话,根据本领域中通常已知的方法学(例如,萃取、结晶和色谱法)进行进一步纯化。更加显然,以多于一种对映异构体的形式存在的反应产物可以通过已知的技术(特别地是制备型色谱法,例如制备型hplc、手性色谱法)从它们的混合物中分离。还可以通过超临界流体色谱(sfc)获得单独的非对映异构体或单独的对映异构体。
[0293]
起始材料和中间体是可商购的化合物或可以根据本领域中通常已知的常规反应程序制备的化合物。
[0294]
分析部分
[0295]
lc-ms(液相色谱/质谱法)
[0296]
一般程序
[0297]
使用lc泵、二极管阵列(dad)或uv检测器以及如在对应的方法中所指定的柱进行高效液相色谱法(hplc)测量。如果必要的话,包括另外的检测器(参见以下方法表格)。
[0298]
将来自柱的流带至配置有大气压离子源的质谱仪(ms)。以下在技术人员的知识范围内:设置调谐参数(例如扫描范围、停留时间等)以获得允许进行化合物的标称单一同位素分子量(mw)的鉴定的离子。利用适当的软件进行数据采集。通过其实验保留时间(r
t
)和离子描述化合物。如果未在数据表中不同地指定,那么报告的分子离子对应于[m h]
(质子化的分子)和/或[m-h]-(去质子化的分子)。在该化合物不是直接可电离的情况下,指定加合物类型(即[m nh4]
、[m hcoo]-等)。对于具有多种同位素模式的分子(br、cl等)来说,报告的值是针对最低同位素质量获得的值。获得的所有结果具有通常与所使用的方法相关联的实验不确定性。
[0299]
在下文中,“sqd”意指单四极检测器,“msd”意指质量选择检测器,“rt”意指室温,“beh”意指桥接的乙基硅氧烷/二氧化硅杂合物,“dad”意指二极管阵列检测器,“hss”意指高强度二氧化硅。
[0300]
表:lcms方法代码(以ml/min表示流速;以℃表示柱温(t);以分钟表示运行时间)。
[0301]
[0302][0303]
nmr
[0304]
对于许多化合物,1h nmr光谱如下记录:在以300或400mhz运行的bruker avance iii光谱仪上,在以400mhz运行的bruker avance iii-hd上,在以400mhz运行的bruker avance neo光谱仪上,在以500mhz运行的bruker avance neo光谱仪上,或在以600mhz运行
的bruker avance 600光谱仪上,使用氯仿-d(氘代氯仿,cdcl3)、dmso-d6(氘代dmso,二甲基-d6亚砜)、甲醇-d4(氘代甲醇)、苯-d6(氘代苯,c6d6)或丙酮-d6(氘代丙酮、(cd3)2co)作为溶剂。化学位移(δ)被报告为相对于四甲基硅烷(tms)(用作内部标准)的百万分率(ppm)。
[0305]
熔点
[0306]
值是峰值或熔融范围,并且所获得的值具有通常与这种分析方法相关的实验不确定性。
[0307]
方法a:对于多种化合物,在mettler toledo mp50上的开口毛细管中确定熔点。使用10℃/分钟的温度梯度来测量熔点。最高温度是300℃。从数字显示器读取熔点数据并且从视频记录系统进行检查。
[0308]
方法b:对于多种化合物,用dsc823e(梅特勒
·
托利多公司(mettler toledo))装置来测定熔点。使用10℃/分钟的温度梯度来测量熔点。标准最高温度为300℃。
[0309]
实验部分
[0310]
术语“m.p.”表示熔点,“aq.”表示水性,“r.m.”表示反应混合物,“rt”表示室温,“dipea”表示n,n-二异-丙基乙胺,“dipe”表示二异丙基醚,“thf”表示四氢呋喃,“dmf”表示二甲基甲酰胺,“dcm”表示二氯甲烷,“etoh”表示乙醇,“etoac”表示乙酸乙酯,“acoh”表示乙酸,“iproh”表示异丙醇,“iprnh
2”表示异丙胺,“mecn”或“acn”表示乙腈,“meoh”表示甲醇,“pd(oac)
2”表示二乙酸钯(ii),“rac”表示外消旋,“sat.”表示饱和,“sfc”表示超临界流体色谱,“sfc-ms”表示超临界流体色谱/质谱,“lc-ms”表示液相色谱/质谱,“gcms”表示气相色谱/质谱,“hplc”表示高效液相色谱,“rp”表示反相,“uplc”表示超高效液相色谱,“r
t”(或“rt”)表示保留时间(以分钟为单位),“[m h]
”表示化合物的游离碱的质子化质量,“dast”表示二乙氨基三氟化硫,“dmtmm”表示4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓氯化物,“hatu”表示o-(7-氮杂苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓六氟磷酸盐(1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐),“xantphos”表示(9,9-二甲基-9h-呫吨-4,5-二基)双[二苯基膦],“tbat”表示四丁基三苯基二氟硅酸铵,“tfa”表示三氟乙酸,“et2o”表示二乙醚,“dmso”表示二甲基亚砜,“sio
2”表示二氧化硅,“xphos pd g3”表示(2-二环己基膦-2',4',6'-三异丙基-1,1'-联苯)[2-(2
′‑
氨基-1,1'-联苯)]钯(ii)乙磺酸盐,“cdcl
3”表示氘代氯仿,“mw”表示微波或分子量,“min”表示分钟,“h”表示小时,“rt”表示室温,“quant”表示定量,“nt”表示未测试,“cpd”表示化合物,“pocl
3”表示氧氯化磷(v)。
[0311]
对于关键中间体、连同一些最终化合物,手性中心的绝对构型(表示为r和/或s)经由与已知构型的样品进行比较或使用适用于测定绝对构型的分析技术如vcd(振动圆二色法)或x-射线结晶学来确定。当手性中心的绝对构型是未知的时,任意地指定为r*。
[0312]
实例-实例a
[0313]
中间体的制备
[0314]
6-氯-1-甲基-1h-吡唑并[3,4-b]吡啶i-133的合成
[0315][0316]
向6-氯-1h-吡唑并[3,4-b]吡啶[63725-51-9](1g,6.51mmol)在乙腈(10ml)中的
溶液中添加cs2co3[534-17-8](4.24g,13mmol),并将混合物在室温搅拌30min。然后将反应混合物冷却至0℃并逐滴添加碘甲烷[74-88-4](1.63ml,2.28g/ml,26.15mmol)。添加后,将反应混合物在室温搅拌1小时,然后在mw中在150℃加热10分钟并在室温搅拌两天。将反应用冰水淬灭并用etoac萃取。将合并的有机萃取物用盐水洗涤,经mgso4干燥,过滤并在真空中浓缩。将粗残余物通过fcc(庚烷/etoac 0至30%)纯化,以得到呈黄色固体的i-133(500mg,46%)。
[0317]
n-(1-甲基-1h-吡唑并[3,4-b]吡啶-6-基)乙酰胺i-132的合成
[0318][0319]
将6-氯-1-甲基-1h-吡唑并[3,4-b]吡啶i-133(500mg,2.98mmol)在1,4-二噁烷(5ml)中的溶液用氮气喷雾15min。添加乙酰胺[60-35-5](212mg,3.59mmol)、pd(oac)2[3375-31-3](67mg,0.3mmol)、xanthphos[161265-03-8](86.5mg,0.15mmol)和碳酸铯[534-17-8](1.95g,5.97mmol)。将反应混合物在100℃加热5小时。将溶剂在真空下浓缩,将粗混合物悬浮于dcm中并过滤。将滤液在真空下浓缩并将获得的固体通过fcc(庚烷/etoac 0至20%)纯化,以获得呈固体的i-132(458mg,81%)。
[0320]
1-甲基-1h-吡唑并[3,4-b]吡啶-6-胺i-131的合成
[0321][0322]
将37%hcl水溶液[7647-01-0](23ml,1.18g/ml,274.8mmol)添加到n-(1-甲基-1h-吡唑并[3,4-b]吡啶-6-基)乙酰胺i-132(458mg,2.41mmol)在水(25ml)中的悬浮液中。将所得溶液在回流下加热3小时。将挥发物在真空下蒸发,以得到呈黄色固体的粗品i-131(444mg,100%),将其不经进一步纯化而使用。
[0323]
5-氯-3-甲基-3h-咪唑并[4,5-b]吡啶i-135和5-氯-1-甲基-1h-咪唑并[4,5-b]吡啶i-134的合成
[0324][0325]
在0℃,将nah[7646-69-7](在矿物油中的60%分散体,1.5g,37.5mmol)分批添加到5-氯-3h-咪唑并[4,5-b]吡啶[52090-89-8](5g,32.56mmol)在无水dmf(70ml)中的搅拌混合物中。使所得混合物升温至室温并搅拌30min,之后逐滴添加mei[74-88-4](2.3ml,2.28g/ml,36.945mmol)。在室温搅拌2小时后,将混合物小心地用水淬灭。添加etoac和更多的水。将有机层分离,用盐水洗涤(x5),干燥(mgso4),过滤并在减压下蒸发。将粗混合物通过fcc(dcm/meoh 1%至3%)纯化,以提供呈浅橙色固体的i-135(3.7g,68%)和呈白色固体的i-134(880mg,16%)。
[0326]
1-乙基-5-硝基吲哚啉i-148的合成
[0327][0328]
将5-硝基吲哚啉[32692-19-6](3g,18.27mmol)溶解于无水dmf(60ml)中,同时在氮气气氛下搅拌。在室温,经10min的时间段,分批添加nah[7646-69-7](在矿物油中的60%分散体,1.46g,36.55mmol)。将反应混合物在室温搅拌20min。经10min的时间段,逐滴添加碘乙烷[75-03-6](4.66g,29.88mmol)在dmf(10ml)中的溶液,然后将混合物在75℃加热并在该温度搅拌过夜。使其冷却至室温,通过添加水淬灭并用etoac萃取。将有机萃取物合并并用盐水洗涤,经mgso4干燥并蒸发。将残余物通过fcc(庚烷/dcm 0至80%)纯化,产生呈橙色固体的i-148(2.24g,64%)。
[0329]
根据以上程序合成结构类似物。
[0330][0331][0332]
1-乙基吲哚啉-5-胺i-145的合成
[0333][0334]
在室温,将1-乙基-5-硝基吲哚啉i-148(1g,5.2mmol)和pd/c(10%wt.pd,0.1g,0.094mmol)在meoh(25ml)中的混合物置于氢气气氛下并搅拌直至观察到吸收了3当量的氢气。将催化剂过滤并将滤液在减压下浓缩。将获得的残余物通过fcc(dcm/meoh 0至5%)纯化,产生呈紫色油状物的i-145(440mg,52%)。
[0335][0336]
1-(二氟甲基)-6-硝基-1h-吲唑i-157的合成
[0337][0338]
将nah[7646-69-7](在矿物油中的60%分散体,0.1g,2.54mmol)添加到6-硝基-2h-吲唑[65750-02-9](500mg,3.06mmol)和氯二氟乙酸钠[1895-39-2](0.78g,5.09mmol)在n-甲基吡咯烷酮(8.5ml)中的溶液中。此后,将反应混合物在室温搅拌15min,并进一步在100℃搅拌30min。将其用乙酸乙酯稀释,并依次用水和盐水洗涤。将合并的有机萃取物经mgso4干燥,过滤并将溶剂在真空下蒸发。将残余物通过fcc(庚烷/etoac 0至20%)纯化,以获得呈固体的i-157(130mg,20%)。
[0339]
1-(二氟甲基)-1h-吲唑-6-胺i-156的合成
[0340][0341]
在100-ml氢化烧瓶中,将pd/c(10%wt.pd,68.09mg,0.064mmol)添加到i-157(120mg,0.56mmol)在乙酸乙酯(5ml)中的溶液中。将反应吹扫三次(氢气/真空)并置于氢气气氛下。将反应在室温搅拌过夜。将其经硅藻土(decalite)过滤,用etoac充分洗涤并将溶剂在减压下浓缩,以提供呈浅粉色固体的i-156(103mg,100%)。
[0342]
1-(二氟甲基)-1h-吲唑-5-胺i-158的合成
[0343][0344]
在mw小瓶中,将铁粉[7439-89-6](513.4mg,9.19mmol)添加到2-甲基-5-硝基-2h-吲唑[5228-48-8](228.5mg,1.29mmol)、氯化铵[12125-02-9](209.5mg,3.92mmol)在etoh(11ml)和di水(11ml)混合物中的混合物中。将小瓶密封,将反应混合物剧烈搅拌并在100℃加热1小时。将粗混合物经硅藻土过滤并将滤饼用etoac(约30ml)洗涤。将滤液再次经微孔
过滤器过滤并将滤饼用etoac(约20ml)洗涤。将过滤物用水(约10ml)洗涤,经mgso4干燥并在减压下在50℃浓缩,以提供呈棕色油状物的i-158(193mg,97%),将其不经进一步纯化而用于下一步骤。
[0345]
3-溴-2-肼基-5-硝基吡啶i-161的合成
[0346][0347]
将3-溴-2-氯-5-硝基吡啶[5470-17-7](1.5g,6.32mmol)溶解于1,4-二噁烷(81ml)中,将溶液冷却至0℃并在0℃快速(《15秒)添加水合肼[7803-57-8](9.2ml,1.03g/ml,0.19mol)。添加后,将混合物在0℃剧烈搅拌1.5小时,然后使其升温至室温并再搅拌一小时。将混合物在旋转蒸发器上浓缩成约20ml的深红色混合物。然后使其冷却至0℃并添加di水(150ml)。将沉淀出的固体经烧结漏斗滤出,同时用约5 5ml的di水洗涤烧瓶和固体。在烘箱中在50℃在真空下干燥16小时后,分离出呈浅灰色固体的i-161(1.21g,82%,约96%-97%纯度)。
[0348]
8-溴-6-硝基-[1,2,4]三唑并[4,3-a]吡啶i-160的合成
[0349][0350]
在easymax压力管中,将3-溴-2-肼基-5-硝基吡啶i-161(4g,17.2mmol)悬浮于原甲酸三甲酯[149-73-5](28.2ml,0.97g/ml,0.26mol)中。将管用螺旋盖密封并将混合物在100℃加热2.5小时。使反应冷却至室温,然后冷却至0℃持续约30min,并将悬浮物滤出,同时用庚烷/etoac的1:1混合物(10ml)洗涤反应小瓶和所过滤的固体,以产生呈浅棕色固体的i-160(3.66g,》98%纯度,88%)。
[0351]
8-溴-[1,2,4]三唑并[1,5-a]吡啶-6-胺i-159的合成
[0352][0353]
将8-溴-6-硝基-[1,2,4]三唑并[4,3-a]吡啶吡啶i-160(500mg,2.06mmol)、nh4cl[12125-02-9](880.4mg,16.5mmol)和铁粉[7439-89-6](804.3mg,14.4mmol)放入装有磁力搅拌棒的螺旋盖小瓶中并悬浮于etoh(8ml)中。将悬浮液剧烈搅拌并在85℃加热64小时。将悬浮液经烧结漏斗过滤并将滤液在真空中浓缩,重吸收于mecn(约20ml)中并再次浓缩,以得到棕褐色固体(423mg,含有大量的盐/铁)。将147mg的该物质在饱和nahco3水溶液(50ml)和dcm/meoh 95:5(25ml)之间分配。收集有机层并将水层重新用dcm/meoh 95:5(15 10ml)萃取。将合并的有机层在减压下浓缩,以得到呈微绿色固体的i-159(78mg,18%)。
[0354]
8-溴-[1,2,4]三唑并[4,3-a]吡啶-6-胺i-162的合成
[0355]
[0356]
将8-溴-6-硝基-[1,2,4]三唑并[4,3-a]吡啶i-160(1g,4.11mmol,1当量)和铁粉[7439-89-6](1.38g,24.7mmol)放入螺旋盖试管中并添加acoh[64-19-7](18.8ml)。将混合物在室温剧烈搅拌3小时。将绿色浓稠悬浮液用di水(30-40ml)稀释。将由此获得的深色混合物在真空中浓缩至约10ml的体积。将残余物通过缓慢添加80ml的饱和nahco3和k2co3水溶液的1:1混合物(添加约10-15ml后,冒泡停止了,然后形成了固体,在添加更多的碱性溶液后其重新溶解)而中和。然后将混合物用dcm/meoh 95:5(5x150ml)萃取。将合并的有机萃取物经na2so4干燥,过滤并将滤液在真空中浓缩,以提供呈浅棕褐色固体的i-162(450mg,51%)。
[0357]
叔丁基(5-羟基-1-甲基哌啶-3-基)氨基甲酸酯i-164的合成
[0358][0359]
将苄基3-((叔丁氧基羰基)氨基)-5-羟基哌啶-1-甲酸酯[1785642-46-7](1g,2.85mmol)和pd/c(10%wt.pd,3.04g,2.85mmol)在meoh(20ml)中的混合物在室温在氢气的大气压下氢化,直至观察到完全转化。将悬浮液不经纯化而用于下一步骤。向该悬浮液中添加pd/c(10%wt.pd,3.04g,2.85mmol)和多聚甲醛[30525-89-4](0.5g,16.67mmol),并将混合物置于氢气下直至观察到完全转化。将催化剂过滤并将滤液在真空中浓缩。将获得的残余物通过fcc(dcm/meoh:nh3(7n)0至7%)纯化,产生呈白色固体的i-164(537mg,82%)。
[0360]
叔丁基(r)-(1-乙基哌啶-3-基)氨基甲酸酯i-1001的合成
[0361][0362]
将三乙酰氧基硼氢化钠[56553-60-7](19.84g,93.62mmol)分批添加到(r)-3-(boc-氨基)哌啶[309956-78-3](12.5g,62.41mmol)和乙醛(5m,在thf中)[75-07-0](14.98ml,5m,74.89mmol)的混合物中。将混合物在室温搅拌4h。添加水和nahco3并将混合物用dcm萃取。将合并的有机萃取物经mgso4干燥,过滤并在真空中浓缩。将粗品通过快速柱色谱法(二氧化硅;meoh-nh3在dcm中,0至4%)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的叔丁基(r)-(1-乙基哌啶-3-基)氨基甲酸酯i-1001。
[0363]
3-氨基-5-羟基-1-甲基哌啶盐酸盐i-163的合成
[0364][0365]
将在1,4-二噁烷中的4m hcl溶液(6.89ml,27.6mmol)添加到(5-羟基-1-甲基哌啶-3-基)氨基甲酸酯i-164(520mg,2.26mmol)在1,4-二噁烷(6.9ml)中的溶液中。将所得混合物在室温搅拌3小时。将挥发物浓缩,以产生呈白色固体的i-163(461mg,定量),将其不经进一步纯化而使用。
[0366]
根据以上程序合成结构类似物。
[0367][0368]
叔丁基(1-乙酰基-5-羟基哌啶-3-基)氨基甲酸酯i-166的合成
[0369][0370]
将ac2o[108-24-7](229μl,2.43mmol)逐滴添加到叔丁基n-(5-羟基哌啶-3-基)氨基甲酸酯[1502766-14-4](0.5g,2.31mmol)和三乙胺[121-44-8](417μl,3.01mmol)在无水dcm(10ml)中的溶液中,并将混合物在室温搅拌过夜。将反应混合物用饱和nahco3溶液洗涤,经mgso4干燥并蒸发,以产生呈白色泡沫的i-165(525mg,88%)。
[0371]
叔丁基(r)-(1-环丙基哌啶-3-基)氨基甲酸酯i-201的合成
[0372][0373]
在压力管中在氮气下,将氰基硼氢化钠[25895-60-7](667mg,10.61mmol)添加到
(r)-3-boc-氨基哌啶[309956-78-3](1g,4.99mmol)、(1-乙氧基环丙氧基)三甲基硅烷[27374-25-0](1ml,4.99mmol)和乙酸(3ml,52.4mmol)在meoh(40ml)中的悬浮液中。将反应混合物在70℃搅拌过夜。将粗混合物通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、meoh)纯化,以提供呈白色固体的i-201(600mg,50%)。
[0374]
3-氯-1-(二氟甲基)-5-硝基吡啶-2(1h)-酮i-168的合成
[0375][0376]
在氮气惰性气氛下在室温,将3-氯-2-羟基-5-硝基吡啶[22353-38-4](2g,11.46mmol)在dmso(20ml)中的溶液置于easymax压力管中。在室温,将nah[7646-69-7](在矿物油中的60%分散体,0.5g,12.5mmol)添加到该混合物中,使其反应15min。将氯二氟乙酸钠[1895-39-2](2g,13.12mmol)添加到混合物中并将所得溶液在60℃搅拌过夜。使反应混合物冷却至室温并通过添加di水淬灭。将所得溶液用etoac萃取三次。将合并的有机萃取物经mgso4干燥,过滤并在真空下浓缩。将残余物通过fcc(庚烷/etoac 0至40%)纯化,以获得呈白色固体的i-168(390mg,15%)。
[0377]
5-氨基-3-氯-1-(二氟甲基)吡啶-2(1h)-酮i-167的合成
[0378][0379]
在密封mw小瓶中,将3-氯-1-(二氟甲基)-5-硝基吡啶-2(1h)-酮i-168(100mg,0.45mmol)、铁粉[7439-89-6](73.8mg,1.32mmol)和饱和氯化铵水溶液[12125-02-9](0.42ml,约7.2m,约3.01mmol)在etoh(1.7ml)中的混合物在氮气下在80℃加热过夜。使反应混合物冷却至室温并经硅藻土(dicalite)过滤,同时用etoh充分洗涤。将滤液在减压下蒸发,悬浮于dcm中并再次过滤。将滤液浓缩并通过fcc(dcm/meoh 0至5%)纯化,以获得呈黑色膜状物的i-167(37mg,43%)。
[0380]
6-氯咪唑并[1,2-b]哒嗪-2-甲醛i-172的合成
[0381][0382]
将甲基6-氯咪唑并[1,2-b]哒嗪-2-甲酸酯[572910-59-9](2g,9.45mmol)放入装有磁力搅拌棒的100-ml rb烧瓶中。将烧瓶置于氮气(3个真空/氮气循环)下。添加无水dcm(26ml),将混合物剧烈搅拌并冷却至-78℃。然后,经10分钟,逐滴添加在环己烷中的1m dibal溶液[1191-15-7](15.6ml,15.6mmol)。将所得混合物在-78℃搅拌1小时。然后将其通过添加饱和nh4cl水溶液(35ml)淬灭,添加di水(15ml)并将混合物用dcm(5x60ml)萃取。将合并的有机萃取物经na2so4干燥,过滤并在真空中浓缩。将粗固体通过fcc(庚烷/etoac 4:1至1:4)纯化,以得到呈无色固体的i-172(795mg,46%)。
[0383]
6-氯-2-(二氟甲基)咪唑并[1,2-b]哒嗪i-171的合成
[0384][0385]
将6-氯咪唑并[1,2-b]哒嗪-2-甲醛i-172(795mg,4.38mmol)悬浮于无水dcm(15ml)中,将悬浮液冷却至0℃并在0℃逐滴添加dast[38078-09-0](1.74ml,13.1mmol)。将所得混合物在0℃搅拌5min,然后升温至室温并搅拌2小时。逐滴添加另一份dast[38078-09-0](0.87ml,6.57mmol)并将所得悬浮液在室温搅拌16小时。将粗混合物冷却至0℃并通过缓慢添加饱和nahco3水溶液(50ml)淬灭。将两相混合物在室温搅拌直至冒泡停止,并将产物用dcm(3x20ml)萃取。将合并的有机萃取物经na2so4干燥,过滤并在真空中浓缩。将获得的残余物通过fcc(庚烷/etoac 9:1至1:1)纯化,以提供呈无色结晶固体的i-171(790mg,89%)。
[0386]
n-(2-(二氟甲基)咪唑并[1,2-b]哒嗪-6-基)-1,1-二苯基甲亚胺i-170的合成
[0387][0388]
将6-氯-2-(二氟甲基)咪唑并[1,2-b]哒嗪i-171(250mg,1.23mmol)、pd2dba3[51364-51-3](112.5mg,0.12mmol)、binap[98327-87-8](152.9mg,0.25mmol)和nao
t
bu[865-48-5](188.8mg,1.96mmol)放入20-ml mw小瓶中。将小瓶密封并置于氮气(3个真空/氮气循环)下。然后,添加二苯甲酮亚胺[1013-88-3](309μl,1.08g/ml,1.84mmol)在脱气1,4-二噁烷[123-91-1](9ml)中的溶液。然后将混合物剧烈搅拌并在80℃加热3小时。将混合物在真空中浓缩并将残余物通过fcc(庚烷/etoac 3:1至3:7)纯化,以提供呈橙色固体的i-170(300mg,约95%纯度,67%),将其不经进一步纯化而使用。
[0389]
根据以上程序合成结构类似物。
[0390][0391][0392]
2-(二氟甲基)咪唑并[1,2-b]哒嗪-6-胺盐酸盐i-169的合成
[0393][0394]
将n-(2-(二氟甲基)咪唑并[1,2-b]哒嗪-6-基)-1,1-二苯基甲亚胺i-170(300mg,约95%纯度,67%)溶解于thf(5ml)中并添加1m hcl水溶液[7647-01-0](7.5ml,7.5mmol)。将混合物在室温剧烈搅拌2小时。将混合物在真空中在50℃浓缩,重新吸收于乙腈(3x15ml)中并浓缩(3x),以提供黄色固体,将它用mecn(1.5ml)研磨,以得到呈浅黄色固体的i-169(135mg,约85%-90%纯度,约45%),将其不经进一步纯化而使用。
[0395]
根据以上程序合成结构类似物。
[0396][0397]
(ez)-n'-((5-溴吡啶-2-基)亚甲基)-4-甲基苯磺酰肼i-176的合成
[0398][0399]
将4-甲基苯磺酰肼[1576-35-8](1.0g,5.38mmol)添加到5-溴吡啶-2-甲醛[31181-90-5](1.0g,5.376mmol)在dcm(10ml)和meoh(10ml)中的溶液中。将反应在室温搅拌1小时。将挥发物在减压下除去并将获得的固体i-176(1.90g,定量)按原样用于下一步骤。
[0400]
6-溴-[1,2,3]三唑并[1,5-a]吡啶i-175的合成
[0401][0402]
将粗(ez)-n'-((5-溴吡啶-2-基)亚甲基)-4-甲基苯磺酰肼i-176(1.90g,5.36mmol)和吗啉[110-91-8](10ml,115.9mmol)的混合物在90℃搅拌1小时。将反应冷却至室温,然后冷却至0℃并用dipe处理直至形成沉淀物。弃掉固体并将滤液在减压下蒸发。将
粗产物通过fcc(庚烷/etoac 0至60%)纯化,以获得呈白色固体的i-175(970mg,91%)。
[0403]
6-氨基咪唑并[1,2-a]吡啶-2-甲酰胺i-180的合成
[0404][0405]
在压力管中在90℃,将乙基6-氨基咪唑并[1,2-a]吡啶-2-甲酸酯[158980-21-3](1g,4.87mmol)在氨水[7664-41-7](28%,在h2o中,20ml)中的混合物搅拌并加热3小时。将挥发物在真空下蒸发并将粗产物i-180(0.86g,定量)不经任何纯化而用于下一步骤。
[0406]
n-(2-氰基咪唑并[1,2-a]吡啶-6-基)-2,2,2-三氟乙酰胺i-179的合成
[0407][0408]
在氮气下在0℃,将tfaa[407-25-0](0.28ml,1.51g/ml,1.99mmol)添加到6-氨基咪唑并[1,2-a]吡啶-2-甲酰胺i-180(100mg,0.57mmol)和三乙胺[121-44-8](0.39ml,0.73g/ml,2.84mmol)在无水thf(3ml)中的溶液中。将反应在0℃再搅拌一小时,然后在室温搅拌2小时。将反应混合物通过添加水淬灭并用dcm萃取。将合并的有机萃取物在mgso4上干燥,过滤并在真空中蒸发。将获得的固体i-179(140mg,定量)不经进一步纯化而用于下一步骤。
[0409]
6-氨基咪唑并[1,2-a]吡啶-2-甲腈i-178的合成
[0410][0411]
将n-(2-氰基咪唑并[1,2-a]吡啶-6-基)-2,2,2-三氟乙酰胺i-179(150mg,0.59mmol)和k2co3[584-08-7](163.1mg,1.18mmol)在di水(3.11ml)和meoh(3.11ml)中的溶液在室温搅拌过夜。将反应混合物用水(20ml)稀释并将其用2-methf萃取,用盐水洗涤,在mgso4上干燥,过滤并在真空下浓缩,以产生呈棕色/绿色固体的i-178(93mg,定量)。
[0412]
乙基6-碘咪唑并[1,5-a]吡啶-1-甲酸酯i-186的合成
[0413][0414]
在氮气下,将无水dmf(50ml)添加到装有nah[7646-69-7](在矿物油中的60%分散体,2,24g,56.06mmol)的小瓶中。将混合物冷却至0℃并逐滴添加异氰基乙酸乙酯[2999-46-4](6.13ml,56.06mmol)。在0℃保持30min后,分三个批次添加2-氟-5-碘-吡啶[171197-80-1](10.0g,44.85mmol)。使反应升温至室温然后在60℃加热16小时。将反应冷却至室温并用etoac(500ml)和水(300ml)稀释。将有机层分离并用盐水(2x100ml)洗涤。将合并的水层用etoac(200ml)萃取。将合并的有机层经mgso4干燥,过滤并在真空中浓缩。将粗产物通过fcc(庚烷/etoac 0至70%)纯化,以获得呈灰白色固体的i-186(2.94g,21%)。
[0415]
6-碘咪唑并[1,5-a]吡啶-1-甲酸i-185的合成
[0416][0417]
将1m naoh水溶液[1310-73-2](36ml,36mmol)添加到乙基6-碘咪唑并[1,5-a]吡啶-1-甲酸酯i-186(3.75g,11.86mmol)在thf(35ml)中的溶液中。将反应混合物在60℃搅拌2小时。将挥发物在减压下浓缩,并将水性残存物用1m hcl水溶液处理直至ph呈微酸性。将沉淀出的固体滤出,用水洗涤,然后在50℃在真空下干燥,以产生呈灰白色固体的i-185(3.25g,95%)。
[0418]
6-碘咪唑并[1,5-a]吡啶-1-甲酰胺i-184的合成
[0419][0420]
将socl2[7719-09-7](4.1ml,56.5mmol)逐滴添加到6-碘咪唑并[1,5-a]吡啶-1-甲酸i-185(3.25g,11.28mmol)在无水乙腈(30ml)中的悬浮液中。将反应在60℃搅拌1小时。将挥发物在减压下除去。将粗产物溶解于无水dcm(50ml)中,将溶液冷却至0℃并分批添加nh3的水溶液[7664-41-7](28%,在水中,50ml,740mmol),使混合物升温至室温并在该温度搅拌1小时。将反应混合物过滤,将固体滤饼用水洗涤并干燥,以获得呈淡棕色固体的i-184(2.29g,71%)。
[0421]
6-碘咪唑并[1,5-a]吡啶-1-甲腈i-183的合成
[0422][0423]
在0℃,在搅拌下,将pocl3[10025-87-3](0.82ml,8.78mmol)逐滴添加到6-碘咪唑并[1,5-a]吡啶-1-甲酰胺i-184(2.29g,7.98mmol)在无水dmf(23ml)中的溶液中。使反应升温至室温并搅拌30min。将反应用冰水(约50ml)淬灭并用etoac(400ml)和水(150ml)稀释。将有机层分离并将水层用etoac(100ml)萃取。将合并的有机萃取物经mgso4干燥,过滤并在减压下浓缩,以产生呈棕色固体的所希望物质i-183(2.15g,97%)。
[0424]
6-氯-2-(2-甲氧基乙基)哒嗪-3(2h)-酮i-189的合成
[0425][0426]
将6-氯哒嗪-3-醇[19064-67-6](1g,7.66mmol)、k2co3[584-08-7](1.27g,9.18mmol)和四丁基溴化铵[1643-19-2](0.049g,0.15mmol)放入50-ml压力管中,并添加乙腈(12.5ml)和2-溴乙基甲基醚[6482-24-2](1.08ml,1.48g/ml,11.49mmol)。将反应介质在115℃搅拌5小时,然后在室温搅拌16小时。然后将其倒入di水(50ml)中并用dcm萃取(2x)。将合并的有机萃取物经mgso4干燥,过滤并在真空下浓缩。将获得的粗物质通过fcc(庚烷/etoac 0至60%)纯化,以获得呈白色固体的i-189(1.13g,78%)。
[0427]
叔丁基(1-(2-甲氧基乙基)-6-氧代-1,6-二氢哒嗪-3-基)氨基甲酸酯i-188的合成
[0428][0429]
平行地运行两个相同的反应,其中在氮气下,将双(二亚苄基丙酮)钯[32005-36-0](86.12mg,0.15mmol)添加到6-氯-2-(2-甲氧基乙基)哒嗪-3(2h)-酮i-189(565mg,3mmol)、氨基甲酸叔丁酯[4248-19-5](421mg,3.59mmol)、4,5-双(二苯基膦基)-9,9-二甲基呫吨[161265-03-8](173.3mg,0.3mmol)和碳酸铯[534-17-8](2.44g,7.49mmol)在脱气1,4-二噁烷(20ml)中的搅拌悬浮液中。将混合物在密封管中在95℃搅拌两天。
[0430]
将两种反应混合物合并并在真空下浓缩。将粗物质在dcm和水之间分配,将有机层分离并将水层用dcm反萃取。将合并的有机层经mgso4干燥,过滤并在真空下蒸发。将获得的粗固体悬浮于dcm(20ml)中,过滤并将滤液在真空下浓缩,将获得的残余物通过fcc(庚烷/etoac 100:0至0:100)纯化,以获得呈黄色固体的i-188(842mg,52%)。
[0431]
根据以上程序合成结构类似物。
[0432][0433]
6-氨基-2-(2-甲氧基乙基)哒嗪-3(2h)-酮盐酸盐i-187的合成
[0434][0435]
将在1,4-二噁烷中的4m hcl溶液[7647-01-0](14.25ml,57mmol)添加到叔丁基(1-(2-甲氧基乙基)-6-氧代-1,6-二氢哒嗪-3-基)氨基甲酸酯i-188(842mg,3mmol)在1,4-二噁烷(14.5ml)中的溶液中,并将混合物在室温搅拌一天。将混合物在真空中浓缩,以产生呈黑色油状物的i-187(617mg,95%),将其不经进一步纯化而使用。
[0436]
根据以上程序合成结构类似物。
[0437][0438]
6-氨基-2-(2-甲氧基乙基)哒嗪-3(2h)-酮盐酸盐i-192的合成
[0439][0440]
将4-溴-6-氯-2-甲基哒嗪-3(2h)-酮[1178884-53-1](1.024g,4.58mmol)、cs2co3[534-17-8](2.481g,7.62mmol)和1,1'-双(二苯基膦基)二茂铁-二氯钯(ii)二氯甲烷复合物[95464-05-4](88mg,0.108mmol)放入vlt管中并置于氮气(3个真空/氮气循环)下。然后添加三甲基硼氧六环[823-96-1](0.5ml,3.58mmol)、1,4-二噁烷(17ml)和di水(1ml)。将反应混合物在110℃搅拌2.5小时。将反应混合物冷却至室温,用di水(约50ml)稀释并将粗物质用dcm(3x20ml)萃取。将合并的有机层经mgso4干燥,过滤并在减压下浓缩。通过fcc(庚烷/etoac 0至50%)进行纯化,提供了呈白色粉末的i-192(532mg,73%)。
[0441]
6-氯-2-(2-(二甲基氨基)乙基)哒嗪-3(2h)-酮i-195的合成
[0442][0443]
在氮气下,将6-氯哒嗪-3-醇[19064-67-6](500mg,3.83mmol)添加到dmea[108-01-0](0.46ml,0.89g/ml,4.6mmol)在无水甲苯(10ml)中的搅拌溶液中。添加tsunoda试剂[157141-27-0](1.41ml,0.92g/ml,5.36mmol)并将所得棕色溶液在100℃加热4小时。使反应混合物冷却至室温并将溶剂蒸发。将残余物通过fcc(dcm/nh3(10%,在meoh中)0至5%)纯化,以提供呈深棕色油状物的i-195(660mg,80%)。
[0444]
9-甲基-9h-嘌呤-2-胺i-196的合成
[0445][0446]
将6-氯-9-甲基-9h-嘌呤-2-胺[3035-73-2](500mg,2.72mmol)、pd/c(10%wt.pd,
289.8mg,0.27mmol)在meoh(30ml)和thf(50ml)中的混合物在室温氢化过夜。将催化剂过滤并将滤液浓缩,以获得呈红色固体的i-196(780mg,85%),将其不经进一步纯化而使用。
[0447]
9-甲基-9h-嘌呤-2-胺i-199的合成
[0448][0449]
将氨水[7664-41-7](28%,在水中,10.53ml,0.9g/ml,155.76mmol)添加到3,6-二氯-[1,2,4]三唑并[4,3-b]哒嗪[33050-38-3](2g,10.58mmol)在1,4-二噁烷(10.5ml)中的混合物中,将其在压力管中在90℃搅拌并加热4小时。使反应混合物冷却至室温,将固体过滤,用水和庚烷洗涤并干燥,以产生呈棕色固体的i-199(1.6g,产率89%)。
[0450]
3-氧代环丁烷-1-甲酸酯i-1003的合成
[0451][0452]
在室温,向3-氧代环丁烷-1-甲酸[23761-23-1](10g,87.64mmol)在dcm(400ml)中的混合物中,添加et3n[121-44-8](18.3ml,0.728g/ml,131.46mmol)和dmap[1122-58-3](1.07g,8.764mmol)。然后,将混合物在0℃冷却并逐滴添加氯甲酸苄酯[501-53-1](13.76ml,1.195g/ml,96.4mmol)。将混合物在室温搅拌24h。添加水并将混合物用dcm萃取,分离有机层。将合并的有机层干燥(na2so4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(meoh在dcm中0/100至3/97)纯化。收集所希望的级分,将溶剂在真空中蒸发,以产生3-氧代环丁烷-1-甲酸酯i-1003(9g,产率50%)。
[0453]
苄基3-乙基-3-羟基环丁烷-1-甲酸酯i-1004的合成
[0454][0455]
在-60℃,向苄基3-氧代环丁烷-1-甲酸酯i-1003(1500mg,7.345mmol)在thf(20ml)中的混合物中添加乙基溴化镁[925-90-6](3ml,3m,9mmol)。将混合物在-50℃搅拌2小时。添加饱和nh4cl溶液并将粗品用acoet(2x10ml)萃取,将合并的有机层干燥并在真空中蒸发,以提供p1。将粗品通过快速色谱法(二氧化硅,acoet在庚烷中0/100至20/80)纯化,将相应的层在真空中蒸发,以产生呈油状物的苄基3-乙基-3-羟基环丁烷-1-甲酸酯i-1004(750mg,产率44%)。
[0456]
1h nmr(400mhz,氯仿-d)d 7.31-7.40(m,1h),5.14(s,1h),2.73(quin,j=8.38hz,1h),2.20-2.45(m,5h),1.62(q,j=7.40hz,2h),1.28(br s,1h),0.95(t,j=7.40hz,3h)
[0457]
苄基3-((叔丁基二甲基甲硅烷基)氧基)-3-乙基环丁烷-1-甲酸酯i-1005的合成
乙基环丁-1-胺i-1008(200mg,产率79%)。
[0467]
1h nmr(400mhz,氯仿-d)d 2.86-2.99(m,1h),2.39-2.51(m,2h),1.69-1.82(m,2h),1.50(q,j=7.24hz,4h),0.81-0.93(m,12h),0.05-0.12(m,6h)
[0468]
使用相同的程序合成结构类似物。
[0469][0470]
2-(苄氧基)乙酰氯i-1009的合成
[0471][0472]
在0℃,将亚硫酰氯[7719-09-7](588μl,7.82mmol)添加到苄氧基乙酸[30379-55-6](1g,6.02mmol)在无水dcm(20ml)中的搅拌溶液中。经3h,将混合物从45℃搅拌至室温。将溶剂在真空中蒸发,以产生呈米色固体的2-(苄氧基)乙酰氯i-1009(1.12g,定量)。
[0473]
1-氟环丙烷-1-碳酰氯i-111的合成
[0474][0475]
将草酰氯[79-37-8](3.08ml,1.48g/ml,35.9mmol)逐滴添加到1-氟环丙烷羧酸[137081-41-5](4.14g,39.8mmol)和dmf(100μl,0.94g/ml,1.29mmol)在无水dcm(150ml)中的溶液中。将反应混合物在室温搅拌16小时然后在真空下蒸发(36℃,400毫巴),并将获得的粗酰氯i-111不经进一步纯化而使用。
[0476]
将粗品不经进一步纯化而用于下一步骤。
[0477]
(三丁基甲锡烷基)甲醇i-117的合成
[0478][0479]
在-20℃,将在己烷中的2.5m n-buli溶液[109-72-8](5.4ml,13.5mmol)逐滴添加到二异丙胺[108-18-9](2ml,0.72g/ml,14.26mmol)在无水thf(45ml)中的搅拌溶液中。将反应混合物在-20℃搅拌30min。然后将其冷却至-78℃并逐滴添加三丁基氢化锡[688-73-3](3.53ml,1.08g/ml,12.73mmol)。使混合物升温至0℃,保持30min。然后将其冷却至-78℃并分批添加多聚甲醛[30525-89-4](463mg,5.09mmol)。添加后,经30min,使反应从-78℃缓慢升温至室温并在室温再搅拌30min。将混合物用水稀释并用et2o萃取。将有机层分离,干燥(mgso4),过滤并将挥发物在真空中蒸发。将粗产物通过fcc(庚烷/etoac 0至10%)纯化,以产生呈无色油状物的i-117(2.8g,68%)。
[0480]
甲基4-(1-氟环丙基)苯甲酸酯i-1010的合成
[0481][0482]
将meoh(10ml)和无水thf(30ml)的混合物用氮气脱气。添加三乙胺[121-44-8](6.48ml,46.5mmol)、4,5-双(二苯基膦基)-9,9-二甲基呫吨[161265-03-8](430.48mg,0.74mmol)、1-溴-4-(1-氟环丙基)苯[1783975-92-7](4g,18.6mmol)和乙酸钯(ii)[3375-31-3](83.51mg,0.37mmol)。然后将混合物在120℃在20巴一氧化碳下在高压釜中搅拌24h。将溶剂蒸发,吸收于饱和nahco3溶液中,用dcm萃取,在mgso4上干燥并蒸发。将残余物在硅胶柱上纯化,洗脱液:在庚烷中的etoac,从0至10%。将纯的级分蒸发,产生呈无色油状物的甲基4-(1-氟环丙基)苯甲酸酯i-1010(2.92g,产率81%),其在冷却至室温后固化成白色固体。
[0483]
3-氟-4-(三氟甲基)苯甲酸i-93的合成
[0484][0485]
向3-氟-4-(三氟甲基)苯甲腈[231953-38-1](1g,5.29mmol)在etoh(20ml)中的混合物中添加2m naoh水溶液[1310-73-2](4ml,8mmol)。将混合物在80℃加热16小时。将混合物在室温下冷却并添加hcl水溶液(2m)直至ph=2。将混合物用etoac(2x5ml)萃取,将合并的有机层经mgso4干燥并在真空中蒸发,以提供呈白色固体的i-93(0.9g,82%)。
[0486]
2-氟-6-碘-4-(三氟甲基)苯甲酸i-97的合成
[0487][0488]
在氮气下,将碘[7553-56-2](2.68g,10.6mmol)添加到2-氟-4-(三氟甲基)苯甲酸[115029-24-8](2g,9.61mmol)、pida[3240-34-4](3.4g,10.6mmol)和pd(oac)2[3375-31-3](107.8mg,0.48mmol)在dmf(30ml)中的搅拌溶液中。将混合物在100℃搅拌16小时。添加etoac和1m hcl水溶液,将有机层分离,经mgso4干燥,过滤并将挥发物在真空中除去,以产生呈棕色黏性固体的粗品i-97,将其不经进一步纯化而使用。
[0489]
将粗品不经进一步纯化而用于下一反应步骤。
[0490]
叔丁基3-氟-4-(三氟甲基)苯甲酸酯i-94的合成
[0491][0492]
将2-甲基-2-丙醇[75-65-0](4.6g,62.5mmol)、n,n'-二环己基碳二亚胺[538-75-0](13.9g,67.3mmol)和4-(二甲基氨基)吡啶[1122-58-3](0.587g,4.8mmol)添加到3-氟-4-(三氟甲基)苯甲酸i-93(10g,48mmol)在thf(250ml)中的混合物中。将混合物在室温搅拌16小时。将粗混合物过滤并用冷的thf洗涤。弃掉固体并将滤液在真空中蒸发。将残余物通过fcc(dcm)纯化,以提供i-94(9g,71%)。
[0493]
甲基4-溴-2-碘苯甲酸酯i-1011的合成
[0494][0495]
在室温,将碳酸铯[534-17-8](5.98g,18.35mmol)和碘甲烷[74-88-4](1.14ml,2.28g/ml,18.35mmol)添加到4-溴-2-碘苯甲酸[1133123-02-0](5g,15.29mmol)在dmf(9ml)中的搅拌溶液中,保持16h。将反应混合物用etoac稀释并通过滤纸过滤。将有机层在真空中浓缩。将粗品通过快速柱色谱法(二氧化硅;etoac在庚烷中从0/100至20/80)纯化。收集所希望的级分并在真空中浓缩,以产生呈无色油状物的甲基4-溴-2-碘苯甲酸酯i-1011(4.27g,80%)。
[0496]
根据以上程序合成结构类似物。
[0497][0498]
叔丁基3-氟-2-异丁酰基-4-(三氟甲基)苯甲酸酯i-95的合成
[0499][0500]
在-78℃,将2m lda溶液(在thf/n-庚烷中)[4111-54-0](18.9ml,2m,37.8mmol)添加到叔丁基3-氟-4-(三氟甲基)苯甲酸酯i-94(5g,18.9mmol)在无水thf(125ml)中的混合物中。将混合物在-78℃搅拌45min,然后在-78℃,逐滴添加异丁酰氯[79-30-1](2.1ml,1.017g/ml,20.8mmol)。将反应在-78℃搅拌2小时。将反应通过添加饱和nh4cl水溶液淬灭,在-78℃添加。使粗混合物升温至室温并用etoac(2x25ml)萃取,将有机层分离,干燥并在真空中蒸发。将残余物通过fcc(dcm)纯化,产生呈油状物的i-95(5.2g,82%)。
[0501]
甲基2-(2-(苄氧基)乙酰基)-4-溴苯甲酸酯i-1012的合成
[0502][0503]
在-78℃,将异丙基氯化镁溶液(2m,在thf中)[1068-55-9](807μl,1.61mmol)逐滴
添加到甲基4-溴-2-碘苯甲酸酯i-1011(500mg,1.47mmol)在无水thf(7.5ml)中的溶液中,并将所得混合物在0℃在氮气气氛下搅拌30min。然后添加无水溴化锌[7699-45-8](367mg,1.61mmol)并将反应混合物在0℃搅拌15min。添加2-(苄氧基)乙酰氯i-1009(325mg,1.76mmol)和四(三苯基膦)钯(0)[14221-01-3](86mg,0.073mmol)并将所得混合物在60℃在氮气气氛下搅拌2h。将反应混合物用饱和nh4cl水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;dcm在庚烷中0/100至100/0)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色黏性固体的甲基2-(2-(苄氧基)乙酰基)-4-溴苯甲酸酯i-1012(468mg,82%)。
[0504]
根据以上程序合成结构类似物。
[0505][0506][0507]
5-(三氟甲氧基)异苯并呋喃-1(3h)-酮i-1016的合成
[0508][0509]
在密封管中在氮气下,将4-(三氟甲氧基)苯甲酸[330-12-1](2000mg,9.7mmol)和二溴甲烷(37ml)添加到磷酸氢二钾[7758-11-4](5070mg,29.11mmol)和pd(oac)2[3375-31-3](218mg,0.97mmol)中。将混合物在140℃搅拌24h。将反应混合物通过短硅藻土垫过
滤,并将溶剂在真空中除去。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至20/80)纯化。收集所希望的级分并在真空中浓缩,以产生呈微黄色固体的5-(三氟甲氧基)异苯并呋喃-1(3h)-酮i-1016(771mg,36%)。
[0510]
使用相同的程序合成结构类似物。
[0511][0512][0513]
3,5-二溴异苯并呋喃-1(3h)-酮i-1021的合成
[0514][0515]
将n-溴代琥珀酰亚胺[128-08-5](10g,56.33mmol)和偶氮二异丁腈(aibn)[78-67-1](231mg,1.41mmol)添加到5-溴苯酞[64169-34-2](10g,46.94mmol)在dce(200ml)中的搅拌溶液中。将混合物在80℃搅拌18h。将反应混合物在室温下冷却并将溶剂浓缩。将粗品通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至20/80)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的3,5-二溴异苯并呋喃-1(3h)-酮i-1021(10.9g,73%)。
[0516]
使用相同的程序合成结构类似物。
[0517][0518][0519]
5-溴-3-羟基异苯并呋喃-1(3h)-酮i-1026的合成
[0520][0521]
将3,5-二溴异苯并呋喃-1(3h)-酮i-1021(7.34g,25.14mmol)悬浮于水(300ml)中并在100℃加热1h。将混合物在室温下冷却并用dcm:meoh 9:1萃取三次。将合并的有机层经无水mgso4干燥,过滤并在减压下浓缩,以产生呈白色固体的5-溴-3-羟基异苯并呋喃-1(3h)-酮i-1026(5.9g,99%)。
[0522]
使用相同的程序合成结构类似物。
[0523][0524][0525]
2-(4-甲氧基苯基)-4,4-二甲基-5h-噁唑(i-1)的合成
[0526][0527]
在30min内,在氮气下,将2-氨基-2-甲基-1-丙醇[124-68-5](53.03g,0.59mol)在无水dcm(200ml)中的溶液逐滴添加到乙基4-甲氧基苯甲酰氯[100-07-2](51.18g,0.3mol)在无水dcm(254ml)中的搅拌溶液中,使用冰/水浴将温度保持在约18℃。搅拌3h后,将沉淀物通过硅藻土过滤并用dcm洗涤。将有机相在氮气下在2℃搅拌并逐滴添加到亚硫酰氯[7719-09-7](65.29ml,0.9mol)中,保持温度低于10℃。逐滴添加结束时,将反应混合物在室温搅拌18h,然后在真空下浓缩。将残余物通过柱色谱法(二氧化硅,meoh在dcm中0/100至1/99)纯化。收集所希望的级分并将溶剂在真空中蒸发,以产生呈黄色油状物的2-(4-甲氧基苯基)-4,4-二甲基-5h-噁唑(i-1)(42g,68%)。
[0528]
[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]-2-甲基-丙-1-醇(i-2)的合成
[0529][0530]
在室温,将在thf/甲苯中的1m 2,2,6,6-四甲基哌啶基氯化镁氯化锂复合物溶液[898838-07-8](60ml,60mmol)逐滴添加到2-(4-甲氧基苯基)-4,4-二甲基-5h-噁唑(i-1)(5.29g,25.78mmol)在无水thf(100ml)中的搅拌溶液中。搅拌4h后,将反应混合物冷却至0℃并逐滴添加异丁醛[78-84-2](7ml,76.69mmol)在无水thf(10ml)中的溶液。将所得反应混合物在室温搅拌2h。将溶剂在真空中部分除去,将残余物用饱和nh4cl水溶液稀释并用etoac萃取。将合并的有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,etoac在庚烷中0/100至50/50)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色油状物的[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]-2-甲基-丙-1-醇(i-2)(6.3g,89%)。
[0531]
使用相同的程序合成结构类似物。
[0532][0533]
1-[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]-2-甲基-丙-1-酮(i-4)的合成
[0534][0535]
在室温,将戴斯-马丁高碘烷[87413-09-0](4.07g,9.59mmol)添加到[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]-2-甲基-丙-1-醇(i-2)(1.98g,7.14mmol)在无水dcm(120ml)中的搅拌溶液中。搅拌2.5h后,再添加戴斯-马丁高碘烷[87413-09-0](1.35g,3.18mmol)并将所得混合物再搅拌1h。将混合物过滤并向滤液中添加20%na2s2o3水溶液。将产物用dcm萃取并将有机层进一步用饱和nahco3水溶液和盐水洗涤。将有机层干燥(mgso4),过滤并将溶剂在真空中蒸发,以产生呈黄色固体的1-[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]-2-甲基-丙-1-酮(i-4)(0.50g,48%纯度),将其不经进一步纯化而用于下一步骤。
[0536]
使用相同的程序合成结构类似物。
[0537][0538]
乙基4-甲氧基-2-丙酰基-苯甲酸酯(i-6)的合成
[0539][0540]
将h2so4[7664-93-9](2.5ml,46.9mmol)逐滴添加到1-[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]丙-1-酮(i-5)(2g,7.65mmol)在水(3ml)和etoh(57ml)混合物中的搅拌溶液中。将混合物在90℃搅拌20h。将溶剂在真空中浓缩,将残余物用水稀释并用et2o萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发,以产生乙基4-甲氧基-2-丙酰基-苯甲酸酯(i-6)(1.52g,84%),将其不经进一步纯化而用于下一步骤。
[0541]
5-甲基-2-苯基-异吲哚啉-1,3-二酮(i-7)的合成
[0542][0543]
将苯胺[62-53-3](1.24ml,13.57mmol)逐滴添加到5-甲基异苯并呋喃-1,3-二酮[19438-61-0](2g,12.33mmol)在acoh[64-19-7](12.3ml)中的搅拌溶液中。将混合物在140℃搅拌2h。向冷却的混合物中添加水并将所得反应混合物在室温搅拌2h。将形成的固体过滤并再用水洗涤,以产生呈白色固体的5-甲基-2-苯基-异吲哚啉-1,3-二酮(i-7)(2.77g,95%),将其不经进一步纯化而用于下一步骤。
[0544]
使用相同的程序合成结构类似物。
[0545][0546]
5-溴-3-乙基-3-羟基-2-苯基-异吲哚啉-1-酮(i-9)的合成
[0547][0548]
在0℃并在氮气下,将3m乙基溴化镁溶液[925-90-6](1.65ml,4.96mmol)逐滴添加到5-溴-2-苯基-异吲哚啉-1,3-二酮[82104-66-3](1g,3.31mmol)在thf(20ml)中的搅拌溶液中。搅拌5min后,将混合物通过添加水淬灭。将溶剂在真空中蒸发,以产生呈微黄色油状物的5-溴-3-乙基-3-羟基-2-苯基-异吲哚啉-1-酮(i-9)(1.1g,定量),将其不经进一步纯化而用于下一步骤。
[0549]
使用相同的程序合成结构类似物。
[0550][0551][0552]
4-异丙基-6-甲氧基-2h-酞嗪-1-酮(i-12)的合成
[0553][0554]
将水合肼[7803-57-8](0.15ml,3.09mmol)添加到1-[2-(4,4-二甲基-5h-噁唑-2-基)-5-甲氧基-苯基]-2-甲基-丙-1-酮(i-4)(0.50g,48%纯度)在acoh[64-19-7](3ml)中的搅拌溶液中。将混合物在80℃搅拌18h。将混合物用水稀释并用dcm萃取。将水相用饱和nahco3水溶液碱化并用dcm萃取。将合并的有机层干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,etoac在庚烷中0/100至50/50)纯化。收集所希望的级分并将溶剂在真空中浓缩,以产生4-异丙基-6-甲氧基-2h-酞嗪-1-酮(i-12)(103mg,54%)。
[0555]
使用相同的程序合成结构类似物。
[0556][0557]
6-溴-4-乙基-2h-酞嗪-1-酮(i-14)的合成
[0558][0559]
在密封管中,将水合肼[7803-57-8](0.52ml,16.56mmol)添加到5-溴-3-乙基-3-羟基-2-苯基-异吲哚啉-1-酮(i-9)(1.1g,3.31mmol)在etoh(12ml)中的搅拌溶液中。将混合物在80℃搅拌16h。再添加水合肼[7803-57-8](0.52ml,16.56mmol)并将混合物在80℃搅拌5h。再添加水合肼[7803-57-8](1.03ml,33.11mmol)并将混合物在80℃搅拌3天。混合物冷却后,将形成的固体过滤并在真空下干燥,以产生6-溴-4-乙基-2h-酞嗪-1-酮(i-14)(500mg,60%)。
[0560]
使用相同的程序合成结构类似物。
[0561]
[0562]
[0563][0564]
4-异丙基-1-氧代-6-(三氟甲基)-1,2-二氢酞嗪-5-甲腈i-85的合成
[0565][0566]
在室温,将kcn[151-50-8](130.6mg,2.01mmol)添加到5-氟-4-异丙基-6-(三氟甲基)酞嗪-1(2h)-酮(i-86)(500mg,1.82mmol)在dmso(20ml)中的溶液中。将混合物在mw辐照下在150℃加热40min。使粗混合物冷却至室温,用水稀释并用etoac(3x5ml)萃取。将有机层在真空中蒸发并通过fcc(dcm/meoh 0至5%)纯化,以提供呈固体的(i-85)(420mg,产率82%)。
[0567]
4,6-二溴酞嗪-1(2h)-酮i-1044的合成
[0568][0569]
将k2co3[584-08-7](3.07g,22.22mmol)添加到6-溴酞嗪-1(2h)-酮i-1035(2.5g,11.11mmol)在dmf(28ml)中的混合物中。将悬浮液在室温搅拌10min。然后添加苄基三甲基三溴化铵[111865-47-5](8.66g,22.22mmol)。将反应混合物在40℃搅拌5h。添加na2s2o3饱和溶液(直至ph 7)。将混合物用dcm萃取。将有机层分离并经无水mgso4干燥,过滤并将溶剂在真空中浓缩。将产物与甲苯共蒸馏(x5),以产生呈米色固体的4,6-二溴酞嗪-1(2h)-酮i-1044(685mg,19%)。将水层用dcm:meoh(9:1)萃取。将有机层分离并经无水mgso4干燥,过滤并将溶剂在真空中浓缩。将产物与甲苯共蒸馏,以产生呈米色固体的4,6-二溴酞嗪-1(2h)-酮i-1044(2.32g,67%)。
[0570]
使用相同的程序合成结构类似物。
[0571]
[0572][0573]
4-异丙基-1-氧代-6-(三氟甲基)-1,2-二氢酞嗪-5-甲腈i-105的合成
[0574][0575]
将4-溴-6-(三氟甲基)酞嗪-1(2h)-酮i-82(4g,13.65mmol)在1,4-二噁烷(89.7ml)和di水(29.9ml)中的溶液放入easymax压力管中并用氮气脱气15min。然后,添加4,4,5,5-四甲基-2-(丙-1-烯-2-基)-1,3,2-二氧代环戊硼烷[126726-62-3](3.89ml,0.89g/ml,20.67mmol)、k3po4[7778-53-2](8.85g,41.69mmol)和ruphos pd g3[1445085-77-7](0.6g,0.71mmol)并将反应混合物在100℃搅拌4小时。将反应用盐水(约400ml)淬灭并将产物用etoac(3x200ml)萃取。将合并的有机萃取物经mgso4干燥,过滤并在减压下在40℃浓缩。将粗化合物通过fcc(庚烷/etoac 0至30%)纯化,以提供呈白色固体的i-105(3g,86%)。
[0576]
使用相同的程序合成结构类似物。
[0577][0578]
4-异丙基-1-氧代-6-(三氟甲基)-1,2-二氢酞嗪-5-甲腈i-104的合成
[0579][0580]
在氮气气氛下,将在己烷中的1m二乙基锌溶液[557-20-0](22.6ml,22.6mmol)添加在含有无水dcm(20ml)的烧瓶中并将其0℃搅拌。经20min,经由注射泵逐滴添加tfa[76-05-1](1.73ml,1.49g/ml,22.6mmol)。将反应混合物在0℃搅拌20min,并经20min经由注射泵添加二碘甲烷[75-11-6](1.82ml,3.33g/ml,22.6mmol)。将混合物在0℃再搅拌20min,并经20min经由注射泵添加4-(丙-1-烯-2-基)-6-(三氟甲基)酞嗪-1(2h)-酮i-105(500mg,1.97mmol)在dcm(10ml)中的溶液。添加后,使反应混合物升温至室温并再搅拌2小时。将反应通过添加饱和nh4cl水溶液淬灭并将固体过滤。将有机层分离,在mgso4上干燥并浓缩。经由制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)进行纯化,产生呈白色固体的i-104(302mg,57%)。
[0581]
4-(1-乙氧基乙烯基)-6-(三氟甲基)酞嗪-1(2h)-酮i-116的合成
[0582][0583]
在氮气下在密封管中,将双(三苯基膦)二氯化钯(ii)[13965-03-2](244.4mg,0.34mmol)和三丁基(1-乙氧基乙烯基)锡[97674-02-7](1.43ml,1.07g/ml,4.1mmol)添加到4-溴-6-(三氟甲基)酞嗪-1(2h)-酮i-82(1g,3.41mmol)在无水1,4-二噁烷中的搅拌溶液中。将混合物在100℃搅拌16小时。将混合物用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将挥发物在真空下浓缩。将粗产物通过fcc(庚烷/etoac 0至30%)纯化,以产生呈浅黄色固体的i-116(948mg,95%纯度,93%)。
[0584]
4-乙酰基-6-(三氟甲基)酞嗪-1(2h)-酮i-115的合成
[0585][0586]
在0℃,将6m hcl水溶液[7647-01-0](2.77ml,16.6mmol)逐滴添加到4-(1-乙氧基乙烯基)-6-(三氟甲基)酞嗪-1(2h)-酮i-116(945mg,3.32mmol)在1,4-二噁烷中的搅拌溶液中。将混合物在室温搅拌1小时。然后将其用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将挥发物在真空中蒸发,以产生呈白色固体的i-115(842mg,95%纯度,94%)。
[0587]
4-(2-羟基丙-2-基)-6-(三氟甲基)酞嗪-1(2h)-酮i-114的合成
[0588][0589]
在0℃在氮气下,将在thf中的1.4m甲基溴化镁溶液[75-16-1](7ml,9.83mmol)逐滴添加到4-乙酰基-6-(三氟甲基)酞嗪-1(2h)-酮i-115(839mg,3.28mmol)在无水thf(21.3ml)中的搅拌溶液中。然后将混合物在5℃搅拌1小时。将其用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将挥发物在真空中蒸发。将粗产物通过fcc(庚烷/etoac 0至30%)纯化,以产生呈灰白色固体的i-114(576mg,97%-98%纯度,64%)。
[0590][0591]
4-(1-甲氧基环丙基)-6-(三氟甲基)酞嗪-1(2h)-酮i-126的合成
[0592][0593]
将4-(1-氟环丙基)-6-(三氟甲基)酞嗪-1(2h)-酮i-109(300mg,1.1mmol)和在meoh中的0.5m naome[124-41-4](22ml,11mmol)的混合物在氮气气氛下搅拌并在压力管中在120℃加热12小时。将溶剂蒸发并将残余物吸收于di水中。添加nh4cl[12125-02-9](1g)并将混合物用etoac萃取。将合并的有机萃取物用盐水洗涤,在mgso4上干燥并蒸发。将残余物通过fcc(dcm/meoh 0至5%)纯化,产生呈白色固体的i-126(200mg,64%)。
[0594]
乙基2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)乙酸酯(i-21)的合成
[0595][0596]
在密封管中,将氯乙酸乙酯[105-39-5](80μl,0.85mmol)添加到4-异丙基-6-甲氧基-2h-酞嗪-1-酮(i-12)(148mg,0.68mmol)、18-冠醚-6[17455-13-9](11mg,0.042mmol)、碘化钾[7681-11-0](17mg,0.1mmol)、k2co3[584-08-7](118mg,0.85mmol)在无水acn(6ml)和dcm(2ml)中的搅拌混合物中。将混合物在90℃搅拌5h。将混合物用水和盐水处理然后用
dcm萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,etoac在庚烷中0/100至30/70)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的乙基2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)乙酸酯(i-21)(163mg,79%)。
[0597]
使用相同的程序合成结构类似物。
[0598]
[0599]
[0600]
[0601]
[0602][0603]
乙基2-(7-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-28)的合成
[0604][0605]
在0℃,将溴乙酸乙酯[105-36-2](0.82ml,7.4mmol)添加到7-溴-4-乙基-2h-酞嗪-1-酮(i-14)(2.6g,6.16mmol,60%纯度)和nah[7646-69-7](在矿物油中的60%分散体,0.27g,6.78mmol)在无水dmf(24.6ml)中的搅拌悬浮液中。将混合物在室温搅拌1h。将混合物用水稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,etoac在庚烷中0/100至20/80)纯化。收集所希望的级分并在真空中浓缩,以产生呈浅黄色油状物的乙基2-(7-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-28)(1.15g,55%)。
[0606]
使用相同的程序合成结构类似物。
[0607][0608]
乙基2-(6-溴-4-(羟基甲基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1031的合成
[0609][0610]
在室温在氮气气氛下,将氯三甲基硅烷[75-77-4](1.37ml,0.86g/ml,10.7mmol)和碘化钠[7681-82-5](1.62g,10.7mmol)添加到乙基2-(4-((苄氧基)甲基)-6-溴-1-氧代酞嗪-2(1h)-基)乙酸酯i-1056(2.23g,4.65mmol)在无水乙腈(24ml)中的搅拌溶液中。将混合物在80℃搅拌7h。将混合物用饱和nahco3水溶液(32ml)和10%na2s2o3水溶液(32ml)稀释并用acoet萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;acoet在庚烷中0/100至35/65)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的乙基2-(6-溴-4-(羟基甲基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1031(523mg,33%)。
[0611]
乙基2-(6-溴-4-(1-乙氧基乙烯基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1074的合成
[0612][0613]
在氮气气氛下在密封管中,将双(三苯基膦)二氯化钯(ii)[13965-03-2](92mg,0.13mmol)和三丁基(1-乙氧基乙烯基)锡[97674-02-7](402μl,1.07g/ml,1.15mmol)添加到乙基2-(4,6-二溴-1-氧代酞嗪-2(1h)-基)乙酸酯i-1055(500mg,1.28mmol)在甲苯(10ml)中的搅拌溶液中。将混合物在80℃搅拌4h。将混合物用饱和nahco3水溶液稀释并用acoet萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,acoet in庚烷0/100至20/80)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色固体的乙基2-(6-溴-4-(1-乙氧基乙烯基)-1-氧代酞嗪-2(1h)-基)乙酸
酯i-1074(252mg,41%)。
[0614]
使用相同的程序合成结构类似物。
[0615][0616]
[0617]
乙基2-(6-(1-乙氧基乙基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1077的合成
[0618][0619]
在室温在氮气气氛下,将三(三苯基膦)氯化铑(i)[14694-95-2](31mg,0.034mmol)添加到乙基2-(6-(1-乙氧基乙烯基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1075(116mg,0.34mmol)在乙醇(5ml)中的搅拌溶液中。然后,将氮气气氛替换为h2(气球)并将反应混合物在室温搅拌16h。将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅12g;acoet在庚烷中0/100至20/80)纯化。收集所希望的级分并在真空中浓缩,以产生呈无色黏性固体的乙基2-(6-(1-乙氧基乙基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1077(107mg,90%)。
[0620][0621]
乙基2-(6-羟基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1079的合成
[0622]
[0623]
在室温在氮气气氛下,将氯三甲基硅烷[75-77-4](250μl,1.95mmol)和碘化钠[7681-82-5](2.96mg,1.95mmol)添加到乙基2-(4-异丙基-6-甲氧基-1-氧代酞嗪-2(1h)-基)乙酸酯(i-21)(300mg)在无水乙腈中的搅拌溶液中。将混合物在80℃搅拌5h。添加氯三甲基硅烷[75-77-4](250μl,1.95mmol)和碘化钠[7681-82-5](2.96mg,1.95mmol)并将混合物在80℃搅拌16h。将混合物用饱和nahco3水溶液(18ml)和10%na2s2o3水溶液(18ml)稀释并用acoet萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅25g;acoet在庚烷中0/100至100/0)纯化。收集所希望的级分并在真空中浓缩,以产生呈米色固体的乙基2-(6-羟基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1079(55mg,21%)。
[0624]
乙基2-(4-异丙基-1-氧代-6-(2,2,2-三氟乙氧基)酞嗪-2(1h)-基)乙酸酯i-1080的合成
[0625][0626]
将2,2,2-三氟乙基全氟丁基磺酸酯[79963-95-4](47μl,1.68g/ml,0.21mmol)添加到乙基2-(6-羟基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1079(55mg,0.19mmol)和碳酸铯[534-178](93mg,0.28mmol)在dmf(2ml)中的搅拌溶液中。将混合物在室温搅拌4h。将混合物用饱和nahco3水溶液稀释并用acoet萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅25g;acoet在庚烷中0/100至15/85)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色固体的乙基2-(4-异丙基-1-氧代-6-(2,2,2-三氟乙氧基)酞嗪-2(1h)-基)乙酸酯i-1080(36mg,51%)。
[0627]
乙基2-(4-溴-6-(二甲基氨基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1081的合成
[0628][0629]
在密封管中,将在thf中的二甲胺溶液2m[124-40-3](1.92ml,3.85mmol)添加到乙基2-(4,6-二溴-1-氧代酞嗪-2(1h)-基)乙酸酯i-1055(500mg,1.28mmol)和n,n-二异丙基乙胺[7087-68-5](1.35ml,0.74g/ml,7.69mmol)在dmso中的搅拌溶液中。将混合物在125℃搅拌16h。将混合物用水稀释并用acoet萃取。将有机层用水洗涤(x3),分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;acoet在庚烷中0/100至50/50)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的乙基2-(4-溴-6-(二甲基氨基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1081(142mg,31%)。
[0630]
使用相同的程序合成结构类似物。
[0631][0632]
乙基2-(6-环丙基-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-32)的合成
[0633][0634]
在氮气下,将[1,1'-双(二苯基膦基)二茂铁]二氯钯(ii)与二氯甲烷的复合物[95464-05-4](109mg,0.13mmol)添加到乙基2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-28)(300mg,0.88mmol)、环丙基硼酸[411235-57-9](190mg,2.21mmol)和碳酸铯[534-17-8](0.63g,1.95mmol)在1,4-二噁烷(4ml)和水(1ml)混合物中的搅拌悬浮液中。将混合物在90℃搅拌16h。将冷却的混合物用水稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至50/50)纯化。收集所希望的级分并将溶剂在真空中蒸发,以产生呈黄色油状物的乙基2-(6-环丙基-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-32)(129mg,48%)。
[0635]
乙基2-(4-乙基-6-异丙烯基-1-氧代-酞嗪-2-基)乙酸酯(i-33)的合成
[0636][0637]
在氮气下,将[1,1'-双(二苯基膦基)二茂铁]二氯钯(ii)与二氯甲烷的复合物[95464-05-4](0.15g,0.18mmol)添加到乙基2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-28)(0.4g,1.18mmol)、异丙烯基三氟硼酸钾[395083-14-4](0.44g,2.95mmol)和碳酸铯[534-17-8](0.84g,2.59mmol)在1,4-二噁烷(8ml)和水(2ml)混合物中的搅拌悬浮液中。将混合物在105℃搅拌5h。将冷却的混合物用水稀释并用etoac萃取。将有机层分离,用盐水洗涤,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至50/50)纯化。收集所希望的级分并将溶剂在真空中蒸发,以产生呈黄色油状物的乙基2-(4-乙基-6-异丙烯基-1-氧代-酞嗪-2-基)乙酸酯(i-33)(148mg,87%)。
[0638]
使用相同的程序合成结构类似物。
[0639][0640]
[0641]
乙基2-(1-氧代-6-(三氟甲基)-4-(3,3,3-三氟丙-1-烯-2-基)酞嗪-2(1h)-基)乙酸酯(i-129)的合成
[0642][0643]
在室温在氮气下,将pd(pph3)4[14221-01-3](76.2mg,0.066mmol)添加到乙基2-(4-溴-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-81、1-(三氟甲基)乙烯基硼酸亚己基乙二醇酯[1011460-68-6](307.4mg,1.38mmol)和na2co3[497-19-8](559.1mg,5.28mmol)在dme/di水(7.6ml/3.8ml)中的搅拌溶液中。将所得混合物在95℃搅拌3.5小时。使反应冷却至室温,用水稀释并用etoac萃取。将有机萃取物用盐水洗涤,经mgso4干燥,过滤并在真空下浓缩。将粗产物通过fcc(庚烷/etoac 0至10%)纯化,以产生呈无色油状物的i-129(287mg,52%)。
[0644]
乙基2-(4-乙基-6-异丙基-1-氧代-酞嗪-2-基)乙酸酯(i-35)的合成
[0645][0646]
在0℃并在氮气下,将钯/活性炭(10%)[7440-05-3](31mg)添加到乙基2-(4-乙基-6-异丙烯基-1-氧代-酞嗪-2-基)乙酸酯(i-33)(309mg,1.03mmol)在etoh(13ml)中的溶液中。将所得混合物在氢气气氛下搅拌16h。将反应混合物通过硅藻土过滤,用meoh洗涤并将溶剂在真空中浓缩,以产生呈白色固体的乙基2-(4-乙基-6-异丙基-1-氧代-酞嗪-2-基)乙酸酯(i-35)(300mg,95%),将其不经进一步纯化而用于下一步骤。
[0647]
使用相同的程序合成结构类似物。
[0648][0649][0650]
乙基2-(4-乙基-6-甲酰基-1-氧代-酞嗪-2-基)乙酸酯(i-37)的合成
[0651][0652]
将四氧化锇[20816-12-0](2.61ml,0.1mmol)添加到乙基2-(4-乙基-1-氧代-6-乙烯基-酞嗪-2-基)乙酸酯(i-34)(0.74g,2.57mmol)和高碘酸钠[7790-28-5](1.1ml,5.13mmol)在thf(10ml)和水(10ml)混合物中的搅拌溶液中。将混合物在室温搅拌16h。将混合物用水稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至15/85)纯化。收集所希望的级分并将溶剂在真空中蒸发,以产生呈白色固体的(i-37)(0.5g,68%)。
[0653]
乙基2-(4-甲酰基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯(i-119)的合成
[0654][0655]
在0℃,将戴斯-马丁高碘烷[87413-09-0](1.1g,2.51mmol)分批添加到乙基2-(4-(羟基甲基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-118(645mg,1.93mmol)在无水dcm(19.4ml)中的搅拌溶液中。将混合物在室温搅拌1小时。将反应用饱和nahco3水溶液(64ml)和10%na2s2o3水溶液(10ml)稀释并用dcm萃取。将有机层分离,干燥(mgso4),过滤并将挥发物在真空中浓缩。将粗产物通过fcc(庚烷/etoac 0至42%)纯化,以产生呈白色固体的i-119(381mg,92%纯度,55%)。
[0656]
乙基2-[6-(二氟甲基)-4-乙基-1-氧代-酞嗪-2-基]乙酸酯(i-38)的合成
[0657][0658]
在-10℃并在氮气下,将dast[38078-09-0](0.7ml,5.27mmol)逐滴添加到乙基2-(4-乙基-6-甲酰基-1-氧代-酞嗪-2-基)乙酸酯(i-37)(0.5g,1.76mmol)在无水dcm(10ml)中的搅拌溶液中。将混合物在室温搅拌16h。将混合物用饱和nahco3水溶液稀释并用dcm萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至15/85)纯化。收集所希望的级分并将溶剂在真空中蒸发,以产生呈白色固体的(i-38)(0.39g,71%)。
[0659]
使用相同的程序合成结构类似物。
[0660]
[0661][0662]
乙基2-(1-氧代-4-(2,2,2-三氟-1-羟基乙基)-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯(i-125)的合成
[0663][0664]
在0℃在氮气下,将在thf中的1m tbaf溶液[429-41-4](152μl,015mmol)逐滴添加到乙基2-(4-甲酰基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-119(250mg,0.76mmol)和2-三氟甲基三甲基硅烷[81290-20-2](230μl,1.52mmol)在无水thf(6.25ml)中的搅拌溶液中。将反应在0℃搅拌15min,然后在室温搅拌12小时。将混合物冷却至0℃,添加在thf中的1m tbaf溶液[429-41-4](1.5ml,1.5mmol)并将混合物在室温搅拌20min。将混合物用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将挥发物在真空中蒸发。将粗产物通过fcc(庚烷/etoaco0至30%)纯化,以产生呈白色泡沫状固体的i-125(235mg,75%)。
[0665]
乙基2-(6-溴-4-(1-甲氧基乙基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1092的合成
[0666][0667]
将三甲基氧鎓四氟硼酸盐[420-37-1](161mg,1.09mmol)和2,6-二-叔丁基-4-甲基吡啶[38222-83-2](298mg,1.45mmol)添加在mw小瓶中,密封并置于氮气(3个真空/氮气循环)下。然后,添加乙基2-(6-溴-4-(1-羟基乙基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1093(129mg,0.36mmol)在无水二氯甲烷(14ml)中的悬浮液,并将反应混合物在室温搅拌1.5小时。然后,将反应通过添加饱和nahco3水溶液淬灭并用dcm萃取(x3)。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,acoet在庚烷中0/100至50/50)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色油状物的乙基2-(6-溴-4-(1-甲氧基乙基)-1-氧代酞嗪-2(1h)-基)乙酸酯i-1092(61mg,43%)。
[0668]
使用相同的程序合成结构类似物。
[0669][0670]
乙基2-(6-(二甲基氨基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1095的合成
[0671][0672]
在密封管中,将在thf中的二甲胺溶液2m[124-40-3](0.64ml,1.23mmol)和碳酸铯[534-17-8](1107mg,3.4mmol)添加到乙基2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯(i-31)(300mg,0.85mmol)在1,4-二噁烷(10ml)中的搅拌溶液中。将混合物用n2鼓泡10min。将ruphos pd g4[1599466-85-9](145mg,0.17mmol)添加到混合物中并将反应在70℃搅拌16h。然后,添加在thf中的二甲胺溶液2m[124-40-3](0.64ml,1.23mmol)、碳酸铯[534-17-8](277mg,0.85mmol)和ruphos pd g4[1599466-85-9](145mg,0.17mmol)并将混合物在75℃搅拌16。将混合物用盐水稀释并用etoac萃取。将有机层用盐水洗涤,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至50/50)纯化。收集所希望的级分并在真空中浓缩,以产生呈棕色油状物的乙基2-(6-(二甲基氨基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1095(208mg,73%)。
[0673]
乙基2-(4-(仲丁基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-102的合成
[0674][0675]
将0.5m仲丁基溴化锌溶液[171860-66-5](12.7ml,6.33mmol)逐滴添加到乙基2-(4-溴-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-81(0.8g,2.11mmol)和双(三-叔丁基膦)钯(0)[53199-31-8](107.8mg,0.21mmol)在无水thf(20ml)中的搅拌溶液中。将所得混合物在40℃搅拌6小时。将反应混合物通过添加饱和nh4cl水溶液淬灭。将挥发物在真空下除去并将水相用etoac萃取。将有机萃取物用盐水洗涤并经mgso4干燥。将粗化合物通过fcc(庚烷/etoac 0/100至25/75)纯化,以产生白色固体,将其进一步通过制备型sfc(固定
相:chiralpak daicel ic 20x250mm,流动相:co2、etoh 0.4iprnh2)纯化,产生呈白色固体的i-102(65mg,9%)。
[0676]
使用相同的程序合成结构类似物。
[0677][0678]
乙基2-(8-碘-4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-84的合成
[0679][0680]
将乙基2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯(i-29)(600mg,1.74mmol)、[cp*rh(mecn)3](sbf6)2[59738-27-1](144.5mg,0.17mmol)、nis[516-12-1](780.8mg,3.47mmol)和naoac[127-09-3](28.5mg,0.35mmol)放入20-ml mw小瓶中。将小瓶密封并添加无水1,2-dce(10ml),然后将悬浮液在120℃加热16小时。将粗混合物用dcm(30ml)稀释并通过添加饱和na2s2o3水溶液(50ml)淬灭。在室温剧烈搅拌两相混合物5min后,将两层分离并将水层用dcm(2x20ml)反萃取。将合并的有机萃取物经na2so4干燥,过滤并在真空中浓缩。将获得的残余物通过fcc(庚烷/etoac 93:7至7:3)纯化,以提供呈浅粉色固体的i-84(360mg,44%)。
[0681]
乙基2-(8-羟基-4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-74的合成
[0682][0683]
将乙基2-(8-碘-4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯(i-74)(200mg,0.43mmol)、pdcl2(dppf)[72287-26-4](31.3mg,0.043mmol)、koac[127-08-2](125.8mg,1.28mmol)和双(频哪醇合)二硼[73183-34-3](217mg,0.85mmol)放入20-ml mw小瓶中。将小瓶密封并置于氮气(3个真空/氮气循环)下,并且添加无水dmso(4ml)。然后将悬浮液在80℃加热16小时。将粗混合物用盐水(30ml)稀释并用dcm(4x25ml)萃取。将合并的有机层经na2so4干燥,过滤并在真空中浓缩。将获得的深棕色残余物(粗乙基2-(4-异丙基-1-氧代-8-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯)和高硼酸钠四水合物(265mg,1.72mmol)放入螺旋盖试管中并添加thf/di水的1:1混合物(2ml),并且将混合物在室温剧烈搅拌2小时。添加另一部分的高硼酸钠四水合物(265mg,1.72mmol,4当量)并将混合物在室温再搅拌14小时。
[0684]
将混合物用di水(5ml)稀释,将ph酸化至ph《3并将混合物用dcm(3x15ml)萃取。将合并的有机萃取物经na2so4干燥,过滤并在真空中浓缩。将所得棕色残余物通过fcc(庚烷/etoac 95:5至4:1)纯化,以提供呈无色结晶固体的i-74(85.5mg,55%)。
[0685]
乙基2-(4-(2-氟丙-2-基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-112的合成
[0686][0687]
在-78℃并在氮气下,将dast[38078-09-0](394μl,2.84mmol)逐滴添加到乙基2-(4-(2-羟基丙-2-基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-113(508mg,1.42mmol)在无水dcm(13.7ml)中的搅拌溶液中。使反应混合物经1小时从-78℃升温至0℃并在室温搅拌16小时。将混合物用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过fcc(庚烷/etoac 0至16%)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的i-112(458mg,89%)。
[0688]
使用相同的程序合成结构类似物。
[0689][0690]
乙基2-(6-环丁基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1097的合成
[0691][0692]
将乙基2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯(i-31)(894mg,2.531mmol)和环丁基三氟硼酸钾[395083-14-4](451mg,2.784mmol)添加到pd(oac)2[3375-31-3](57mg,0.253mmol)、catacxium a[321921-71-5](91mg,0.253mmol)和碳酸铯[534-17-8](2.47g,7.593mmol)在甲苯(20ml)和水(2ml)中的搅拌溶液中,同时鼓泡n2。将所得混合物在100℃加热16h。将反应混合物用h2o稀释并将有机层用dcm萃取,用无水mgso4干燥,过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅25g;etoac在庚烷中0/100至15/85)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色油状物的乙基2-(6-环丁基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1097(176mg,21%)。
[0693]
4-异丙基-1-氧代-1,2-二氢酞嗪-6-甲腈i-1069的合成
[0694][0695]
将tbuxphos[564483-19-8]和pd2(dba)3[51364-51-3]添加到dma中,同时将溶剂在45℃通过鼓泡氮气而脱气。将混合物在氮气下在45℃搅拌5分钟。在氮气下在45℃,添加zn[7440-66-6]和氰化锌[557-21-1]。在氮气下在45℃,添加6-溴-4-异丙基酞嗪-1(2h)-酮(i-15)。将混合物在密封管中在120℃搅拌16h。将混合物冷却至室温,然后用饱和nahco3稀释并用acoet萃取。将有机层分离,用水洗涤,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅25g;acoet在庚烷中0/100至70/30)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的4-异丙基-1-氧代-1,2-二氢酞嗪-6-甲腈i-1069(100mg,31%)。
[0696]
乙基2-(6-(1,1-二氟乙基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1098的合成
[0697][0698]
在室温,向乙基2-(6-乙酰基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1071(0.3g,0.94mmol)在dcm(8ml)中的混合物中添加二乙基氨基三氟化硫[38078-09-0](1ml,1.22g/ml,7.56mmol)和三乙胺三氢氟酸盐[73602-61-6](0.47ml,0.989g/ml,2.85mmol)。将混合物在50℃搅拌两天。将粗品在0℃下冷却并用饱和nahco3溶液淬灭(逐滴)。将粗品用ch2cl2(2x5ml)萃取,将有机层干燥并在真空中蒸发,以提供油状物,将其通过柱色谱法(sio2,ch2cl2)纯化。将所希望的级分浓缩,以产生呈油状物的乙基2-(6-(1,1-二氟乙基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1098(189mg,产率59%)。
[0699]
乙基2-(6-(2,2-二氟环丙基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1099的
合成
[0700][0701]
氟磺酰基二氟乙酸甲酯在室温,将[680-15-9](0.99ml,1.52g/ml,7.84mmol)添加到乙基2-(4-异丙基-1-氧代-6-乙烯基酞嗪-2(1h)-基)乙酸酯i-1073(589mg,1.96mmol)和碘化钾[7681-11-0](1.3g,7.84mmol)在丙腈(6ml)中的搅拌溶液中。将混合物在密封管中在50℃搅拌120h。在冷却至室温后,将混合物用水淬灭并用庚烷萃取(3x)。将有机层分离,合并,用饱和nahco3水溶液和盐水洗涤,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅25g;etoac在庚烷中0/100至40/60)纯化。收集所希望的级分并在真空中浓缩,以产生呈黄色油状物的乙基2-(6-(2,2-二氟环丙基)-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸酯i-1099(200mg,29%)。
[0702]
2-(6-乙基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸i-1100的合成
[0703][0704]
向2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸(i-53)(250mg,0.7688mmol)在thf(7ml)中的混合物中添加三乙基硼烷[97-94-9](2.3ml,1m,2.306mmol)、cs2co3[534-17-8](751.5mg,2.31mmol)和[1,1'-双(二苯基膦基)二茂铁]二氯钯(ii)[72287-26-4](56.65mg,0.077mmol),将混合物用n2鼓泡10min。将反应在95℃搅拌16h。将粗品用水处理并用hcl(2n,至ph=2-3)酸化,将有机层用acoet(2x5ml)萃取,将有机层干燥并在真空中蒸发,以提供呈固体的2-(6-乙基-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸i-1100(130mg,产率62%)。
[0705]
2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)乙酸(i-39)的合成
[0706][0707]
将氢氧化锂[1310-65-2](45mg,1.88mmol)添加到乙基2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)乙酸酯(i-21)(163mg,0.54mmol)在thf(2ml)和水(0.5ml)中的搅拌溶液中。将混合物在室温搅拌2h。将溶剂在真空中部分除去,将残余物用水稀释并用1n hcl水溶液酸化直至ph=5。将形成的固体过滤并在真空下在50℃干燥2h,以产生呈白色固体的2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)乙酸(i-39)(116mg,78%),将其不经进一步纯化而用于下一步骤。
[0708]
使用相同的程序合成结构类似物。
[0709]
[0710]
[0711]
[0712][0713]
2-(4-乙基-6-溴-1-氧代-酞嗪-2-基)乙酸(i-44)的合成
[0714][0715]
将1n naoh水溶液[1310-73-2](2ml,2mmol)添加到乙基2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-31)(200mg,0.59mmol)在meoh(3ml)中的搅拌溶液中。将混合物在70℃搅拌1.5h。将混合物用1n hcl水溶液酸化直至ph=1,然后用dcm萃取。将有机层分离,干燥(na2so4),过滤并将溶剂在真空中蒸发,以产生呈白色固体的2-(4-乙基-6-溴-1-氧代-酞嗪-2-基)乙酸(i-44)(150mg,82%),将其不经进一步纯化而用于下一步骤。
[0716]
注意:该反应大多数时候在室温进行2小时。
[0717]
使用相同的程序合成结构类似物。
[0718]
[0719]
[0720]
[0721]
[0722][0723]
2-(4-乙酰基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸i-61的合成
[0724][0725]
将1m naoh水溶液[1310-73-2](4.3ml,4.3mmol)逐滴添加到乙基2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯i-80(797mg,2.15mmol)在meoh(14.2ml)中的搅拌溶液中并将混合物在室温搅拌16小时。将混合物用hcl水溶液[7647-01-0]酸化至ph 3-4。然后将其用etoac萃取并将有机萃取物经mgso4干燥,过滤并在真空中浓缩。将粗固体溶解于1,4-二噁烷(20ml)中并在10℃逐滴添加6m hcl水溶液[7647-01-0](1.79ml,10.8mmol)。添加后,将混合物在室温搅拌1小时。将其用etoac萃取并将有机萃取
物经mgso4干燥,过滤并在真空中浓缩,以得到呈棕色固体的i-61(652mg,96%)。
[0726][0727]
n-(2,3-二氢-1h-茚-4-基)新戊酰胺i-1122的合成
[0728][0729]
在0℃,将新戊酰氯[3282-30-2](1.85ml,0.98g/ml,15.02mmol)逐滴添加到2,3-二氢-1h-茚-4-胺[32202-61-2](2g,15.02mmol)在dcm(25ml)和三乙胺[121-44-8](3.14ml,0,73g/ml,22.52mmol)中的搅拌溶液中。然后将反应在室温搅拌1h。将水添加到混合物中并将其用dcm萃取(x3)。将有机层分离,经无水mgso4干燥,过滤并在真空中浓缩。将产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至80/0)纯化,以产生呈白色固体的n-(2,3-二氢-1h-茚-4-基)新戊酰胺i-1122(3g,91%)。
[0730]
n-(5-溴-2,3-二氢-1h-茚-4-基)新戊酰胺i-1123的合成
[0731][0732]
将4-甲基苯磺酸[104-15-4](1.19g,6.9mmol)、乙酸钯(ii)[3375-31-3](155mg,0.69mmol)和nbs[128-08-5](2.21g,12.42mmol)添加到n-(2,3-二氢-1h-茚-4-基)新戊酰胺i-1122(3g,13.81mmol)在甲苯(23ml)中的搅拌溶液中。将混合物在室温搅拌16h。添加水并将混合物用dcm萃取(x3)。将有机层分离,经无水mgso4干燥,过滤并在真空中浓缩。将产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至80/0)干燥。收集所希望的级分并在真空中浓缩,以产生呈白色固体的n-(5-溴-2,3-二氢-1h-茚-4-基)新戊酰胺i-1123(4.7g,98%)。
[0733]
5-溴-2,3-二氢-1h-茚-4-胺i-1124的合成
[0734][0735]
将n-(5-溴-2,3-二氢-1h-茚-4-基)新戊酰胺i-1123(3.47g,9.37mmol)溶解于乙
醇(22ml)中并在室温搅拌。然后将硫酸[7664-93-9](22.05ml,390.81mmol)缓慢添加到水(22ml)中并将该混合物添加到反应混合物中。将混合物在100℃搅拌过周末。将混合物冷却至室温,然后用2m naoh水溶液碱化。将粗混合物用二氯甲烷萃取,然后将有机层经无水mgso4干燥,过滤并在真空中浓缩,以产生呈棕色油状物的5-溴-2,3-二氢-1h-茚-4-胺i-1124(1895mg,94%)。
[0736]
5-(2-甲氧基吡啶-4-基)-2,3-二氢-1h-茚-4-胺i-1125的合成
[0737][0738]
将5-溴-2,3-二氢-1h-茚-4-胺i-1124(1895mg,7.15mmol)溶解于二噁烷(29ml)中,然后添加在水(5.7ml)中的碳酸钾[584-08-7](2.17g,15.73mmol)和(2-甲氧基吡啶-4-基)硼酸[762262-09-9](1.31g,8.58mmol)。将混合物用氮气脱气15min,然后添加pd(dppf)cl2
·
ch2cl2[95464-05-4](293mg,0.36mmol)。将反应混合物加热至80℃,持续3小时。将混合物冷却至室温并将其用acoet和水萃取。将有机相分离,经无水mgso4干燥,过滤并在真空中浓缩。将粗产物通过快速柱色谱法(二氧化硅;etoac在庚烷中0/100至80/20)纯化,以产生呈米色固体的5-(2-甲氧基吡啶-4-基)-2,3-二氢-1h-茚-4-胺i-1125(1650mg,95%)。
[0739]
1-异丙基-3-硝基-1h-吡唑i-1086的合成
[0740][0741]
将碳酸钾[584-08-7](59.9g,433.34mmol)和2-碘丙烷[75-30-9](28ml,1.7g/ml,279.46mmol)添加到3-硝基-1h-吡唑[26621-44-3](20g,176.87mmol)在乙腈(200ml)中的溶液中。将混合物在45℃搅拌24h。将溶剂在真空(80%-90%)中除去。然后,添加乙腈(31ml)和mtbe(124ml)的混合物并将反应混合物在室温搅拌30min。将反应混合物过滤并用乙腈和mtbe(2:8)洗涤。将溶剂在真空中在45℃除去并在45℃与mtbe共蒸馏。使粗产物静置12h,以产生固体晶体。将晶体用庚烷(124ml)溶解并在室温搅拌1.5h,将混合物过滤并用庚烷洗涤,干燥4.5h,以产生呈白色固体的1-异丙基-3-硝基-1h-吡唑i-1086(20.2g,73%)。
[0742]
2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(嘧啶-4-基)乙酰胺i-79的合成
[0743][0744]
将1m naoh水溶液[1310-73-2](2.33ml,2.33mmol)添加到乙基2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯(i-80)在meoh(7.7ml)中的溶液中,并
将反应混合物在室温搅拌16小时。将挥发物在真空中蒸发,以产生呈淡棕色固体的粗2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸钠(i-79),将其不经进一步纯化而使用。
[0745]
2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(嘧啶-4-基)乙酰胺i-78的合成
[0746][0747]
在室温在氮气下,将三乙胺[121-44-8](486μl,3.47mmol)添加到2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸钠(i-79)(474mg,1.16mmol)和4-氨基嘧啶[591-54-8](135mg,1.39mmol)在无水dmf(12.6ml)中的搅拌溶液中。将混合物在室温搅拌5min,然后添加t3p溶液[68957-94-8](50%wt,在etoac中,1.03ml,1.74mmol)并将混合物在室温搅拌18h。将其用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(dcm/meoh 0至1%)纯化,以产生呈黄色固体的i-78(56mg,11%)。
[0748]
2-(4-乙酰基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(嘧啶-4-基)乙酰胺i-76的合成
[0749][0750]
在0℃,将6m hcl水溶液[7647-01-0](107μl,0.64mmol)逐滴添加到2-(4-(1-乙氧基乙烯基)-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(嘧啶-4-基)乙酰胺(i-78)(54mg,0.13mmol)在1,4-二噁烷(2.9ml)中的搅拌溶液中。将混合物在室温搅拌1小时。将其用饱和nahco3水溶液稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发,以产生呈白色固体的i-76(42mg,82%),将其不经进一步纯化而使用。
[0751][0752]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺i-77的合成
[0753][0754]
将t3p溶液[68957-94-8](3.61ml,50%wt.,在etoac中,6.05mmol)添加到2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸(i-29)(950mg,3.02mmol)、在1,4-二噁烷中的0.5m nh3溶液[7664-41-7](12ml,6.05mmol)和三乙胺[121-44-8](1.68ml,0.728g/ml,12.09mmol)在12ml无水1,4-二噁烷中的搅拌混合物中。在室温24小时后,添加水和乙酸乙酯。将有机层分离,用盐水洗涤,干燥(mgso4),过滤并在减压下蒸发,以提供呈白色固体的i-77(948mg,87%),将其不经进一步纯化而使用。
[0755]
通常,将中间体用前缀“i
‑”
标记,将最终化合物按原样标示并且可以具有前缀“x
‑”
。
[0756]
最终化合物的制备
[0757]
实例a1
[0758]
2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)-n-嘧啶-4-基-乙酰胺(最终化合物2)的合成
[0759][0760]
在0℃在氮气下,将在thf中的2m异丙基氯化镁溶液[1068-55-9](0.325ml,0.65mmol)添加到乙基2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸酯(i-28)(0.1g,0.29mmol)和4-氨基嘧啶[591-54-8](31mg,0.34mmol)在无水thf(4ml)中的搅拌溶液中。将混合物在室温搅拌3h。将混合物用0℃的水稀释并用etoac萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,meoh在dcm中0/100至10/90)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)-n-嘧啶-4-基-乙酰胺(最终化合物2)(18mg,15%)。
[0761]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0762][0763]
实例a2
[0764]
n-([1,2,4]三唑并[4,3-b]哒嗪-6-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(最终化合物x2)的合成
[0765][0766]
将[1,2,4]三唑并[4,3-b]吡嗪-6-胺[19195-46-1](42.6mg,0.32mmol)放入干的装有磁力搅拌棒的mw小瓶中,并将该装置置于氮气(3个真空/氮气循环)下。添加无水dmf(0.9ml)并将溶液冷却至0℃。在0℃保持10分钟后,逐滴添加lihmds溶液(1.0m,在thf中,0.56ml,0.56mmol)并将所得溶液在0℃搅拌15分钟。然后,在0℃,逐滴添加乙基2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酸酯(i-29)(80mg,0.23mmol)在无水thf(0.76ml)中的溶液。使所得混合物经1小时从0℃升温至室温,同时剧烈搅拌,并在室温再搅拌3小时。将混合物在真空(降至60毫巴,在50℃)中浓缩。将获得的玻璃状残余物通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)纯化,以得到呈无色固体的x2(51mg,51%)。
[0767]
注意:在一些实例的合成中,使用thf和甲苯作为溶剂代替dmf,然而,dmf是通常能对难溶性胺具有更好效果的溶剂。
[0768]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0769]
[0770]
[0771]
[0772]
[0773][0774]
实例a3
[0775]
2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)-n-嘧啶-4-基-乙酰胺(最终化合物10)的合成
[0776][0777]
将4-氨基嘧啶[591-54-8](28mg,0.29mmol)添加到2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)乙酸(i-39)(56mg,0.2mmol)、1-丙烷膦酸酐[68957-94-8](0.3ml,0.47mmol)和三乙胺[121-44-8](0.1ml,0.72mmol)在无水dcm(3ml)中的搅拌溶液中。将混合物在室温搅拌4h。将混合物用饱和na2co3水溶液稀释并用dcm萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,etoac在庚烷中0/100至100/0)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的2-(4-异丙基-6-甲氧基-1-氧代-酞嗪-2-基)-n-嘧啶-4-基-乙酰胺(最终化合物10)(45mg,63%)。
[0778]
注意:dcm和dmf可以作为溶剂不加区别地用于该反应中。
[0779]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0780]
[0781]
[0782]
[0783]
[0784]
[0785]
[0786]
[0787]
[0788]
[0789]
[0790]
[0791]
[0792]
[0793]
[0794]
[0795]
[0796]
[0797]
[0798]
[0799]
[0800]
[0801]
[0802]
[0803]
[0804]
[0805]
[0806]
[0807]
[0808]
[0809]
[0810]
[0811]
[0812]
[0813]
[0814][0815]
注意:x12和x43通过x24的手性sfc分离而分离;x41通过x68的手性sfc分离而分离;x55和x106通过x89的手性sfc分离而分离;x56和x149通过x91的手性sfc分离而分离;x124和x115通过x95的手性sfc分离而分离;x122和x130通过x133的手性sfc分离而分离;x163通过x68的手性sfc分离而分离;x166通过x152的手性sfc分离而分离
[0816]
实例a4
[0817]
反式-2-(6-甲基-4-乙基-1-氧代-酞嗪-2-基)-n-(4-羟基环己基)乙酰胺(最终化合物26)的合成
[0818][0819]
将1-羟基苯并三唑[123333-53-9](84.9mg,0.72mmol)添加到2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)乙酸(i-44)(150mg,0.48mmol)和反式-4-氨基环己醇[27489-62-9]
(85.5mg,0.62mmol)在无水dcm(5ml)中的搅拌溶液中。将混合物在室温搅拌15min。然后,添加1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐[25952-53-8](120mg,0.62mmol)并将混合物在室温搅拌5h。将混合物用水稀释并用dcm萃取。将有机层分离,干燥(mgso4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,meoh在dcm中0/100至10/90)纯化。收集所希望的级分并在真空中浓缩,以产生呈白色固体的反式-2-(6-溴-4-乙基-1-氧代-酞嗪-2-基)-n-(4-羟基环己基)乙酰胺(最终化合物23)(29.2mg,15%)。
[0820]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0821][0822][0823]
实例a5
[0824]
2-(6-溴-4-(1-羟基乙基)-1-氧代酞嗪-2(1h)-基)-n-(1-异丙基-1h-吡唑-3-基)乙酰胺x-1079的合成
[0825][0826]
在0℃,将硼氢化钠[16940-66-2](4mg,0.1mmol)添加到2-(4-乙酰基-6-溴-1-氧代酞嗪-2(1h)-基)-n-(1-异丙基-1h-吡唑-3-基)乙酰胺x-1014(45mg,0.1mmol)在thf(3ml)和水(1ml)中的搅拌溶液中。将所得混合物在室温搅拌30min。添加nahco3饱和水溶液和etoac并将有机层分离,经无水mgso4干燥,过滤并将溶剂在真空中浓缩。将粗品通过快速柱色谱法(二氧化硅,dcm/meoh(9:1)/dcm从0/100至100/0)纯化。收集所希望的级分并在真空中浓缩。将产物用dipe研磨,以产生呈白色固体的2-(6-溴-4-(1-羟基乙基)-1-氧代酞嗪-2(1h)-基)-n-(1-异丙基-1h-吡唑-3-基)乙酰胺x-1079(34mg,74%)。
[0827]
使用相同的程序合成结构类似物。
[0828]
[0829][0830]
实例a6
[0831]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(9h-嘌呤-2-基)乙酰胺(最终化合物x1)的合成
[0832][0833]
在室温,将pd/c(10%wt pd,14.8mg,0.014mmol)添加到n-(6-氯-9h-嘌呤-2-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺x11(57mg,0.12mmol)和三乙胺(20μl,0.15mmol)在thf(10ml)中的搅拌溶液中。向反应容器中装入氢气(3个真空/氢气循环)并将混合物在氢气气氛下在室温搅拌5小时。将悬浮液经硅藻土(decalite)过滤,用thf充分地洗涤并将滤液在真空中浓缩。将获得的粗无色固体进一步通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)纯化,随后通过制备型sfc(固定相:chiralpak daicel ig20x250mm,流动相:co2,etoh 0.4iprnh2)纯化,以提供呈无色固体的x1(5mg,9%)。
[0834]
实例a7
[0835]
n-((3s,4r)-4-羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺盐酸盐(i-136)的合成
[0836][0837]
在100-ml rb烧瓶中,将在1,4-二噁烷中的4m hcl溶液[7647-01-0](7ml,28mmol)添加到叔丁基(3s,4r)-4-羟基-3-(2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺基)哌啶-1-甲酸酯(i-143)(936mg,1.826mmol)在1,4-二噁烷(6ml)中的悬浮液中。将反应混合物在室温搅拌1小时。将溶剂除去,以提供呈白色粉末的粗n-((3s,4r)-4-羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(i-136)(880mg,98%),将其不经进一步纯化而使用。
[0838]
使用类似的反应条件,使用适当的底物,获得另外的类似物。
[0839][0840][0841]
n-(4-羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(i-139)的合成
[0842][0843]
在氮气下,将pd/c(10%wt.pd,75mg,0.07mmol)添加到i-151(450mg,0.82mmol)在etoh(30ml)中的溶液中。将反应容器置于氢气气氛下并将反应混合物在室温搅拌16小时。将其在氮气下经硅藻土过滤并将滤液在减压下在40℃浓缩。将粗物质溶解于dcm(50ml)中并在teflon过滤器上过滤,将滤液在减压下在40℃浓缩,以提供呈白色固体的i-139(347mg,98%),将其不经进一步纯化而使用。
[0844]
n-((3s,4r)-1-乙基-4-羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(最终化合物x6)的合成
[0845][0846]
在干的mw小瓶中,将碘乙烷[75-03-6](0.1ml,1.244mmol)添加到n-((3s,4r)-4-羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺盐酸盐(i-136)(203mg,0.412mmol)和三乙胺[121-44-8](0.6ml,4.328mmol)在无水mecn(4ml)中的悬浮液中。将反应混合物在室温搅拌过夜。将粗混合物用meoh(约18ml)稀释并通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、meoh)纯化,以提供呈白色粉末的x6(149mg,82%)。
[0847]
使用类似的反应条件,使用适当的底物,获得另外的类似物。
[0848]
[0849]
[0850][0851]
实例a8
[0852]
n-((3s,4r)-1-环丙基-4-羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(最终化合物x15)的合成
[0853][0854]
在mw小瓶中在氮气下,将氰基硼氢化钠[25895-60-7](30mg,0.477mmol)添加到(i-136)(105mg,0.213mmol)、(1-乙氧基环丙氧基)三甲基硅烷[27374-25-0](45μl,0.225mmol)和乙酸(0.15ml,2.62mmol)在meoh(1.5ml)中的悬浮液中。将反应混合物在70℃搅拌过夜。将粗混合物通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液,meoh)纯化,以提供呈白色粉末的x15(68mg,71%)。
[0855]
使用类似的反应条件,使用适当的底物,获得另外的类似物。
[0856][0857]
实例a9
[0858]
n-((3s,4r)-1-环丙基-4-羟基-1-((r*)-3-羟基丁基)哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺、n-((3s,4r)-4-羟基-1-((s*)-3-羟基丁基)哌啶羟基哌啶-3-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(最终化合物x70、x81和x83)的合成
[0859][0860]
在mw小瓶中在氮气下,将氰基硼氢化钠[25895-60-7](25mg,0.398mmol)添加到(i-136)(95mg,0.193mmol)和3,3-二氟环丁酮[1273564-99-0](50mg,0.471mmol)在meoh(1.5ml)中的悬浮液中。将反应混合物在40℃搅拌过夜。将粗混合物通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、meoh)纯化,以提供呈灰白色固体的x70(8mg,9%)、呈灰白色固体的x81(11mg,12%)以及呈无色固体的x83(46mg,48%)。
[0861]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0862][0863]
实例a10
[0864]
n-([1,2,4]三唑并[4,3-a]吡嗪-6-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(x14)的合成
[0865][0866]
将tcfh[207915-99-9](268mg,0.95mmol)添加到(i-52)(150mg,0.48mmol)、[1,2,4]三唑并[4,3-a]吡嗪-6-胺[2111465-25-7](97mg,0.72mmol)和1-甲基咪唑[616-47-7](0.19ml,1.03g/ml,2.39mmol)在无水mecn(3.7ml)中的溶液中。将反应混合物在室温搅拌16小时。将粗混合物通过制备型hplc纯化(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)纯化,以提供呈浅棕褐色固体的x14(101mg,49%)。
[0867]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0868]
[0869][0870]
实例a11
[0871]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(2-(三氟甲基)咪唑并[1,2-a]吡啶-6-基)乙酰胺(x64)的合成
[0872][0873]
在干的小瓶中在氮气下,将1-氯-n,n,2-三甲基-1-丙烯胺[26189-59-3](104μl,0.78mmol)添加到羧酸(i-29)(80mg,0.25mmol)在二噁烷(2ml)中的混合物中。将混合物在室温搅拌1小时。然后添加2-(三氟甲基)咪唑并[1,2-a]吡啶-6-胺[1343040-93-6](61.5mg,0.31mmol),随后添加吡啶[110-86-1](70μl,0.98g/ml,0.87mmol)。将混合物在室温搅拌5小时。添加水并将粗产物用etoac(3x5ml)萃取,将合并的有机层干燥,过滤并在真空中蒸发。经由制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)进行纯化,以产生呈深绿色固体的酰胺x64(46mg,36%)。
[0874]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0875][0876]
实例a12
[0877]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(8-甲基-[1,2,4]三唑并[4,3-a]吡啶-6-基)乙酰胺(x28)的合成
[0878][0879]
将n-(8-溴-[1,2,4]三唑并[4,3-a]吡啶-6-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺x52(50mg,0.1mmol,1当量)和双(三-叔丁基膦)钯(0)[53199-31-8](20mg,0.039mmol,40mol%)放入干的8-ml mw小瓶中。将小瓶密封,置于氮气(3个真空/氮气循环)下并用冰浴冷却至0℃。添加无水thf(1ml),将混合物在0℃搅拌2分钟并经2min逐滴添加mezncl溶液[5158-46-3](2m,在thf中,147μl,0.29mmol,3当量)。将所得溶液在室温剧烈搅拌18小时。将粗混合物通过添加0.2m hcl水溶液(约5ml)淬灭并用etoac(4x5ml)萃取。将合并的有机层经na2so4干燥,过滤并在真空中浓缩。将获得的残余物通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)纯化,以得到呈无色固体的x28(20mg,46%)。
[0880]
实例a13
[0881]
n-(8-氰基-[1,2,4]三唑并[4,3-a]吡啶-6-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(x72)的合成
[0882][0883]
将
t
buxphos pd g3[1447963-75-8](15.2mg,19μmol)、zn(cn)2[557-21-1](27mg,0.23mmol)和n-(8-溴-[1,2,4]三唑并[4,3-a]吡啶-6-基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺x52(65mg,0.13mmol)放入干的mw小瓶中。将小瓶密封并置于氮气(3个真空/氮气循环)下,并添加thf/di水的1.4ml脱气1:2混合物。将小瓶在55℃剧烈
搅拌18h。然后对混合物进行超声处理直至观察到微细粒悬浮液,将其在60℃再加热24小时。
[0884]
将混合物在di水(10ml)和dcm(10ml)之间分配。收集有机层,并将水层重新用dcm(2x10ml)、然后用dcm/meoh 95:5(4x10ml)萃取。将合并的有机层经na2so4干燥,过滤并在真空中浓缩。将获得的残余物进一步通过制备型hplc(固定相:rp xbridge prep c18 obd-5μm,50x250mm,流动相:0.25%nh4hco3水溶液、ch3cn)纯化,随后通过制备型sfc(固定相:chiralpak daicel id 20x250mm,流动相:co2、etoh 0.4iprnh2)纯化,以得到呈浅棕褐色固体的x72(9mg,15%)。
[0885]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0886][0887]
实例a14
[0888]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-((1s,3s)-3-甲氧基-3-甲基环丁基)乙酰胺(x29)的合成
[0889][0890]
将n-((1s,3s)-3-羟基-3-甲基环丁基)-2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺x25(108mg,0.27mmol,1当量)放入20-ml小瓶中并溶解于无水dcm(10.4ml)中。将溶液在冰浴中冷却至0℃并置于氮气下。在0℃保持10min后,顺序地添加2,6-二-叔丁基-4-甲基吡啶[38222-83-2](223.2mg,1.09mmol,4当量)和三甲基氧鎓四氟硼酸盐[420-37-1](120.6mg,0.82mmol,3当量)。将小瓶用氮气冲洗并密封,将混合物在0℃搅拌5min,然后使其升温至室温并搅拌2小时。然后将混合物通过添加饱和nahco3水溶液(约5ml)淬灭并用dcm(2x8ml)萃取。将合并的有机萃取物浓缩并通过fcc(庚烷/etoac 4:1至0:1)纯化,以提供呈无色固体的标题酰胺x29(74mg,66%)。
[0891]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0892][0893][0894]
实例a15
[0895]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(3-甲基-3h-咪唑并[4,5-b]吡啶-5-基)乙酰胺(x47)的合成
[0896][0897]
在微波容器中,将2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)乙酰胺(i-77)(20mg,0.119mmol)、5-氯-3-甲基-3h-咪唑并[4,5-b]吡啶(i-135)(50mg,0.16mmol)、k2co3(36mg,0.26mmol)、cui(1.2mg,0.0063mmol)和反式-n,n'-二甲基环己烷-1,2-二胺[67579-81-1](1.2mg,8.4μmol)悬浮于2ml的无水1,4-二噁烷中。将所得混合物用氮气脱气5min,然后在密闭容器中在170℃加热18h。将混合物经ptfe过滤器过滤,用meoh洗涤并通过制备型hplc(固定相:rp xbridge prep c18 obd-10μm,50x250mm,流动相:0.25%nh4hco3水
溶液、meoh)纯化,以提供呈白色固体的x47(9mg,17%)。
[0898]
使用类似的反应条件,使用适当的试剂,获得另外的类似物。
[0899][0900]
实例a16
[0901]
2-(4-异丙基-1-氧代-6-(三氟甲基)酞嗪-2(1h)-基)-n-(2-(三氟甲基)咪唑并[1,2-a]吡啶-6-基)乙酰胺(x109)的合成
[0902][0903]
在氮气下,对羧酸(i-29)(100mg,0.32mmol)和1-甲基-1h-吡唑并[3,4-b]吡啶-6-胺(i-131)(88.1mg,0.48mmol)在无水吡啶[110-86-1](8ml)中的混合物进行超声处理10min,然后在室温搅拌40min。在室温,逐滴添加在dcm中的1m氯化钛溶液(iv)[7550-45-0](1.27ml,1.27mmol)。将混合物在室温搅拌1小时,然后在80℃加热30小时。将溶剂在真空中蒸发并将粗混合物用1m hcl水溶液处理直至ph《7。将粗产物用acoet萃取,将合并的有机层经mgso4干燥,过滤并在真空中蒸发。将该粗化合物从15ml热乙腈中重结晶,以产生呈无色固体的x109(90mg,64%)。
[0904]
实例a17
[0905]
叔丁基(3s,4r)-3-(2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酰胺基)-4-羟基哌啶-1-甲酸酯i-1132的合成
[0906]
[0907]
在室温,将hatu[148893-10-1](1.17g,3.08mmol)添加到2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酸i-1131(500mg,1.54mmol)在dmf(3.5ml)中的搅拌溶液中,随后添加叔丁基(3s,4r)-3-氨基-4-羟基哌啶-1-甲酸酯[1820579-78-9](379.54mg,1.75mmol)和dipea[7087-68-5](2.65ml,0.75g/ml,15.38mmol)。将混合物在室温搅拌18h。添加水,并将反应混合物再搅拌30分钟,然后将白色固体过滤并用水洗涤。将固体在烘箱中在50℃干燥过夜,以获得呈白色固体的叔丁基(3s,4r)-3-(2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)乙酰胺基)-4-羟基哌啶-1-甲酸酯i-1132(570mg,产率67%)。
[0908][0909]
2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)-n-((1r,3s)-3-乙基-3-羟基环丁基)乙酰胺x-1090的合成
[0910][0911]
在室温,向2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)-n-((1r,3s)-3-((叔丁基二甲基甲硅烷基)氧基)-3-乙基环丁基)乙酰胺x-1089(120mg,0.2236mmol)在dcm(5ml)中的混合物中添加tfa[76-05-1](0.1711ml,1.49g/ml,2.2364mmol)。将混合物搅拌16h。将粗品在真空中蒸发,用饱和na2co3稀释并将混合物在室温搅拌30min。然后,将混合物用dcm萃取,将有机层分离,并将水相进一步用另外的dcm萃取(2x)。将合并的有机层干燥(na2so4),过滤并将溶剂在真空中蒸发。将粗产物通过快速柱色谱法(二氧化硅,meoh在dcm中0/100至3/97)纯化。收集所希望的级分,将溶剂在真空中蒸发,以产生呈白色固体的2-(6-溴-4-异丙基-1-氧代酞嗪-2(1h)-基)-n-((1r,3s)-3-乙基-3-羟基环丁基)乙酰胺x-1090(57mg,产率60%)。
[0912]
表征数据-lc-ms和熔点
[0913]
lcms:[m h]
意指化合物的游离碱的质子化质量,r
t
意指保留时间(以分钟计),方法是指用于lcms的方法。
[0914]
[0915]
[0916]
[0917]
[0918]
[0919]
[0920]
[0921][0922]
表征数据-化合物 nmr
[0923]
这描绘于下表中:
[0924]
[0925]
[0926]
[0927]
[0928]
[0929]
[0930]
[0931]
[0932]
[0933]
[0934]
[0935]
[0936]
[0937]
[0938]
[0939]
[0940]
[0941]
[0942]
[0943]
[0944]
[0945]
[0946]
[0947]
[0948][0949]
实例b-药物组合物
[0950]
将本发明的化合物(例如,实例的化合物)与药学上可接受的载体缔合,从而提供包含这样的活性化合物的药物组合物。在制备药物组合物的过程中,将治疗有效量的本发明的化合物(例如,实例的化合物)与药学上可接受的载体紧密混合。
[0951]
实例c-生物学实例
[0952]
根据本发明的化合物的活性可以通过体外方法进行评估。本发明的化合物表现出有价值的药理学特性,例如对抑制nlrp3活性敏感的特性,例如如以下测试所示,因此适用于与nlrp3炎症小体活性相关的疗法。
[0953]
pbmc测定
[0954]
从健康个体收集外周静脉血,并通过ficoll-histopaque(西格玛奥德里奇公司(sigma-aldrich),a0561)密度梯度离心从血液中分离人外周血单核细胞(pbmc)。分离后,将pbmc储存在液氮中以备后用。解冻后,在生长培养基(补充有10%胎牛血清、1%pen-strep和1%l-谷氨酰胺的rpmi培养基)中测定pbmc细胞活力。在dmso中以1:3的系列稀释度点样化合物,并在96孔板(falcon,353072)中的30μl培养基中稀释至最终浓度。将pbmc以
7.5
×
104个细胞/孔的密度添加,并在5%co2培养箱中于37℃孵育30min。lps刺激是通过添加100ng/ml lps(最终浓度,英杰公司(invivogen),tlrl-smlps)进行6小时,然后收集细胞上清液并经由msd技术根据制造商的指南(msd,k151a0h)分析il-1β(μm)和tnf细胞因子水平(μm)。
[0955]
ic
50
值(对于il-1β)和ec
50
值(tnf)是在本发明/实例的化合物上获得的,并且描述于下表:
[0956]
[0957]
[0958]
[0959]
[0960]
[0961]
[0962]
[0963]
[0964]
[0965]
[0966]
[0967]
[0968]
[0969]
[0970]
[0971]
[0972]
[0973]
[0974]
[0975]
[0976]
[0977]
[0978]
[0979]
[0980]
[0981]
[0982]
[0983]
[0984]
[0985]
[0986]
[0987]
[0988]
[0989]
[0990]
[0991]
[0992]
[0993]
[0994]
[0995][0996]
实例d-进一步测试
[0997]
本发明的一种或多种化合物(包括最终实例的化合物)在许多其他方法中进行测试,以评估渗透性、稳定性(包括代谢稳定性和血液稳定性)和溶解度等其他特性。
[0998]
渗透性测试
[0999]
体外被动渗透性和作为p-糖蛋白(p-gp)转运底物的能力使用mdr1稳定转导的mdck细胞进行测试(这可以在提供adme、pk服务的商业组织中进行,例如cyprotex公司)。渗透性实验以单一浓度(5μm)在transwell系统中一式两份进行,孵育120min。测量存在和不存在p-gp抑制剂gf120918时的顶端到基底外侧(a至b)转运和不存在p-gp抑制剂时的基底外侧到顶端(b至a)转运并计算测试化合物的渗透速率(表观渗透性)(p
app x10-6
cm/sec)。
[1000]
肝微粒体代谢稳定性测试
[1001]
通过使用来自与1μm测试化合物在37℃孵育长达60分钟的人类和临床前物种的肝微粒体(0.5mg/ml蛋白质)来对测试化合物的代谢稳定性进行测试(这可以在提供adme、pk服务的商业组织中进行,例如cyprotex公司)。
[1002]
体外代谢半衰期(t
1/2
)使用来自母体化合物剩余百分比相对于时间关系(κ)的对
数线性回归的斜率来计算,
[1003]
t
1/2
=-ln(2)/κ。
[1004]
体外内在清除率(cl
int
)(ml/min/mg微粒体蛋白)使用以下公式计算:
[1005][1006]
其中:v
inc
=孵育体积,
[1007]wmicprot,inc
=孵育中微粒体蛋白的重量。
[1008]
肝脏肝细胞代谢稳定性测试
[1009]
使用来自与1μm测试化合物在37℃孵育长达120分钟的人类和临床前物种的肝脏肝细胞(1milj细胞)来对测试化合物的代谢稳定性进行测试。
[1010]
体外代谢半衰期(t
1/2
)使用来自母体化合物剩余百分比相对于时间关系(κ)的对数线性回归的斜率来计算,
[1011]
t
1/2
=-ln(2)/κ。
[1012]
体外内在清除率(cl
int
)(μl/min/百万细胞)使用以下公式计算:
[1013][1014]
其中:v
inc
=孵育体积,
[1015]
#细胞
inc
=孵育中的细胞数(x106)
[1016]
溶解度测试
[1017]
测试/测定一式三份进行,并使用tecan fluent对所有液体处理进行半自动化,通用步骤如下:
[1018]-将20μl 10mm原液分配到500μl 96孔板中
[1019]-蒸发dmso(genevac公司)
[1020]-添加搅拌棒和400μl的缓冲液/生物相关介质。
[1021]-将溶液搅拌72h(ph 2和ph 7)或者24h(fassif和fessif)
[1022]-过滤溶液
[1023]-并使用三点校准曲线通过uplc/uv对滤液进行定量
[1024]
lc条件是:
[1025]-waters acquity uplc
[1026]-流动相a:在h2o中的0.1%甲酸,b:在ch3cn中的0.1%甲酸
[1027]-柱:waters hss t3 1.8μm 2.1x50mm
[1028]-柱温:55℃
[1029]-注射体积:2μl
[1030]-流速:0.6ml/min
[1031]-uv波长:250_350nm
[1032]-梯度:0min:0%b,0.3min:5%b,1.8min:95%b,2.6min:95%b
[1033]
血液稳定性测定
[1034]
本发明/实例的化合物以一定浓度掺入来自同意的临床前物种的血浆或血液中;然后在孵育至预定时间和条件(37℃、0℃(冰)或室温)后,可以用lcms/ms确定血液或血浆
基质中的测试化合物浓度。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。