一种防晒剂a-plus的中控及成品分析方法
技术领域
1.本发明属于化学品分析检测技术领域,具体涉及防晒剂a-plus中控及成品分析方法。
背景技术:
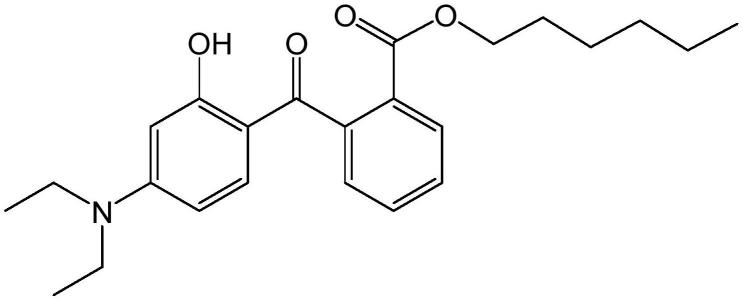
2.二乙氨基羟苯甲酰基苯甲酸己酯(uvinul-a plus,简称a-plus),是一种能吸收uva的有机化合物,为basf公司开发出的一种新型uva(长波紫外线,uva包含uva1(340~400nm)和uva2(320~340nm)两个波段)吸收剂,并以uvinul a plus为商品名上市,其结构式如下所示:
[0003][0004]
a-plus吸收紫外线高达400nm,其中对于波长354nm的紫外线吸收率最大。a-plus的紫外吸收波段和传统的丁基甲氧基二苯甲酰甲烷(avb)相近,但是更注重于uva1波段(340~400nm)的防护,而且光化学稳定性好,与其它油脂的复配性佳,可作为防晒剂在化妆品中被广泛使用。该产品具有可靠的长效的防老化,皱纹以及皮肤癌效果,也易于和其他的有机以及无机紫外过滤剂结合使用,用于防晒以及其他化妆品中。
[0005]
目前,美国食品和药物管理局(fda)、欧盟经济委员会(eec)和我国卫生部规定其在化妆品中的最大添加限量(质量分数)为10%。巴斯夫(basf)公司总结出a-plus在防晒产品中所扮演的功能性包括:对uva1有高的吸收效果;对紫外线产生的自由基有很强的防护效果;可以提升uvb防晒成分的spf值;很好的光稳定性,可以长时间维持效能。
[0006]
由于a-plus合成过程中反应复杂,其中的副产物直接影响测定结构的准确性,检测时间长,严重影响了a-plus的生产效率,因此,合成a-plus的中控分析很关键,产品品质中含量测定也很关键。
[0007]
目前对于a-plus的制备,basf采用gc面积百分比法测定其成品品质,方法重现性及稳定性不好。
[0008]
因此,亟需研究一种快速、准确的a-plus中控及成品的检测,对a-plus的生产和工艺的开发均具有重要的意义。
技术实现要素:
[0009]
为了克服上述问题,本发明人对a-plus的制备及其中控和成品品质分析方法进行
了锐意研究,研究出一种防晒剂a-plus的中控及成品分析方法。以3-二乙氨基苯酚和邻苯二甲酸酐为原料,经偶合反应制得中间体4-二乙氨基酮酸,4-二乙氨基酮酸再与正己醇发生酯化反应制备a-plus。其中,在制得a-plus成品之前,包括偶合反应阶段、酯化反应阶段、酯化后的析晶、重结晶阶段之一或多个阶段所获取的样品或对制得的a-plus用高效液相色谱法分析反应进行的程度或a-plus纯度,不仅实现了a-plus生产过程中的在线快速检测,还能检测制得的a-plus的品质。本发明提供的a-plus的中控和成品品质分析方法精密度高,重现性好,可以避免使用气相色谱法导致的重现性不好的问题,从而完成了本发明。
[0010]
具体来说,本发明的目的在于提供以下方面:
[0011]
一方面,提供一种防晒剂a-plus的制备方法,所述方法包括:以3-二乙氨基苯酚和邻苯二甲酸酐为原料,经偶合反应制备中间体4-二乙氨基酮酸,4-二乙氨基酮酸再与正己醇进行酯化,得到酯化反应混合物,经纯化获得防晒剂a-plus。
[0012]
另一方面,提供一种防晒剂a-plus中控及成品分析方法,所述方法是在第一方面所述的a-plus的制备方法中进行中控及成品分析,优选地,分别取制得a-plus成品之前,包括偶合反应阶段、酯化反应阶段、酯化后的析晶、酯化后的重结晶阶段之一或多个阶段所获得的样品和防晒剂a-plus作为待测样品,测定反应进行的程度或a-plus纯度。
[0013]
本发明所具有的有益效果包括:
[0014]
(1)本发明提供的防晒剂a-plus的制备方法,制得的a-plus收率达到92%以上。
[0015]
(2)本发明提供的防晒剂a-plus中控及成品分析方法,精密度高,重现性好,解决了使用气相色谱法导致的重现性不好的问题。
[0016]
(3)本发明提供的防晒剂a-plus中控及成品分析方法,不仅实现了a-plus生产过程中的在线快速检测,全面准确反映中控待测液中含有的杂质含量,检测反应进行的程度或为制得的a-plus的品质提供指标,还能直接检测制得的a-plus的品质,适合批量、快速分析,实用性非常强。
附图说明
[0017]
图1示出实施例1中hplc谱图;
[0018]
图2示出实施例2中dhba中控待测液液相色谱图;
[0019]
图3示出实施例2中a-plus中控待测液液相色谱图;
[0020]
图4示出实施例4中的线性标准曲线图;
[0021]
图5示出对比例1中a-plus成品待测液液相色谱图。
具体实施方式
[0022]
下面通过附图和实施例对本发明进一步详细说明。通过这些说明,本发明的特点和优点将变得更为清楚明确。
[0023]
在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。尽管在附图中示出了实施例的各种方面,但是除非特别指出,不必按比例绘制附图。
[0024]
一方面,根据本发明提供的一种防晒剂a-plus的制备方法,所述方法包括:以3-二乙氨基苯酚和邻苯二甲酸酐为原料,经偶合反应制备中间体4-二乙氨基酮酸,4-二乙氨基
酮酸再与正己醇进行酯化,经纯化获得防晒剂a-plus。
[0025]
进一步地,所述偶合反应的原料之一为3-二乙氨基苯酚其cas号为91-68-9,也称为间二乙氨基苯酚、间二乙氨苯酚或间羟基-n,n-二乙基苯胺,其和另一原料邻苯二甲酸酐以芳香烃化合物为反应溶剂,所述芳香烃化合物优选选自苯甲腈、甲苯、乙苯、邻二甲苯、间二甲苯、对二甲苯、氯苯中的一种或多种,更优选为甲苯。
[0026]
根据本发明,所述芳香烃化合物既作为惰性溶剂不与偶合反应原料之间反应,又能保证反应体系呈均相,有利于反应匀速进行,反应产生的杂质含量低。
[0027]
根据本发明,所述3-二乙氨基苯酚、邻苯二甲酸酐与芳香烃化合物之间的摩尔量之比为1:(1.01~2):(1.5~5.0),优选为1:(1.05~1.5):(2.0~4.0),更优选为1:(1.1~1.3):(2.5~2.6)。
[0028]
在本发明中,为了使反应彻底,选择一种原料过量,由于邻苯二甲酸酐存在分解的情况,优选邻苯二甲酸酐稍过量;同时,反应使用的芳香烃化合物用量不能太多,避免增加后处理的负担,但也不能过少,会导致偶合反应的原料无法完全溶解于反应溶剂中,导致反应不均匀。
[0029]
进一步地,所述反应在惰性气体中进行,有利于反应持续稳定的发生。所述惰性气体包括稀有气体或氮气,优选廉价易得的氮气。
[0030]
根据本发明,偶合反应控制的反应温度为90~130℃,优选为100~120℃,更优选为110~116℃。
[0031]
其中,所述反应温度过高,反应过于剧烈,还会引发更多的副反应;反应温度过低,反应时间延长,也可能导致副反应的发生,在上述反应温度范围内,副反应少,且反应稳定,得到的4-二乙氨基酮酸的选择性较高。
[0032]
在本发明中,偶合反应控制的反应时间为10~20h,优选为12~18h,例如15~16h。
[0033]
根据本发明,反应时间过短,反应不完全;反应时间过长,不仅不会提高4-二乙氨基酮酸的选择性,反而可能导致副反应的发生,从经济角度出发,控制偶合的反应时间为10~20h较为合适。
[0034]
任选地,反应结束后,将上述反应的产物经析晶、重结晶得到后续酯化反应所需的中间体4-二乙氨基酮酸。
[0035]
根据本发明,偶合反应制备中间体4-二乙氨基酮酸发生的反应如式(1)所示:
[0036][0037]
进一步地,中间体4-二乙氨基酮酸与正己醇发生酯化反应使用的溶剂优选选自浓硫酸、四氢呋喃、二氯乙烷、甲苯中的任意一种或几种,更优选为浓硫酸。
[0038]
在本发明中,浓硫酸参与反应既能作为催化剂,又能作为脱水剂促使反应进行。
[0039]
根据本发明,所述4-二乙氨基酮酸、正己醇与溶剂之间的重量比为(1~6):(8~15):1,优选为(2~5):(10~13):1,更优选为(3.6~4.0):(12~12.5):1。
[0040]
其中,使用过量廉价的正己醇以提高昂贵的4-二乙氨基酮酸的利用率,且减少a-plus产物的分解。
[0041]
在本发明中,酯化反应温度为100~130℃,优选为105~
[0042]
120℃,更优选为108~110℃。
[0043]
进一步地,通过控制酯化反应真空度,使反应高效、稳定进行,所述真空度为-2~2mpa,优选为-1~1mpa,例如-0.086~0.094mpa。
[0044]
根据本发明,酯化反应至4-二乙氨基酮酸≤0.5%(hplc)时停止反应,时间约为6h。
[0045]
反应结束后,将酯化反应产物经水洗、析晶、重结晶的纯化步骤得到a-plus成品。
[0046]
根据本发明,酯化反应制备a-plus发生的反应如式(2)所示:
[0047][0048]
另一方面,根据本发明提供的一种防晒剂a-plus中控及成品分析方法,所述方法是在第一方面所述的a-plus的制备方法中进行中控及成品分析,优选地,分别取制得a-plus成品之前,包括偶合反应阶段、酯化反应阶段、酯化后的析晶、重结晶阶段之一或多个阶段所获得的样品和防晒剂a-plus作为待测样品,测定反应进行的程度或a-plus纯度。
[0049]
进一步地,所述方法采用高效液相色谱法进行纯度测定,包括以下步骤:
[0050]
步骤1,分别配制待测样品溶液及标准样品溶液。
[0051]
在步骤1中,所述待测样品溶液可以是制得a-plus成品之前,包括偶合反应阶段、酯化反应阶段、酯化后的析晶、重结晶阶段之一或多个阶段所获得的样品经配制得到的中控待测液,也可以是合成反应最终得到的a-plus经配制得到的a-plus成品待测液。
[0052]
其中,所述a-plus以3-二乙氨基苯酚和邻苯二甲酸酐为原料,经偶合反应制得中间体4-二乙氨基酮酸,4-二乙氨基酮酸再与正己醇发生酯化反应经水洗、析晶、重结晶制得所述a-plus。
[0053]
在本发明中,当待测样品溶液为中控待测液时,在偶合反应阶段、酯化反应阶段、酯化后的析晶阶段或重结晶阶段取样,经配制得到;当待测样品为a-plus成品待测液时,则将上述反应制得的a-plus成品经配制得到。
[0054]
进一步地,以偶合反应阶段的样品配制中控待测液时,由于此过程4-二乙氨基酮酸慢慢以固体形式析出,形成固液混合样品,需将其中的固体溶解,并保证溶解后的中控样品呈均一相,避免对中控分析造成影响。
[0055]
本发明人经过大量实验,研究发现,有且仅有二甲基亚砜是溶解样品中的固体以及不溶物的最佳选择,虽然n-n-二甲基甲酰胺等溶剂也能产生类似的效果,但有且仅有二
甲基亚砜不仅能将a-plus的偶合反应阶段中控样品中的固体以及不溶物完全溶解,使中控样品呈均一相,最重要的是在测定过程中,不会产生干扰。
[0056]
其中,所述二甲基亚砜的使用量不能太少,否则不能将中控样酯化反应混合物中的杂质完全溶解,当然其使用量也不能太多,避免对中控分析造成影响,具体体现在:色谱分析时,由于二甲基亚砜在液相上会出峰,且与偶合反应原料3-二乙氨基苯酚出峰时间较为接近,二甲基亚砜峰用量大会导致二甲基亚砜峰太大,进而干扰3-二乙氨基苯酚的出峰。
[0057]
优选地,所述二甲基亚砜与偶合反应阶段样品,也就是与3-二乙氨基苯酚和邻苯二甲酸酐反应阶段取的偶合反应混合物的体积比为1:(0.5~2),更优选地,为1:(0.8~1.2),例如1:1。
[0058]
根据本发明,以偶合反应阶段的样品配制中控待测液时,能直接反应偶合反应进行的程度,便于判断进行下一阶段,即酯化反应的可行性,当测得偶合反应阶段的样品中杂质间二乙氨基酚含量≤0.5%(hplc)时,结束偶合反应。
[0059]
其中,当中控待测液为酯化反应阶段、酯化后的析晶阶段或酯化后的重结晶阶段样品时,将所述样品用有机溶剂ⅰ溶解完全得到。
[0060]
进一步地,所述有机溶剂ⅰ优选选自乙腈、n-n-二甲基甲酰胺、甲醇、二甲基亚砜中的任意一种或几种,更优选为乙腈。
[0061]
根据本发明,所述乙腈与酯化反应阶段、酯化后的析晶阶段或酯化后的重结晶阶段样品的流动相相关,不会对检测产生影响。
[0062]
其中,以酯化反应阶段的样品配制中控待测液时,能反映酯化反应进行的程度,判断反应是否达到结束的状态,当酯化反应至4-二乙氨基酮酸≤0.5%(hplc)时,表明可以终止反应;以酯化后的析晶阶段或酯化后的重结晶阶段样品配制中控待测液时,初步判断制得的a-plus成品的品质和纯度。
[0063]
根据本发明,当待测样品为a-plus成品待测液时,将a-plus成品用有机溶剂ⅱ溶解完全,所得a-plus成品待测液中a-plus的浓度为0.3~1.5mg/ml,所述有机溶剂ⅱ的选择优选与配制偶合反应阶段、酯化反应阶段、酯化后的析晶阶段或重结晶阶段使用的有机溶剂ⅰ相同,例如乙腈。
[0064]
其中,当待测样品为a-plus成品待测液时,直接反映制得的a-plus成品的纯度和品质是否满足市场要求。
[0065]
根据本发明,将a-plus标准样品溶解于有机溶剂ⅲ中得到a-plus标准溶液,所述有机溶剂ⅲ的选择优选与溶解a-plus成品使用的有机溶剂ⅱ相同,例如乙腈。
[0066]
进一步地,所述a-plus标准样品的浓度为1mg/ml。
[0067]
步骤2,将步骤1配制的待测样品溶液或标准样品溶液注入高效液相色谱仪,进行色谱分析。
[0068]
步骤2中,色谱分析使用的高效液相色谱仪为安捷伦1260高效液相色谱仪或性能相当的仪器如安捷伦1290高效液相色谱仪,waters h-class等。
[0069]
进一步地,色谱柱填料为十八烷基硅烷键合硅胶,粒径为2~6μm,优选为3~5μm,柱规格为2.0~4.6mm
×
150~300mm,例如4.6mm
×
250mm。
[0070]
根据本发明,色谱的分析时,控制色谱柱温度为25~35℃,例如30℃;进样量为1~20μl,优选为5~12μl,例如10μl;检测波长为225nm。
[0071]
其中,色谱的分析时,以乙腈、蒸馏水、甲醇、磷酸中的任意一种或几种组成流动相,优选以乙腈、蒸馏水和磷酸组成流动相,例如以乙腈和浓度为0.1%磷酸组成流动相。
[0072]
根据本发明,乙腈和浓度为0.1%磷酸组成流动相,流速为0.5~5ml/min,优选为1~2ml/min,乙腈和浓度为0.1%磷酸的体积比为(20~85):(15~80)。
[0073]
进一步地,按如下方式进行梯度洗脱:
[0074][0075]
在本发明中,当检测的中控待测样为偶合反应阶段的样品时,将步骤1中配制的中控待测液用有机溶剂如乙腈稀释至控制液相色谱分析时的主峰峰高为1500~2500,否则会导致主峰出现过载的情况,各物质纯度偏差较大。
[0076]
其中,检测样品为制得a-plus成品之前,包括偶合反应阶段、酯化反应阶段、酯化后的析晶、重结晶阶段的样品配制的得到的中控待测液时,能全面准确反映中控待测液中含有的杂质含量,检测反应进行的程度或为制得的a-plus的纯度或品质提供指标;检测样品为a-plus成品待测液时,直接反映制得的a-plus的纯度或品质。
[0077]
实施例
[0078]
以下通过具体实例进一步描述本发明,不过这些实例仅仅是范例性的,并不对本发明的保护范围构成任何限制。
[0079]
实施例1
[0080]
a-plus的中控样品及a-plus成品的制备
[0081]
氮气保护下,于1l的四口烧瓶中加入165.2g 3-n,n-二乙氨基间苯酚、235.8g甲苯和177.7g邻苯二甲酸酐,之后,调节反应升温至110~116℃,保温反应15h时,取中控样品(记为:dhba中控样品),之后经析晶、重结晶,制得4-二乙氨基酮酸;
[0082]
取上述制得的4-二乙氨基酮酸164.88g,置于另一个1l的四口烧瓶中,并在其中加入508.68g正己醇和41.65g浓硫酸,之后控制水泵真空度为-0.086~0.094mpa,调节反应升温至108~110℃,回流分水,反应6h时,取中控样品(记为a-plus中控样品),此时4-二乙氨基酮酸≤0.5%(hplc),之后经水洗、析晶、重结晶得到a-plus(dhhb),其收率为92.75%,其hplc谱图如图1所示,纯度为99.69%。
[0083]
实施例2
[0084]
中控待测液的分析
[0085]
(1)确定高效液相色谱条件
[0086]
仪器:安捷伦1260高效液相色谱仪或性能相当的仪器
[0087]
色谱柱:型号c18(色谱柱填料:十八烷基硅烷键合硅胶),粒径:5um柱规格:4.6mm
×
250mm)
[0088]
柱温:30℃
[0089]
进样量:10μl
[0090]
检测波长:225nm
[0091]
流速:1ml/min
[0092]
流动相洗脱梯度:
[0093][0094]
(2)中控待测液的配制
[0095]
用二甲基亚砜以1:1(v:v)比例将实施例1获得的dhba中控样品中的固体或杂质全部溶解,取1滴用乙腈稀释至10ml,得到dhba中控待测液;
[0096]
将实施例1获得的a-plus中控样品用乙腈溶解并稀释定容,得到a-plus中控待测液。
[0097]
(3)色谱分析
[0098]
按上述色谱条件,将dhba中控待测液注入色谱仪,记录色谱图,所得液相色谱图如图2所示,图中可以看出,在3.58min时被检测出dhba中控待测液中含有3-二乙氨基苯酚,其含量为0.44%,说明偶合反应能够结束,其中dhba出峰时间为18.16min;
[0099]
同理,按上述色谱条件,将a-plus中控待测液注入色谱仪,记录色谱图,所得液相色谱图如图3所示,图中可以看出,dhba含量为0.25%,说明酯化反应能够结束,a-plus出峰时间为27.7min,纯度达到98.3%。
[0100]
实施例3
[0101]
a-plus成品待测液的重复性实验及分析
[0102]
(1)确定高效液相色谱条件
[0103]
实施例3中的高效液相色谱条件与实施例2中的高效液相色谱条件相同。
[0104]
(2)a-plus成品待测液的配制
[0105]
称取50mg实施例1制得的a-plus成品于50ml容量瓶中,用乙腈溶解稀释定容,得到浓度为1mg/ml的a-plus成品待测液。
[0106]
(3)色谱分析
[0107]
按上述色谱条件,将a-plus成品待测液进行6次平行实验,所得峰面积(分别用a1、a2、a3、a4、a5、a6)及其相对标准偏差(rsd)结果如表1所示。
[0108]
表1:
[0109][0110]
由表1可知,进样6次,所得峰面积的rsd值小于2.0%,所述检测a-plus成品的高效液相色谱方法具有良好的重复性。
[0111]
实施例4
[0112]
a-plus成品待测液的线性实验及成品含量测定
[0113]
(1)确定高效液相色谱条件
[0114]
实施例4中的高效液相色谱条件与实施例2中的高效液相色谱条件相同。
[0115]
(2)a-plus标准溶液及a-plus成品待测液的配制
[0116]
准确称取a-plus标准样品(购自于basf公司)230mg置于50.0ml洁净的容量瓶中,用乙腈溶解稀释定容,混匀,得到a-plus标准溶液。
[0117]
分别精密量取a-plus标准溶液1.0mll、1.6ml、2.0ml、2.4ml、3.0ml于10.0ml容量瓶中,用乙腈稀释定容,混匀,分别得到相当于a-plus成品待测溶液浓度60%、80%、100%、120%、140%的a-plus标准溶液。
[0118]
称取50mg实施例1制得的a-plus成品于50ml容量瓶中,用乙腈溶解,稀释定容,得到浓度为1mg/ml的a-plus成品待测液。
[0119]
(3)色谱分析
[0120]
按上述色谱条件,将相当于a-plus成品待测溶液浓度60%、80%、100%、120%、140%的a-plus标准溶液注入色谱仪,记录色谱图中的峰面积,以浓度对峰面积建立标准曲线;所得结果如表2所示,绘制的线性标准曲线图如图4所示。
[0121]
表2:
[0122][0123]
结合表2和图4可知,实施例1制得的a-plus成品的浓度在0.4677~1.430mg/ml范围内与其峰面积成正比,线性方程为y=24841x 256.37,r2=0.9991。
[0124]
按上述色谱条件,将浓度为1mg/ml的a-plus成品待测液注入色谱仪,记录色谱图中的峰面积,代入表2中的线性方程计算,得到测定浓度,与配制的浓度的比值(1mg/ml)即为a-plus的含量,按此步骤平行2次实验(分别记为含量1和含量2),此次实验中使用的a-plus成品记为批号21dhhb002;以与实施例1相相同的方式制备2批(分别记为21dhhb003和21dhhb004)a-plus成品,之后将其分别配制成1mg/ml的a-plus成品待测液,按上述方式进行分别2次a-plus的含量测定实验,三批样品结果如表3所示。
[0125]
表3:
[0126][0127][0128]
上述结果表明,该方法进行a-plus成品含量测定,稳定可靠。
[0129]
实施例5
[0130]
a-plus成品准确度检测
[0131]
以与实施例4相同的方式配制相当于a-plus成品待测溶液浓度80%的a-plus标准溶液3份,将实施例1制得的a-plus成品溶于乙腈中,分别与上述配制的3份相当于a-plus成品待测溶液浓度80%的a-plus标准溶液混合,得到待测液3份,分别记为80%-1、80%-2、80%-3;同理,制备100%待测液三份,分别用100%-1、100%-2、100%-3表示;制备120%待测液三份,分别用120%-1、120%-2、120%-3表示。
[0132]
以与实施例2中相同的高效液相色谱条件检测上述9种样品中的本底量、测得量、加入量和回收率,相应计算回收率的平均值和回收率的相对标准偏差(rsd),所得结果如表3所示:
[0133]
表3:
[0134][0135]
其中,本底量是指待测液中含有的实施例1制得的a-plus成品的重量,测得量是指待测液中加入a-plus标准溶液后测定的a-plus的重量,加入量是指待测液中加入的a-plus标准溶液中a-plus的重量。
[0136]
由表3可知,实施例1制得的a-plus成品的回收率在98.0%~102.0%范围内,说明方法准确度高,验证了方法的可靠性,9组样品的回收率之间的rsd小于2.0%,说明回收率实验稳定可靠。
[0137]
对比例
[0138]
对比例1
[0139]
气相色谱法(gc-ms)测定a-plus成品品质
[0140]
(1)确定气相色谱条件
[0141]
仪器:安捷伦8860气相色谱仪
[0142]
色谱柱:型号hp-5(柱规格:30m
×
320um
×
0.25um)
[0143]
柱箱:初温150℃,10℃/min升至300℃,保持13min
[0144]
进样口:300℃分流比:70:1
[0145]
检测器:300℃
[0146]
流速:1ml/min
[0147]
进样量:0.6μl
[0148]
洗针溶剂:丙酮。
[0149]
(2)a-plus成品待测液的配制
[0150]
称取500mg实施例1制得的a-plus溶于10ml丙酮,得到a-plus成品待测液。
[0151]
(3)色谱分析
[0152]
按上述色谱条件,将a-plus成品待测液注入色谱仪进行测试,结果发现:主峰出峰时间为17.7min,在15.6min会出现杂质峰,其色谱图如图5所示,经分析,杂质峰处物质的分子量295,推断为a-plus分子内酯化产物,且分析时发现该杂质含量不稳定,推断为gc-ms分析过程中产生。
[0153]
以上结合优选实施方式和范例性实例对本发明进行了详细说明。不过需要声明的是,这些具体实施方式仅是对本发明的阐述性解释,并不对本发明的保护范围构成任何限制。在不超出本发明精神和保护范围的情况下,可以对本发明技术内容及其实施方式进行各种改进、等价替换或修饰,这些均落入本发明的保护范围内。本发明的保护范围以所附权利要求为准。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。