用于治疗egfr突变型nsclc的cbp/p300溴结构域抑制剂和egfr抑制剂的组合
发明领域
1.本发明属于非小细胞肺癌(nsclc)治疗领域。因此,本发明涉及用于治疗患有nsclc的患者的cbp/p300溴结构域抑制剂和egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。
2.发明背景
3.非小细胞肺癌是世界上最常见的恶性肿瘤和癌症死亡的主要原因,其5年生存率不超过5%。
4.hou等人最近报道p300通过诱导非小细胞肺癌细胞中的上皮-间质转化来促进增殖、迁移和侵袭(参见hou等人,bmc cancer(2018)18:641)。这些结果是在通过rnai下调p300的nsclc细胞系中获得的。因此,p300蛋白的表达通过核酸诱导机制下调。
5.p300(也称为ep300和kat3b)是一种具有许多不同结构域的大蛋白,其结合不同的蛋白,包括许多dna结合转录因子。环amp应答元件结合蛋白(creb)结合蛋白cbp(也称为crebbp和kat3a)是一种与p300非常相关的蛋白,鉴于它们广泛的序列同一性和功能相似性,这两种蛋白通常被称为旁系同源物。
6.cbp/p300是赖氨酸乙酰转移酶,已被证明催化乙酰基连接到组蛋白和其他蛋白质的赖氨酸侧链。已经提出cbp/p300激活转录,其中作用机制似乎在于将dna结合转录因子桥接到rna聚合酶机构上或通过帮助装配转录前起始复合物。为此目的,不同的cbp/p300结构域被认为与装配在启动子和增强子上的不同转录因子阵列相互作用,以用于不同基因的转录(参见dyson和wright,jbc vo.291,no.13,pp.6714-6722,图2)。
7.cbp/p300的多个结构域之一是溴结构域。1992年在果蝇中首次鉴定出溴结构域,并在大约10年后被描述为乙酰赖氨酸的结合模块。在人类中,有许多含溴结构域的蛋白质,根据序列和结构的相似性可分为八类。似乎所有含溴结构域的蛋白质都参与转录程序的调节。致癌基因重排表明,靶向含溴结构域的蛋白质,尤其是它们的溴结构域,可能特别有益于癌症的治疗。
8.为此,已经开发了几种候选药物,目前正在进行临床试验,其靶向所谓的“溴结构域和超末端基序”蛋白,通常称为bet蛋白,它们构成一组含溴结构域蛋白质。bet-蛋白靶向药物的实例是incb054329(incyte corporation)、abbv-075(abbvie)和i-bet762(glaxosmithkline)。也有药物选择性地靶向cbp和p300的溴结构域,它们是另一组含溴结构域蛋白质的部分。这种抑制剂包括例如ccs1477(cellcentric),其目前正在进行治疗转移性去势抵抗性前列腺癌和血液恶性肿瘤的临床研究,或ft-7051(forma therapeutics inc.),其目前正在进行治疗转移性去势抵抗性前列腺癌的研究。
9.鉴于上述hou等人的研究结果,有必要进一步阐明p300的作用,特别是其在非小细胞肺癌中的不同结构域,以便提供有效的非小细胞肺癌治疗。
10.发明目的及
技术实现要素:
11.本发明的发明人惊奇地发现,如果与egfr抑制剂联合施用,cbp/p300溴结构域抑
制剂,即选择性结合cbp/p300的溴结构域的溴结构域抑制剂,提供了对显示egfr致癌性改变的nsclc的有效治疗,而如果单独施用,cbp/p300溴结构域抑制剂不影响nsclc细胞的细胞增殖。换句话说,本发明人惊奇地发现,与两种活性物质中的任何一种单独对在egfr中显示致癌性改变的nsclc的作用相比,cbp/p300溴结构域抑制剂和egfr抑制剂的组合在治疗在egfr中显示致癌性改变的nsclc中更有效。因此,如上所述,cbp/p300溴结构域抑制剂在单独给药时没有效果(其中“没有效果”特别是指对于受试者中的靶病变或非靶病变没有recist 1.1应答标准所定义的客观应答),而egfr抑制剂在单独给药时的效果随着时间而降低,这可能是由于对egfr抑制剂产生了耐药性。
12.在第一方面,本发明涉及用于治疗患有nsclc的患者的(i)cbp/p300溴结构域抑制剂和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。第一方面也可称为用于治疗患有nsclc的患者的(i)cbp/p300溴结构域抑制剂和(ii)egfr抑制剂的组合,其中nsclc的特征在于在组合中使用的egfr抑制剂的标记的一个或多个适应症中给出的egfr突变谱,或者其中nsclc的特征在于在临床试验设置中由组合中使用的egfr抑制剂靶向的egfr突变谱。
13.在第一个方面的优选实施方案中,egfr中的致癌性改变导致egfr的过度活化。egfr的致癌性改变甚至可能导致组成型活性egfr(这意味着egfr的酶活性,即蛋白激酶活性,是组成型活性的)。
14.在第一方面的另一个优选实施方案中,egfr中的致癌性改变是由以下引起的:egfr基因外显子18或外显子19或外显子20中的缺失和/或插入;egfr基因中的激酶结构域重复;egfr基因的扩增;导致egfr中的氨基酸取代的egfr基因中至少一个碱基突变,其选自l858r、g719s、g719a、g719c、v765a、t783a、s768i、s768v、l861q、e709x、l819q、a750p及其组合;和任何前述的组合。优选地,致癌性改变是由以下引起的:egfr基因外显子19中的缺失;egfr基因外显子20中的插入;导致egfr中的氨基酸取代的egfr基因中至少一个碱基突变,其选自l858r、g719s、g719a、g719c、v765a、t783a、s768i、s768v、l861q、e709x、l819q、a750p及其组合;和任何前述的组合。还可以优选的是,致癌性改变是由以下引起的:egfr基因外显子19中的缺失;导致egfr中的氨基酸取代l858r的egfr基因中的至少一个碱基突变;和它们的组合。egfr基因外显子18中的缺失和插入特别是导致egfr中e709-t710缺失的缺失和egfr中该位置的d的插入。egfr基因外显子19中的缺失特别是导致egfr中e746-a750或l747-e749缺失的缺失。egfr外显子19中的缺失和插入特别是导致egfr中l747-a750缺失的缺失和在egfr中该位置的p的插入,或导致egfr中l747-t751缺失的缺失和在egfr中该位置的s的插入。egfr基因外显子20中的插入特别是导致氨基酸(意思是任何氨基酸或x)插入egfr中两个氨基酸之间的位置的插入,所述两个氨基酸选自d761-e762、a763-y764、y764-v765、a767-s768、s768-v769、v769-d770、d770-n771、n771-p772、p772-h773、h773-v774、v774-c775、v765-m766及其组合。最优选的是,致癌性改变是由egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变和它们的组合引起的。还非常优选的是,致癌性改变是由egfr基因外显子19中的缺失或egfr基因中的至少一个导致egfr中氨基酸取代l858r的碱基突变引起的。当本文将“x”称为氨基酸时,“x”表示任何氨基酸(但当然是在相应位置不同于野生型氨基酸的氨基酸,如果适用的话,例如e709x)。
15.在第一方面的一个实施方案中,非小细胞肺癌在egfr中没有显示额外的抗性改变。因此,用于本发明的组合将用作一线治疗,并且组合中的egfr抑制剂可以是用于(或指示用于)治疗egfr中显示致癌性改变的nsclc的任何egfr抑制剂。
16.在第一方面的另一个实施方案中,非小细胞肺癌还表现出在egfr中的抗性改变。egfr中的抗性改变尤其可由egfr基因中的至少一个碱基突变引起,该突变导致egfr中选自t790m、c797x(主要是c797s)、l792x、g796x、l718q、l718v、g724s、d761y、v834l、t854a及其组合的氨基酸取代。可以优选的是,egfr中的抗性改变是由egfr基因中的至少一个碱基突变引起的,该突变导致egfr中选自t790m、c797x(主要是c797s)、l718q、l718v、t854a及其组合的氨基酸取代。最优选的是,egfr中的抗性改变是由egfr基因中的至少一个碱基突变引起的,该突变导致egfr中t790m的氨基酸取代。当本文将“x”称为氨基酸时,“x”表示任何氨基酸(但当然是在相应位置不同于野生型氨基酸的氨基酸,如果适用的话,例如c797x)。
17.当非小细胞肺癌还表现出在egfr中的抗性改变时,患者先前用(第一种)egfr抑制剂治疗,该抑制剂最初是有效的,然后由于耐药性的发展,特别是由于egfr抗性改变的发展而变得无效。重要的是要理解,在用于本发明的组合中,这种情况下的egfr抑制剂不是先前施用的(第一种)egfr抑制剂,而是(第二种或第三种)egfr抑制剂,尽管单独施用时至少有一种抗性改变,但该抑制剂最初是治疗有效的。我们在这里称之为“初始治疗效果”,因为常见的观察结果是对这种(第二种或第三种)egfr抑制剂产生进一步的耐药性,使得这种(第二种或第三种)egfr抑制剂最终也无效。在这种情况下,用于本发明的组合将用作二线或三线治疗。举例来说,吉非替尼之前可能已经被施用于(单独作为一线治疗)患有表现出致癌性改变的nsclc的患者,随着时间的推移(通常在约10至约12个月后)吉非替尼治疗变得无效,并且发现(例如,通过活组织检查和相应的测试以检测egfr突变)在吉非替尼治疗期间肿瘤中出现egfr t790m抗性改变。在这种情况下,吉非替尼不会用于本发明的组合中,但特别是已经显示(并且表明)奥西替尼在治疗egfr t790m突变阳性非小细胞肺癌患者中有效,其疾病在egfr酪氨酸激酶抑制剂(tki)治疗过程中或治疗后发展。
18.考虑到上述情况,在一个实施方案中,本发明涉及用于治疗患有nsclc的患者的(i)cbp/p300溴结构域抑制剂和(ii)egfr抑制剂的组合,其中nsclc在egfr中显示致癌性改变,条件是,如果nsclc由于先前施用egfr抑制剂而在egfr中另外显示抗性改变,该组合的egfr抑制剂不是以前施用的egfr抑制剂,而特别是以下egfr抑制剂,所述egfr抑制剂是治疗有效的,尽管egfr中的抗性改变(即使得以前施用的egfr抑制剂治疗无效的抗性改变)。人们还可以参考根据本发明第一方面使用的组合,条件是,如果非小细胞肺癌由于先前施用egfr抑制剂而另外表现出egfr中的抗性改变,则该组合的egfr抑制剂不是先前施用的egfr抑制剂,而是以下egfr抑制剂,其如果单独施用(尽管有抗性改变),但在第一个治疗周期中是治疗有效的,或者条件是,如果由于先前施用egfr抑制剂,非小细胞肺癌还表现出egfr中的抗性改变,则该组合的egfr抑制剂不是先前施用的egfr抑制剂,而是指示用于治疗另外表现出egfr中的抗性改变的非小细胞肺癌的egfr抑制剂。
19.举例来说,当考虑两种特定的egfr抑制剂(即“x”和“组合的egfr抑制剂”)时,以上段落被理解为在一个实施方案中涉及用于治疗患有nsclc的患者的(i)cbp/p300溴结构域抑制剂和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变,条件是,如果由于先前施用egfr抑制剂x,非小细胞肺癌还表现出egfr中的抗性改变,则该组合的egfr抑制
剂不是egfr抑制剂x。注意,尽管egfr中的抗性改变(即,使得先前施用的egfr抑制剂x治疗无效的抗性改变),该组合的egfr抑制剂仍是治疗有效的。
20.在第一方面的另一个实施方案中,cbp/p300溴结构域抑制剂是小分子抑制剂。因此,在这样的实施方案中,cbp/p300溴结构域抑制剂不是基于核酸的抑制剂,例如针对cbp和/或p300的shrna或rnai。
21.在第一方面的另一个实施方案中,egfr抑制剂是小分子抑制剂或抗体。因此,在这样的实施方案中,egfr抑制剂不是基于核酸的抑制剂,例如针对egfr的shrna或rnai。在第一方面的另一个实施方案中,egfr抑制剂是小分子抑制剂。在第一方面的另一个实施方案中,egfr抑制剂抑制egfr的酪氨酸激酶活性。
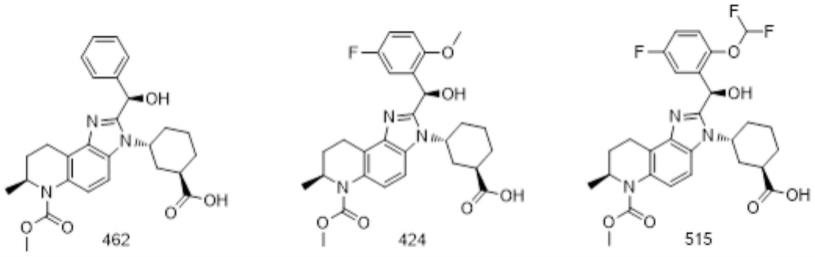
22.cbp/p300溴结构域抑制剂可选自化合物a、化合物c、化合物00030、化合物00071、ccs1477、gne-781、gne-049、sgc-cbp30、cpi-637、ft-6876、化合物462、化合物424和化合物515。这些化合物是可商购的或公开公布的,如下文进一步概述,或者它们的合成和结构在本技术的实施例中示出。可以优选的是,cbp/p300溴结构域抑制剂选自化合物a、化合物c、ccs1477、gne-781、gne-049、cpi-637、化合物462、化合物424和化合物515。
23.egfr抑制药可选自abbv-321、艾维替尼(abivertinib)、阿法替尼、艾氟替尼(alflutinib)、阿美替尼(almonertinib)、阿帕替尼、azd3759、布加替尼(brigatinib)、d 0316、d 0317、d 0318、达可替尼(dacomitinib)、dzd9008、厄洛替尼、fcn-411、吉非替尼、埃克替尼(icotinib)、拉帕替尼(lapatinib)、拉泽替尼(lazertinib)、莫博替尼(mobocertinib)、纳扎替尼(nazartinib)、来那替尼(neratinib)、奥拉夫替尼(olafertinib)、奥希替尼(osimertinib)、波奇替尼(poziotinib)、吡咯替尼(pyrotinib)、瑞齐替尼(rezivertinib)、tas6417、凡德他尼、伐利替尼(varlitinib)、xzp-5809、埃万妥单抗(amivantamab)、cdp1、西妥昔单抗、gc1118、hlx07、jmt101、m1231、耐昔妥珠单抗(necitumumab)、尼妥珠单抗(nimotuzumab)、马妥珠单抗(matuzumab)、帕尼单抗(panitumumab)、sct200、si-b001、syn004、扎芦木单抗(zalutumumab)和其组合。egfr抑制剂也可以选自西妥昔单抗、帕尼单抗、扎芦木单抗、尼妥珠单抗、马妥珠单抗、吉非替尼、厄洛替尼、拉帕替尼、来那替尼、凡德他尼、耐昔妥珠单抗、奥希替尼、阿法替尼、达可替尼、布加替尼、波奇替尼和其组合。在一个优选的实施方案中,egfr抑制剂选自艾维替尼,阿法替尼、艾氟替尼、阿美替尼、阿帕替尼、azd3759、布加替尼、d 0316、d 0317、d 0318、达可替尼、dzd9008、厄洛替尼、fcn-411、吉非替尼、埃克替尼、拉帕替尼、拉泽替尼、莫博替尼、纳扎替尼、来那替尼、奥拉夫替尼、奥希替尼、波奇替尼、吡咯替尼、瑞齐替尼、tas6417、凡德他尼、伐利替尼、xzp-5809和其组合。在一个更优选的实施方案中,egfr抑制剂为吉非替尼或奥希替尼。最优选的egfr抑制剂是奥希替尼。
24.在第一方面的一个优选实施方案中,在每个治疗周期中给患者施用该组合。
25.在第一方面的另一个实施方案中,在第一个治疗周期中,egfr抑制剂作为唯一的活性剂给药,然后在随后的治疗周期中,额外给药cbp/p300溴结构域抑制剂,其中在第一个治疗周期中(即在给药本发明的组合之前),还没有响应单独给药egfr抑制剂而出现egfr的抗性改变。如上所述,抗性改变的发展可以通过例如活组织检查和相应的检测来评估,以便检测egfr突变。因为当使用根据本发明使用的组合时可以防止耐药性的产生,所以优选在每个治疗周期期间给药该组合。
26.在第一方面的另一个实施方案中,cbp/p300溴结构域抑制剂和egfr抑制剂作为单独的剂型给药或包含在单一剂型中。如果cbp/p300溴结构域抑制剂和egfr抑制剂以分开的剂型给药,则在每个治疗周期中的给药可以是伴随的或顺序的。这包括首先给药cbp/p300溴结构域抑制剂,然后给药egfr抑制剂的选择。
27.在第一方面的另一个实施方案中,与作为唯一活性剂给药时egfr抑制剂的治疗效果持续时间相比,该治疗导致egfr抑制剂的治疗效果持续时间延长。在另一个实施方案中,与egfr抑制剂当作为唯一的活性剂给药时的治疗功效相比,该治疗导致egfr抑制剂的治疗功效增加。在另一个实施方案中,该治疗导致对egfr抑制剂耐药性的预防。
28.在第一方面的另一个实施方案中,cbp/p300溴结构域抑制剂以约1mg至约3000mg,优选约10mg至约2000mg,更优选约15mg至约1000mg的日剂量给药。优选以约10mg、约15mg、约20mg、约50mg、约100mg、约250mg、约500mg、约1000mg、约1500mg、约2000mg、约2500mg或约3000mg的日剂量给药cbp/p300溴结构域抑制剂。给药可以间歇进行,即不是每天,但是在给药的当天,可以给药上述的日剂量。如果将ccs1477、化合物462、化合物424或化合物515用作cbp/p300溴结构域抑制剂,则可以以约10mg至约600mg的日剂量施用相应的化合物。
29.在第一方面的另一个实施方案中,如果egfr抑制剂作为唯一的活性剂给药,则egfr抑制剂以典型日剂量范围内的日剂量给药(特别是标记中提到的egfr抑制剂的日剂量,如果有的话)。典型的每日用量(或标明的每日用量,如果有的话)取决于将使用的特定egfr抑制剂。因此,吉非替尼可以例如在用于本发明的组合中以约50至约300mg,优选约100mg至约250mg,最优选约150mg至约250mg的日剂量给药。奥希替尼可以例如在用于本发明的组合中以约5至约1500mg,优选约10mg至约100mg,最优选约50mg至约80mg的日剂量给药。厄洛替尼可以例如在用于本发明的组合中以约10mg至约300mg,优选约25mg至约200mg,最优选约100mg至约150mg的日剂量给药。阿法替尼可以例如在用于本发明的组合中以约5mg至约100mg,优选约10mg至约80mg,最优选约20mg至约40mg的日剂量给药。达可替尼可以例如在用于本发明的组合中以约5mg至约100mg、优选约10mg至约80mg、最优选约15mg至约50mg的日剂量给药。
30.在第一方面的另一个实施方案中,如果egfr抑制剂作为唯一的活性剂给药,则egfr抑制剂以低于上述典型日剂量的日剂量给药。换句话说,如果egfr抑制剂不是作为唯一的活性剂给药,而是在根据本发明使用的组合中给药,则egfr抑制剂的给药量可以低于egfr抑制剂作为唯一的活性剂给药时的给药量。这例如意味着对于上面给出的例子,每日量将处于给定范围的下限或甚至低于这些范围。
31.在第一方面的另一个实施方案中,本发明涉及用于治疗非小细胞肺癌(nsclc)患者的(i)化合物a和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。在该实施方案中,优选egfr抑制剂是奥希替尼并且致癌性改变是由以下引起的:egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变;和它们的组合。在egfr抑制剂为奥希替尼的实施方案中,于egfr基因中的至少一个导致对应于egfr中抗性改变的egfr中t790m的氨基酸取代的碱基突变可能存在也可能不存在。
32.在第一方面的另一个实施方案中,本发明涉及用于治疗患有非小细胞肺癌(nsclc)的患者的(i)化合物a或化合物c和(ii)egfr抑制剂的组合,其中nsclc在egfr中表
现出致癌性改变。在该实施方案中,优选egfr抑制剂是奥希替尼并且致癌性改变是由egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变和它们的组合引起的。在egfr抑制剂为奥希替尼的实施方案中,在egfr基因中的至少一个导致对应于egfr中抗性改变的egfr中氨基酸取代t790m的碱基突变可能存在也可能不存在。
33.在第一方面的另一个实施方案中,本发明涉及用于治疗患有非小细胞肺癌(nsclc)的患者的(i)ccs1477和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。在该实施方案中,优选egfr抑制剂为奥希替尼并且致癌性改变是由egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变和它们的组合引起的。在egfr抑制剂为奥希替尼的实施方案中,在egfr基因中的至少一个导致对应于egfr中抗性改变的egfr中t790m的氨基酸取代的碱基突变可能存在也可能不存在。
34.在第一方面的另一个实施方案中,本发明涉及用于治疗患有非小细胞肺癌(nsclc)的患者的(i)gne-781或gne-049和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。在该实施方案中,优选egfr抑制剂为奥希替尼并且致癌性改变是由egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变和它们的组合引起的。在egfr抑制剂为奥希替尼的实施方案中,在egfr基因中的至少一个导致对应于egfr中抗性改变的egfr中t790m的氨基酸取代的碱基突变可能存在也可能不存在。
35.在第一方面的另一个实施方案中,本发明涉及用于治疗患有非小细胞肺癌(nsclc)的患者的(i)cpi-637和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。在该实施方案中,优选egfr抑制剂为奥希替尼并且致癌性改变是由egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变和它们的组合引起的。在egfr抑制剂为奥希替尼的实施方案中,在egfr基因中的至少一个导致对应于egfr中抗性改变的egfr中t790m的氨基酸取代的碱基突变可能存在也可能不存在。
36.在第一方面的另一个实施方案中,本发明涉及用于治疗非小细胞肺癌(nsclc)患者的(i)化合物462或化合物424或化合物515和(ii)egfr抑制剂的组合,其中nsclc在egfr中表现出致癌性改变。在该实施方案中,优选egfr抑制剂为奥希替尼并且致癌性改变是由egfr基因外显子19中的缺失(特别是导致egfr中e746-a750或l747-e749缺失的缺失);egfr基因中的至少一个导致egfr中的氨基酸取代l858r或a750p的碱基突变和它们的组合引起的。在egfr抑制剂为奥希替尼的实施方案中,在egfr基因中的至少一个导致对应于egfr中抗性改变的egfr中t790m的氨基酸取代的碱基突变可能存在也可能不存在。
37.在第二方面,本发明涉及一种在有需要的患者中治疗非小细胞肺癌的方法,所述方法包括给药患者有效量的(i)cbp/p300溴结构域抑制剂和有效量的(ii)egfr抑制剂,其中非小细胞肺癌在egfr中表现出致癌性改变。
38.在第三方面,本发明涉及一种延长egfr抑制剂在有需要的患者中的治疗效果的持续时间的方法,所述方法包括给药患者有效量的(i)cbp/p300溴结构域抑制剂和有效量的(ii)egfr抑制剂,其中非小细胞肺癌在egfr中表现出致癌性改变。换句话说,与在nsclc治
疗中作为唯一活性剂给药时egfr抑制剂的治疗效果的持续时间相比,egfr抑制剂(当以组合形式给药时)的治疗效果的持续时间延长了。
39.在第四个方面,本发明涉及一种增加egfr抑制剂在有此需要的患者中的治疗效果的方法,所述方法包括给药患者有效量的(i)cbp/p300溴结构域抑制剂和有效量的(ii)egfr抑制剂,其中非小细胞肺癌在egfr中表现出致癌性改变。换句话说,与在非小细胞肺癌治疗中作为唯一活性剂给药的egfr抑制剂的治疗功效相比,egfr抑制剂(当以组合形式给药时)的治疗功效增加。
40.在第五个方面,本发明涉及一种阻断nsclc细胞增殖的方法,所述方法包括对细胞给药有效量的(i)cbp/p300溴结构域抑制剂和有效量的(ii)egfr抑制剂,其中nsclc细胞在egfr中表现出致癌性改变。
41.在第六个方面,本发明涉及一种延缓nsclc细胞增殖的方法,所述方法包括对细胞给药有效量的(i)cbp/p300溴结构域抑制剂和有效量的(ii)egfr抑制剂,其中nsclc细胞在egfr中表现出致癌性改变。
42.以上针对第一方面概述的实施方案同样适用于第二至第六方面的方法。
43.附图简述
44.图1:在egfr突变的非小细胞肺癌细胞(nsclc)中,只有结合到溴结构域(化合物a,ccs1477)或hat域(a485)的cbp/p300抑制剂减弱了egfr抑制剂诱导的基因表达,而阻止cbp与β-连环蛋白相互作用的抑制剂(icg001)没有减弱egfr抑制剂诱导的基因表达。在携带或不携带引起耐药性的看守突变t790m的细胞系中使用了两种不同的egfr抑制剂。显示的受调控基因的例子是alpp(碱性磷酸酶,胎盘型;a和c)和hopx(仅同源结构域蛋白;b和d)。数据来自2项使用qpcrs的独立实验,一式两份(平均值
±
sd)。
45.图2:只有结合cbp/p300的溴结构域(brd)的对映异构体(化合物a)而不是不结合cbp/p300的溴结构域的对映异构体(化合物b)以浓度依赖性方式增强egfr抑制剂介导的nsclc细胞增殖抑制。随着时间的推移,监测egfr突变的hcc827细胞的细胞数量。(a)用单独的dmso(实心圆)、单独的20nm egfr抑制剂(吉非替尼;第一代egfr抑制剂,空心圆)或与cbp/p300 brd抑制剂的溴结构域结合对映异构体(化合物a)的组合处理细胞。(b)hcc827细胞在没有egfr抑制剂的情况下暴露于化合物a和b。在没有egfr抑制剂的情况下,化合物a失去其对egfr突变的nsclc细胞增殖的作用,并且表现得像不结合cbp/p300的溴结构域的化合物b。给出的图来自一个实验,每个时间点和条件三次重复(平均值
±
sd)。
46.图3:只有与cbp/p300的溴结构域结合的抑制剂(化合物a,ccs1477)才能加强egfr抑制剂的作用,而不影响egfr抑制剂不存在时的细胞生长。即使没有egfr抑制剂,抑制cbp/p300抑制剂(a485)的组蛋白乙酰转移酶(hat)结构域的cbp/p300抑制剂影响egfr突变的nsclc细胞的细胞增殖。(a)、(b)、(c)、(d)和(e)使用核荧光染色,将egfr突变的nsclc细胞系hcc827的细胞数作为药物处理(图表图例中的符号)随时间[天数]变化的函数作图。图3(a)和(b)和(c)左侧:用化合物a(图3a)、ccs1477(图3b)、sgc-cbp30(图3c)或a485对细胞进行的单剂处理。在不存在egfr抑制剂的情况下,靶向cbp/p300的溴结构域的化合物a ccs1477,sgc-cbp30不影响egfr突变的nsclc细胞的细胞增殖。图3(a)和(b)和(c)右侧和图3(d)和(e):在300nm吉非替尼(egfr抑制剂)存在下,化合物a(a)和ccs1477(cbp/p300 brd-i)(b)和sgc-cbp30(c)和化合物00071(d)和化合物00030(e)和a485(cbp/p300 hat-i)的抗
增殖活性。靶向cbp/p300的溴结构域的化合物a和ccs1477和sgc-cbp30以及化合物00071和化合物00030增强了egfr抑制剂在egfr突变的nsclc细胞中的作用,尽管当这些化合物在没有egfr抑制剂的情况下使用时没有抗增殖作用(左)。描绘的曲线来自一式三份的一个实验(平均值
±
sd)。
[0047]
图4:(a)评估hcc827细胞数随时间[h]的变化。在没有egfr抑制剂的情况下,化合物a不影响egfr突变的nsclc细胞的细胞增殖,但是当与egfr抑制剂组合时,化合物a防止耐药性的发展。治疗:根据图例,dmso、1μm化合物a、300nm吉非替尼或300nm吉非替尼和1μm化合物a的组合。示例图显示了dmso和化合物a的6孔或吉非替尼或吉非替尼 化合物a处理的24孔的细胞数(平均值
±
sd)。(b)用吉非替尼或吉非替尼 化合物a处理0或22天每个孔的细胞数,如点图所示(来自a中的2个实验板,每个条件48个孔)。****p《0.0001,kruksal-wallis检验,用dunn多重比较。(c)用300nm吉非替尼或300nm吉非替尼 1μm化合物a处理的孔的瀑布图,来自2个平板(48个孔/每种条件),如(a)中分析,显示22天后对处理的响应,作为特定孔上初始细胞数的对数倍数变化。从最高到最低的对数倍数变化对孔进行排序。空心柱是吉非替尼处理的孔,实心柱代表用吉非替尼 化合物a处理的孔。尽管细胞对单独的化合物a没有反应,但当egfr抑制剂与化合物a组合时,化合物a显著增加了对egfr抑制剂的反应。
[0048]
图5:在携带t790m-看守突变的egfr突变nsclc细胞中,与cbp/p300溴结构域结合的抑制剂加强了第三代egfr抑制剂的作用,而不影响没有egfr抑制剂时的细胞生长。(a)在dmso、50nm奥希替尼、2μm化合物a或50nm奥希替尼与2.0、0.5或0.125μm化合物a的组合的存在下评估nci-h1975细胞数量随时间[h]的变化。在不存在第三代egfr抑制剂的情况下,化合物a不影响携带t790m-看守突变的egfr突变的nsclc细胞的细胞增殖,但是当与egfr抑制剂组合时,化合物a防止耐药性的发展。(b)在dmso、50nm奥希替尼、2μm ccs1477或50nm奥希替尼与2.0、0.5或0.125μm ccs1477的组合的存在下nci-h1975细胞生长的评估。在不存在第三代egfr抑制剂的情况下,ccs1477不影响携带t790m-看守突变的egfr突变的nsclc细胞的细胞增殖,但当与egfr抑制剂联合使用时,可防止耐药性的产生。(示例图显示了一式两份的每个数据和时间点,并在graphpad prism中计算了逻辑(logistic)生长曲线拟合)。
[0049]
图6:当在没有egfr抑制剂的情况下使用时,结合cbp/p300的溴结构域的抑制剂在体内没有效果,但是当与egfr抑制剂联合使用时,它们增强了egfr抑制剂的效果,从而提供了更好的随时间推移的肿瘤控制和对治疗更好的响应率。(a)将egfr突变的nci-h1975异种移植物的平均肿瘤体积( sem)随时间作图。描述了四个不同的治疗组:媒介物(30% peg300/h2o;交叉圆圈;n=4),20mg/kg ccs1477(空心圆;n=4),2mg/kg奥希替尼(实心圆;n=9)或2mg/kg奥希替尼与20mg/kg ccs1477(半实心圆;n=10)的组合。当在没有egfr抑制剂的情况下使用ccs1477时,它对肿瘤生长没有影响。然而,当它与egfr抑制剂组合时,对治疗的响应增加,并且在治疗过程中肿瘤大小得到更好的控制。(b)所有4个治疗组的最佳平均响应显示在瀑布图中(媒介物为灰色,20mg/kg ccs1477为白色,2mg/kg奥希替尼为黑色,和2mg/kg奥希替尼与20mg/kg ccs1477的组合为方格)。虚线表示初始肿瘤体积减少了30%。当溴结构域结合抑制剂(ccs1477)与egfr抑制剂(奥希替尼)联合用药时,对治疗的响应率增加,尽管对单独使用溴结构域结合抑制剂(ccs1477)无反应。
[0050]
图7:在确定与化合物00004复合的人crebbp的溴结构域的晶体结构时,该模型的
初始fo-fc差分电子密度图(轮廓为4.0σ)是在用refmac5对化合物建模之前对初始模型进行改进而得到的。
[0051]
图8:化合物a与egfr抑制剂吉非替尼的组合介导对hcc4006长期细胞增殖的抑制——细节在实施例9中提供。
[0052]
图9:化合物a、化合物c和结构上不相关的选择性cbp/p300溴结构域抑制剂(ccs1477、ft-6876和gne-781)与egfr抑制剂奥希替尼的组合介导对hcc827长期细胞增殖的抑制——详情见实施例10。
[0053]
图10:化合物a、化合物c和结构上不相关的选择性cbp/p300溴结构域抑制剂(ccs1477、ft-6876和gne-781)与egfr抑制剂奥希替尼的组合介导对hcc4006长期细胞增殖的抑制——详见实施例11。
[0054]
图11:当在没有egfr抑制剂的情况下使用时,结合cbp/p300的溴结构域的抑制剂在体内没有效果,但是当与egfr抑制剂联合使用时,它们加强了egfr抑制剂的效果,从而随着时间推移提供更好的肿瘤控制和对治疗更好的响应率。(a)将egfr突变的nci-h1975异种移植物的平均肿瘤体积( sem)随时间作图。描述了四个不同的治疗组:媒介物(交叉圆圈),90mg/kg化合物c(空心圆圈),2mg/kg奥希替尼(实心圆)和2mg/kg奥希替尼与90mg/kg化合物c组合(半实心圆圈)。(b)所有4个治疗组的最佳平均响应显示在瀑布图中(灰色为媒介物,90mg/kg化合物c为白色,2mg/kg奥希替尼为黑色和2mg/kg奥希替尼与90mg/kg化合物c的组合为方格)。虚线表示初始肿瘤体积减少了30%——详见实施例12。
[0055]
发明详述
[0056]
在更详细地描述本发明之前,先介绍以下定义。
[0057]
1.定义
[0058]
如在说明书和权利要求中所使用的,单数形式的“一”和“一个”也包括相应的复数形式,除非上下文另有明确规定。
[0059]
在本发明的上下文中,术语“约”表示本领域技术人员将理解的精度区间,以仍然确保所讨论的特征的技术效果。该术语通常表示偏离所示数值
±
10%,优选
±
5%。
[0060]
需要理解的是,术语“包括”不是限制性的。出于本发明的目的,术语“由
……
组成”被认为是术语“包含”的优选实施方案。如果在下文中一个组被定义为包括至少一定数量的实施方案,这也意味着包括优选仅由这些实施方案组成的组。
[0061]
本文所用的术语“cbp/p300溴结构域抑制剂”是指一种小分子,它能强烈地和选择性地结合cbp的溴结构域和p300的溴结构域。该术语与术语“选择性结合cbp/p300的溴结构域的溴结构域抑制剂”和“选择性抑制cbp/p300的溴结构域抑制剂”同义。“强结合”在这方面是指当与cbp的溴结构域和p300的溴结构域结合时,kd小于约300nm,优选小于约100nm。在这方面,“选择性结合”是指与任何其它含溴结构域的蛋白质或bromoscan
tm
的溴结构域结合的kd相比,小分子与cbp的溴结构域和p300的溴结构域结合的kd至多为其约20分之一,优选至多为其约30分之一,更优选至多为其约50分之一,最优选至多为其约70分之一,优选地,当如实施例4所示进行bromoscan
tm
时,与本技术的实施例4的表中discoverx基因符号所示的其它含溴结构域的蛋白质或溴结构域进行比较。为了进行比较,将除cbp和p300之外的任何含溴结构域的蛋白质或bromoscan
tm
的溴结构域的最低kd与cbp和p300的最高kd进行比较。因此,如果例如brd4(全长,短iso.)的kd是除cbp和p300之外的所有含溴结构域的蛋白
质或溴结构域中最低的kd,且为7100nm,这与cbp的kd(29nm)相比(并非与p300的kd相比,p300的kd为12nm,因此低于cbp的kd)。在下面的实施例4的表中,对化合物a进行了上述实施例。
[0062]
通过如上所述的强选择性结合,通常通过cbp/p300的溴结构域发生的与细胞中相互作用配偶体(interaction partner)的相互作用被抑制,因此该分子被称为“抑制剂”。术语“抑制相互作用”是指优选在cbp/p300的溴结构域和相互作用配偶体之间不再发生任何相互作用(至少没有达到可检测的水平)。然而,当cbp/p300的溴结构域和相互作用配偶体之间的给定相互作用(设定为100%)大大降低时,例如降低至约50%、约40%、约30%、优选约20%、更优选约10%或最优选约5%或更低的水平,这种降低的相互作用仍包含在术语“抑制性相互作用”中。就抑制相互作用的化合物的医学用途而言,可能不需要完全抑制相互作用来达到足够的治疗效果。因此,需要理解的是,本文所用的术语“抑制”也指相互作用的减少,其足以实现期望的效果。
[0063]
本文所用的术语“egfr”是指蛋白质“表皮生长因子受体”。egfr是一种跨膜蛋白,通过结合其特异性配体(包括表皮生长因子)而被激活。在其生长因子配体的激活下,egfr从无活性的单体形式转变为有活性的同二聚体。除了在配体结合后形成同二聚体,egfr还可以与erbb受体家族的另一个成员配对,如erbb2/her2/neu,形成活化的异源二聚体。egfr二聚化刺激其固有的细胞内蛋白酪氨酸激酶活性。结果,egfr的c-末端结构域中的几个酪氨酸残基发生了自身磷酸化,这引发了与磷酸化的酪氨酸相关的其他几个蛋白质(通过其自身的磷酸酪氨酸结合sh2结构域)的下游激活和信号传导。这些下游信号蛋白启动几种信号转导级联,主要是mapk、akt和jnk途径,导致dna合成和细胞增殖。导致egfr过度激活的突变与包括肺癌在内的多种癌症相关,并可能尤其导致其持续激活,从而导致不受控制的细胞分裂。
[0064]
本文所用术语“egfr抑制剂”是指能够作用于egfr从而抑制最终导致细胞增殖的细胞内下游信号传导的分子。在本文中,术语“抑制”意味着优选地不再发生下游信号传导。然而,当给定的下游信号(设定为100%)被大大降低时,例如降低至约70%、约60%、约50%、约40%、约30%、优选约20%、更优选约10%或最优选约5%或更低的水平,这种降低的下游信号传导仍被术语“抑制细胞内下游信号传导”所涵盖。就抑制下游信号传导的化合物的医学用途而言,可能不需要完全抑制信号传导来达到足够的治疗效果。因此,需要理解的是,本文上下文中使用的术语“抑制”也指下游信号传导的减少,其足以实现期望的效果。egfr抑制剂可以结合并因此阻断egfr的细胞外配体结合结构域。这种egfr抑制剂通常是一种抗体,特别是单克隆抗体,其选自:埃万妥单抗、cdp1、西妥昔单抗、gc1118、hlx07、jmt101、m1231、耐昔妥珠单抗、尼妥珠单抗、马妥珠单抗、帕尼单抗、sct200、si-b001、syn004、扎芦木单抗和其组合。egfr抑制剂也可以结合到受体的胞质侧,从而抑制egfr酪氨酸激酶活性。这种egfr抑制剂通常是小分子,特别是选自以下的小分子:艾维替尼、阿法替尼、艾氟替尼、阿美替尼、阿帕替尼、azd3759、布加替尼、d 0316、d 0317、d 0318、达可替尼、dzd9008、厄洛替尼、fcn-411、吉非替尼、埃克替尼、拉帕替尼、拉泽替尼、莫博替尼、纳扎替尼、来那替尼、奥拉夫替尼、奥希替尼、波奇替尼、吡咯替尼、瑞齐替尼、tas6417、凡德他尼、伐利替尼、xzp-5809和其组合。
[0065]
如本文所用,术语“其中非小细胞肺癌在egfr中表现出致癌性改变”是指非小细胞
肺癌肿瘤具有突变形式的egfr,其中这种突变形式的egfr与非小细胞肺癌的发展有关。换句话说,egfr的突变形式可以被认为与非小细胞肺癌的发展相关或导致非小细胞肺癌的发展,任选存在其它因素。由于egfr基因的改变,egfr的突变形式存在于非小细胞肺癌肿瘤中,其中这种改变特别是egfr基因中的缺失、egfr基因中的插入、egfr基因中的缺失和插入、egfr基因中的复制、egfr基因的扩增和/或导致egfr中氨基酸取代的egfr基因中的至少一个碱基突变。相应的具体改变如上所述。egfr基因中这种改变的组合经常被发现。“egfr的致癌性改变”不是下文定义的“egfr中的抗性改变”。
[0066]
本文所用术语“egfr中的抗性改变”是指在用egfr抑制剂治疗后,nsclc肿瘤已经获得egfr的进一步改变(除了致癌性改变之外),其中egfr中的这种进一步改变使得nsclc对所述egfr抑制剂(即用于治疗且nsclc最初对其敏感的egfr抑制剂)的治疗具有耐药性。耐药性是由egfr基因的改变介导的,特别是egfr基因中至少一个导致egfr中的氨基酸取代的碱基突变。因此,与上面定义的“egfr中的致癌性改变”相反,“抗性改变”不被认为与非小细胞肺癌的初始发展有关或者是其原因。相反,它为nsclc提供了进一步的生长优势,即,它赋予了nsclc对先前施用的特定egfr抑制剂的治疗的耐药性(并且在肿瘤对该治疗的反应发展为抗性改变之前,该抑制剂在治疗nsclc中是有效的)。一个突出的“egfr中的抗性改变”是egfr中的氨基酸取代t790m,这也被称为看守突变。“egfr中的抗性改变”不是上述定义的“egfr中的致癌性改变”。然而,这两种类型的改变当然可以存在于非小细胞肺癌肿瘤的egfr中,并且经常在患者中检测到,并且相应的细胞系作为模型系统存在(参见例如细胞系nci-h1975)。
[0067]
本文所用术语egfr的“过度活化”是指与野生型情况相比,egfr更有活性,特别是在下游活化和信号传导方面更有活性,从而导致癌细胞生长。
[0068]
这里使用的术语“小分子”是指具有低分子量的小有机化合物。本发明上下文中的小分子优选具有小于5000道尔顿的分子量,更优选小于4000道尔顿,更优选小于3000道尔顿,更优选小于2000道尔顿或甚至更优选小于1000道尔顿。在一个特别优选的实施方案中,本发明上下文中的小分子具有小于800道尔顿的分子量。在另一个优选的实施方案中,本发明上下文中的小分子的分子量为50至3000道尔顿,优选100至2000道尔顿,更优选100至1500道尔顿,甚至更优选100至1000道尔顿。
[0069]
本文所用术语“治疗”是指临床干预,以便治愈或改善疾病、防止疾病复发、减轻疾病症状、减少疾病的任何直接或间接病理后果、实现疾病的稳定(即,不恶化)状态、防止转移、降低疾病进展速度和/或与不接受治疗时的预期生存期相比延长生存期。
[0070]
本文所用的术语“治疗周期”是指在对患者的状况进行初步评估后给药一段时间,其中通常在开始另一个治疗周期前对患者的状况进行再评估。
[0071]
本文提及的cbp/p300溴结构域抑制剂的细节如下:化合物a、化合物c、化合物00030和化合物00071的结构如本技术的实施例部分所示。此外,这些化合物的合成路线显示在本技术的实施例部分。ccs1477可商购获得,例如在aobious,其cas编号为2222941-37-7。gne-781可在例如mce(medchemexpress)购得,其cas编号为1936422-33-1。gne-049可在mce(medchemexpress)等公司购得,其cas编号为1936421-41-8。sgc-cbp30可在mce(medchemexpress)等公司购得,其cas编号为1613695-14-9。cpi-637可在mce(medchemexpress)等公司购得,其cas编号为1884712-47-3。ft-6876可在mce
(medchemexpress)等公司购得,其cas编号为2304416-91-7(ft-6876也称为“cbp/p300-in-8”)。化合物462、424和515的结构描述如下,其中这些结构和合成路线在wo 2020/006483中给出(特别参见化合物424于第33和34页,化合物462于第42和43页,以及化合物515于第47和48页):
[0072][0073]
2.发明人的惊人发现
[0074]
本发明人鉴定了与cbp/p300的溴结构域强烈结合的新化合物,并表明与cbp/p300的溴结构域的结合也是选择性的,因为众所周知有许多蛋白质包含溴结构域。
[0075]
cbp/p300已被确定为真核转录调控网络的中心节点,并与400多种转录因子和其他调控蛋白相互作用。cbp/p300调节许多细胞信号通路之间的串扰和干扰,并且被肿瘤病毒靶向以劫持细胞调节机制(参见dyson和wright,同上,第6714页,右栏)。cbp/p300是包含几个结构域的大蛋白,可从dyson和wright的图1中得出,见上文。这些结构域是nrid、taz1、taz2、kix、crd1、brd、ch2(包括phd结构域和ring指结构域(finger domain))、hat、zz和ncbd结构域。从这些蛋白质的大小和它们的不同结构域已经可以明显看出,它们的细胞功能非常多样,例如,由于cbp/p300能够进行多种相互作用,它们可以与许多不同的相互作用配偶体相互作用。cbp/p300作为组蛋白乙酰转移酶的酶活性位于hat结构域。如上所述,这种酶的功能主要涉及转录激活。cbp/p300也易受翻译后修饰,特别是磷酸化。它们自身的酶活性以及蛋白质受到翻译后修饰,给cbp/p300的各种功能和作用带来了另一个层次的复杂性。goodman和smolk,genes&development 2000,14:1553-1577在引言部分很好地总结了,这些功能和效果甚至可以被对抗,其中指出,cbp/p300功能中的一个主要矛盾是,这些蛋白质似乎能够促成截然相反的细胞过程,并且cbp/p300是否促进细胞凋亡或细胞增殖似乎高度依赖于环境。对于疾病,特别是癌症的影响,这意味着特定疾病和特定癌症类型的环境将决定cbp/p300如何参与,如果它们参与的话。
[0076]
鉴于以上所述,在细胞过程中不可能赋予cbp/p300单一功能也就不足为奇了,其可能受到例如一般的“cbp/p300抑制剂”的影响。相反,由于极大水平的复杂性,剖析cbp/p300的各种功能似乎只有在研究cbp/p300的特定结构域时才有可能,即通过分析当例如抑制cbp/p300在其hat结构域中的酶活性时或当通过阻断(或“抑制”)某些结构域使与相互作用配偶体的特定相互作用变得不可能时所达到的效果。此外,如上所述,这必须放在各自的环境下看待,例如特定的疾病或癌症类型。
[0077]
因此,发明人继续研究它们在特定环境中的作用,其中它们的抑制剂使得不可能通过cbp/p300的溴结构域与相互作用配偶体相互作用。目前,已知cbp/p300的溴结构域识别组蛋白尾部和转录因子idr(固有无序区)中的乙酰-赖氨酸残基,包括p53和creb的那些(参见dyson和wright,同上,第6717页,右栏)。鉴于hou等人(hou等人,见上文)最近的出版
物,本发明人着手研究其抑制剂在非小细胞肺癌(nsclc)细胞中的作用。在该出版物中,基于shrna介导的p300的下调得出结论,p300作为关键的肿瘤启动子促进非小细胞肺癌细胞中的细胞增殖、迁移和侵袭。然而,当单独应用抑制剂时,发明人未能看到对测试的nsclc细胞系的增殖的影响。因此,与其他癌症类型如前列腺癌相反,cbp/p300溴结构域抑制剂不能对nsclc细胞的增殖产生影响,靶向cbp/p300不同结构域的抑制剂是否会显示出在nsclc细胞中通过rnai下调完整p300蛋白时所观察到的效果还有待观察。
[0078]
发明人继续测试了cbp/p300溴结构域抑制剂,并惊奇地发现,与单独施用egfr抑制剂相比,他们的cbp/p300溴结构域抑制剂延长了egfr抑制剂在显示egfr致癌性改变的nsclc细胞中的作用。换句话说,尽管未能单独对在egfr中显示致癌性改变的nsclc的增殖产生作用,但本发明人的cbp/p300溴结构域抑制剂显示了与egfr抑制剂一起的作用。换句话说,随着时间的推移,发明人的cbp/p300溴结构域抑制剂和egfr抑制剂的组合导致测试的egfr突变型nsclc细胞随时间的显著增殖抑制。
[0079]
在他们的实验中,发明人使用在egfr基因外显子19中缺失的nsclc细胞系作为致癌改变(具有外显子19中缺失的hcc827导致在egfr中e746到a750的缺失,具有外显子19中缺失的hcc4006导致egfr中l747到e749的缺失),但是egfr中没有抗性改变。因此,这些细胞系可被视为其肿瘤具有“egfr外显子19缺失”的非小细胞肺癌患者进行一线治疗的模型系统。吉非替尼和奥希替尼分别作为egfr抑制剂与cbp/p300溴结构域抑制剂组合使用(参见以下实施例)。本发明人还使用了具有egfr的nsclc细胞系,该egfr表现出致癌改变l858r以及抗性改变t790m(nci-h1975)。众所周知,egfr t790m会对吉非替尼治疗产生耐药性,导致吉非替尼治疗无效。因此,该细胞系可被视为非小细胞肺癌患者二线治疗的模型系统,所述非小细胞肺癌患者的肿瘤已经对初始egfr抑制剂治疗产生耐药性。在该细胞系中,发明人仅使用了奥希替尼与cbp/p300溴结构域抑制剂(因为尽管在egfr中发生抗性改变t790m,奥希替尼仍然显示有效,而其会导致吉非替尼无效)。鉴于t790m突变,吉非替尼与cbp/p300溴结构域抑制剂的联合试验是没有意义的。
[0080]
在所有受试细胞系中观察到的该组合在长期孵育中的显著增殖抑制作用是特别值得注意的,因为—由于耐药性的发展—当单独使用egfr抑制剂时,随着时间的推移增殖抑制作用将不会保持完全。如下文实验部分的数据所示,这不仅是吉非替尼单独应用时的情况,也是奥希替尼单独应用时的情况。因此,虽然奥希替尼最初能够克服由突变egfr t790m提供的耐药性,因此最初是有效的(与吉非替尼相反,非小细胞肺癌已经对吉非替尼耐药),其对奥希替尼的抗性也随着时间的推移而发展,最终导致奥希替尼变得无效。考虑到它们cbp/p300抑制剂的结果,发明人继续研究观察到的效果是否可以同样推广到cbp/p300抑制剂。为此,测试了其他cbp/p300溴结构域抑制剂,即ccs1477、sgc-cbp30、ft-6876和gne-781。值得注意的是,本发明人测试的不同组cbp/p300抑制剂的结构并不相关,因此它们的共同特征仅与这些抑制剂实现的效果相关,即选择性抑制cbp/p300溴结构域。所有测试的cbp/p300抑制剂的结构如下:
[0081][0082][0083]
还应该提到的是,测试的egfr抑制剂吉非替尼和奥希替尼在结构和作用上非常不同(吉非替尼是一种“非共价抑制剂”,而奥希替尼是一种“共价抑制剂”),但具有共同的抑制egfr激酶活性的功能。此外,它们在开发方面非常不同,即第一代用于治疗nsclc的药物(即吉非替尼)和第三代用于治疗nsclc的药物(即奥希替尼)。
[0084]
此外,发明人不仅测试了单一的nsclc细胞系,还使用了三种不同的nsclc细胞系(hcc827、hcc4006和nci-h1975)。这是特别重要的,因为用给定疾病的单个细胞系进行的体外实验可能提供不可靠的结果,而在给定疾病的至少两个不同细胞系中获得相同的结果是所获得结果可靠的更强有力的指标。此外,似乎优选在这种分析中使用最初被egfr抑制剂完全抑制生长的nsclc细胞系,以便能够更可靠地分析cbp/p300溴结构域抑制剂的作用,特别是在治疗几天后。当然,基于异种移植模型且与使用nsclc细胞系时的最初发现一致的结果甚至更好地证实了可从实验中得出的总体结论。发明人也获得了这样的异种移植物数据,如下文实施例部分所示。
[0085]
3.本发明化合物的药物组合物
[0086]“cbp/p300溴结构域抑制剂”和“egfr抑制剂”是用于本文所要求保护的用途的“药物活性剂”。如上所述,它们可以存在于单独的剂型中或者包含在单一剂型中。
[0087]
本文所用的“药物活性剂”是指化合物能有效调节患者(即人或动物的体内)的响应。本文使用的术语“药学上可接受的赋形剂”是指通常包含在药物组合物中的赋形剂,这是技术人员已知的。这种赋形剂列举如下。鉴于上文给出的“药物活性剂”的定义,可药用赋形剂可被定义为无药物活性的。
[0088]
如果市售的egfr抑制剂与cbp/p300溴结构域抑制剂组合使用,优选通过分开的剂型给药,并且egfr抑制剂以经过认可的剂型和给药途径给药。cbp/p300溴结构域抑制剂可以下述剂型给药,或者以目前正在进行临床试验的剂型给药。
[0089]
根据本发明使用的剂型可以配制用于口服、含服、鼻、直肠、局部、透皮或肠胃外应用。口服应用是优选的。肠胃外应用也是优选的,包括静脉内、肌肉内或皮下给药。本发明的剂型也可以称为制剂或药物组合物。
[0090]
通常,根据本发明的药物组合物可以包含各种药学上可接受的赋形剂,这些赋形剂将根据组合物要实现的功能来选择。本发明意义上的“药学上可接受的赋形剂”可以是用于制备药物剂型的任何物质,包括包衣材料、成膜材料、填充剂、崩解剂、改变释放的材料、载体材料、稀释剂、粘合剂和其它佐剂。典型的药学上可接受的赋形剂包括物质如蔗糖、甘露醇、山梨醇、淀粉和淀粉衍生物、乳糖和润滑剂如硬脂酸镁、崩解剂和缓冲剂。
[0091]
术语“载体”表示药学上可接受的有机或无机载体物质,活性成分与其结合以促进应用。合适的药学上可接受的载体包括,例如,水、盐溶液、醇、油,优选植物油、聚乙二醇、明胶、乳糖、直链淀粉、硬脂酸镁、表面活性剂、香料油、脂肪酸单甘油酯和双甘油酯、petroethral脂肪酸酯、羟甲基纤维素、聚乙烯吡咯烷酮等。药物组合物可以被灭菌,并且如果需要,与助剂混合,如润滑剂、防腐剂、稳定剂、湿润剂、乳化剂、用于影响渗透压的盐、缓冲剂、着色剂、调味剂和/或芳香物质等,它们不会与活性化合物发生有害的反应。
[0092]
如果本发明考虑液体剂型,这些剂型可以包括药学上可接受的乳剂、溶液、混悬剂和糖浆剂,它们含有本领域常用的惰性稀释剂如水。这些剂型可以含有例如用于填充的微晶纤维素、作为悬浮剂的海藻酸或海藻酸钠、作为增粘剂的甲基纤维素和甜味剂/调味剂。
[0093]
对于肠胃外应用,特别合适的载体包括溶液,优选油性或水性溶液,以及悬浮液、乳液或植入物。肠胃外给药的药物制剂是特别优选的,并且包括水溶性形式的水溶液。此外,混悬剂可以制成合适的油性注射混悬剂。合适的亲脂性溶剂或载体包括脂肪油如芝麻油,或合成脂肪酸酯如油酸乙酯或甘油三酯,或脂质体。含水注射剂混悬剂可含有增加混悬剂粘度的物质,如羧甲基纤维素钠、山梨醇或葡聚糖。
[0094]
特别优选的剂型是本发明药物组合物的注射制剂。因此,无菌可注射的水性或油性悬浮液可以例如根据已知技术使用合适的分散剂、润湿剂和/或悬浮剂来配制。无菌注射制剂也可以是在无毒的肠胃外可接受的稀释剂或溶剂中的无菌注射溶液或悬浮液。可以使用的可接受的载体和溶剂是水和等渗氯化钠溶液。无菌油通常也用作溶剂或悬浮介质。
[0095]
用于本发明药物组合物直肠给药的栓剂可以通过例如将化合物与合适的无刺激性赋形剂如可可脂、合成甘油三酯和聚乙二醇混合来制备,所述赋形剂在室温下为固体,但在直肠温度下为液体,使得它们在直肠中融化并从所述栓剂中释放活性剂。
[0096]
对于吸入给药,包含本发明化合物的药物组合物可以使用合适的推进剂,(例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其它合适的气体),以气雾剂喷雾的形式从加压包装或喷雾器中方便地递送。在加压气雾剂的情况下,剂量单位可以通过提供阀
门以输送计量的量来确定。用于吸入器或吹入器中的例如明胶的胶囊和药筒可以配制成含有化合物和合适的粉末基质如乳糖或淀粉的粉末混合物。
[0097]
口服剂型可以是液体或固体,包括例如片剂、锭剂、丸剂、胶囊、粉剂、泡腾制剂、糖衣丸和颗粒剂。口服使用的药物制剂可以作为固体赋形剂获得,任选研磨所得混合物,并在加入合适的助剂(如果需要)后加工颗粒混合物,以获得片剂或糖衣丸芯。合适的赋形剂尤其是填充剂,例如糖,包括乳糖、蔗糖、甘露醇或山梨醇;纤维素制剂,例如玉米淀粉、小麦淀粉、大米淀粉、马铃薯淀粉、明胶、黄蓍胶、甲基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮(pvp)。如果需要,可以加入崩解剂,如交联聚乙烯吡咯烷酮、琼脂或海藻酸或其盐,如海藻酸钠。可以配制口服剂型以确保活性剂的立即释放或活性剂的持续释放。
[0098]
4.进一步的公开和实施方案
[0099]
受体酪氨酸激酶(rtk)抑制剂和其他激酶抑制剂的临床抗肿瘤效果不持久。通常会对这些抑制剂产生耐药性。更具体地说,egfr抑制剂(egfri)的临床抗肿瘤效果是不持久的。根据治疗药物和临床情况,对egfr抑制剂的耐药性通常在9至19个月内出现。因此,需要开发一种能够防止癌症患者产生耐药性的癌症治疗模式。历史上,大多数解决耐药性的方法都集中在复发肿瘤的遗传驱动因素上。为了克服已经建立的耐药性,驱动肿瘤再生的新突变蛋白将被单独或与原发性癌症药物联合治疗靶向。egfri治疗的一个耐药机制是egfr蛋白中的看守突变的产生—这种突变会使egfri无效。最常见的这种看守突变是t790m突变。突变特异性抑制剂,如奥希替尼用于克服对第一代egfr抑制剂的已建立的耐药性,该抑制剂不抑制突变的egfr t790m。egfri治疗的另一种耐药机制是旁路信号传导,其通过其他受体酪氨酸激酶激活,例如通过met、erbb2、hgf、erbb3、igf1r、axl、ntrk1、braf、fgfr3或fgfr1的扩增、过表达或激活。抑制旁路信号传导的治疗干预措施已在临床中进行了试验,结果喜忧参半。
[0100]
先前的公开如专利申请wo2018022637描述了cbp/p300抑制剂作为新型癌症疗法的用途,特别是用于治疗含有p300突变的癌症。wo2011085039描述了用于治疗癌症的方法,包括抑制cbp/p300组蛋白乙酰转移酶(hat)的活性,以及cbp/p300 hat抑制剂用于治疗患有癌症的受试者的用途,特别是与破坏dna的化疗抗癌剂组合。
[0101]
需要新的有效方法和组合物来预防癌症耐药性的发展。这尤其由本部分4的实施方案解决。
[0102]
实施方案1:一种用于治疗动物癌症的方法中的cbp/p300溴结构域抑制剂,该方法包括给有此需要的动物施用cbp/p300溴结构域抑制剂和选自以下的受体酪氨酸激酶抑制剂:egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂,或kras(kirsten rat sarcoma)或braf(原癌基因b-raf和v-raf鼠肉瘤病毒癌基因同系物b)抑制剂,其中所述癌症包括相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不会减缓癌症的进展。
[0103]
实施方案2:一种用于延长动物对受体酪氨酸激酶抑制剂或kras或braf抑制剂癌症疗法的响应持续时间的方法中的cbp/p300溴结构域抑制剂,包括对患有癌症的动物给药cbp/p300溴结构域抑制剂或其药学上可接受的盐,其中与没有给药cbp/p300溴结构域抑制剂或其药学上可接受的盐时对癌症治疗的响应持续时间相比,给药cbp/p300溴结构域抑制
剂或其药学上可接受的盐时对癌症治疗的响应持续时间延长,并且其中受体酪氨酸激酶抑制剂选自egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂。
[0104]
实施方案3:一种用于治疗癌症的组合物,所述组合物包含cbp/p300溴结构域抑制剂或其药学上可接受的盐和选自以下的受体酪氨酸激酶抑制剂的协同组合:egfr、alk、met、her2、ros1、ret、ntrk1和axl的抑制剂,或kras或braf抑制剂,其中所述癌症包括相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不会减缓癌症的进展。
[0105]
实施方案4:一种抑制癌细胞生长的方法,包括施用cbp/p300溴结构域抑制剂和选自以下的受体酪氨酸激酶抑制剂:egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂,或kras或braf抑制剂,并且其中癌细胞包含相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不抑制癌细胞的生长。
[0106]
实施方案5:前述任一实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中所述受体酪氨酸激酶或kras或braf的改变为致癌性改变。
[0107]
实施方案6:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中所述受体酪氨酸激酶抑制剂是egfr抑制剂。
[0108]
实施方案7:根据实施方案6所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中所述受体酪氨酸激酶的改变是egfr中的突变。
[0109]
实施方案8:根据前述任一实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中与单独的cbp/p300抑制剂或单独的受体酪氨酸激酶或kras或braf抑制剂相比,cbp/p300溴结构域抑制剂或其药学上可接受的盐与受体酪氨酸激酶抑制剂或kras或braf抑制剂的组合物或组合在治疗癌症中具有协同作用。
[0110]
实施方案9:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300溴结构域抑制剂或其药学上可接受的盐和受体酪氨酸激酶抑制剂或kras或braf抑制剂的组合物或组合延迟或降低了癌症对受体酪氨酸激酶抑制剂或kras或braf抑制剂的耐药性的风险。
[0111]
实施方案10:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300溴结构域抑制剂以有效量给药,以防止癌细胞对受体酪氨酸激酶抑制剂或kras或braf抑制剂产生耐药性。
[0112]
实施方案11:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中所述egfr抑制剂选自:西妥昔单抗、帕尼单抗、扎芦木单抗、尼妥珠单抗、马妥珠单抗、吉非替尼、厄洛替尼、达可替尼、拉帕替尼、来那替尼、凡德他尼、耐昔妥珠单抗、奥希替尼、阿法替尼、ap26113、egfr抑制剂(cas no.879127-07-8)、egfr/erbb2/erbb-4抑制剂(cas no.881001-19-0)、egfr/erbb-2抑制剂(cas no.17924861-4)、egfr抑制剂ii(bibx 1382,cas no.196612-93-8)、egfr抑制剂iii(cas no.733009-42-2)、egfr/erbb-2/erbb-4抑制剂ii(cas no.944341-54-2)或pkcβii/egfr抑制剂(cas no.145915-60-2)。
[0113]
实施方案12:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300抑制剂是式(i)的化合物
[0114][0115]
其中
[0116]
r1选自卤素和-(任选取代的烃基,其包含1至20个碳原子和任选1至15个选自o、n和s的杂原子);
[0117]r21
选自氢、-(任选取代的c
1-6
烷基),其可以在碳原子之间包含1-3个氧原子,和-(任选取代的c
3-6
环烷基);
[0118]
r3选自-(任选取代的杂环基)、-(任选取代的碳环基)、-(任选取代的c
1-6
亚烷基)-(任选取代的杂环基)和-(任选取代的c
1-6
亚烷基)-(任选取代的碳环基);
[0119]
x1、x2和x3各自独立地选自n、ch和cr
x
,其中所述x1、x2和x3中的至少一个是n;
[0120]r31
选自
–
氢、
–c1-6-烷基和-(被一个或多个f取代的c
1-6-烷基);其中r3和任何r
31
可以任选地连接;和
[0121]
e不存在或选自
–
ch2–
、
–
chr
x
–
、
–
cr
x2
–
、
–
nh
–
、
–
nr
x
–
、
–o–
、
–
l1–
l2–
和
–
l2–
l1–
,其中l1选自
–
ch2–
、
–
chr
x
–
、
–
cr
x2
–
、
–
nh
–
、
–
nr
x
–
和
–o–
和l2选自
–
ch2–
、
–
chr
x
–
和
–
cr
x2
–
;
[0122]r6x
为
–
卤素、
–
oh、=o、c
1-6
烷基、c
1-6
卤代烷基、被一个或多个oh取代的c
1-6
烷基、任选被一个或多个r
xb
取代的单环芳基、任选被一个或多个r
xb
取代的单环杂芳基,任选被一个或多个r
xb
取代的单环环烷基,任选被一个或多个r
xb
取代的单环杂环烷基,任选被一个或多个r
xb
取代的单环环烯基,任选被一个或多个r
xb
取代的单环杂环烯基,其中所述r
xb
独立地选自-卤素、-oh、=o、c
1-4
烷基、c
1-2
卤代烷基、被一个或两个oh取代的c
1-2
烷基;
[0123]
其中环a可以进一步被一个或多个r
x
基团取代,其中环a上的任何两个r
x
基团可以任选地连接和/或环a上的任何r
x
基团可以任选地与r
21
连接;和/或其中环a可以进一步被一个基团r
x
取代,以便与r
6x
一起形成具有以下部分结构的双环部分:
[0124][0125]
其中环b为-(任选取代的杂环)或-(任选取代的碳环);
[0126]rx
各自独立地选自
–
卤素、
–
oh、-o-(任选取代的c
1-6
烷基)、-nh-(任选取代的c
1-6
烷基)、-n(任选取代的c
1-6
烷基)2、=o、-(任选取代的c
1-6
烷基)、
–
(任选取代的碳环基)、
–
(任选取代的杂环基)、-(任选取代的c
1-6
亚烷基)
–
(任选取代的碳环基)、-(任选取代的c
1-6
亚烷基)
–
(任选取代的杂环基)、
–
o-(任选取代的c
1-6
亚烷基)
–
(任选取代的碳环基)和
–
o-(任选取代的c
1-6
亚烷基)
–
(任选取代的杂环基)、以及
[0127]
其中任选取代的烃基、任选取代的c
3-6
环烷基、任选取代的杂环基、任选取代的杂
环、任选取代的碳环基、任选取代的碳环和任选取代的c
1-6
亚烷基的任选取代基独立地选自-(任选被一个或多个卤素取代的c
1-6
烷基)、-卤素、-cn、-no2、氧代、-c(o)r*、-coor*、-c(o)nr*r*、-nr*r*、-n(r*)-c(o)r*、-n(r*)-c(o)-or*、-n(r*)-c(o)-nr*r*、-n(r*)-s(o)2r*、-or*、-o-c(o)r*、-o-c(o)-nr*r*、-sr*、-s(o)r*、-s(o)2r*、-s(o)
2-nr*r*、-n(r*)-s(o)
2-nr*r*,任选被卤素或c
1-6
烷基取代的杂环基,和任选被卤素或c
1-6
烷基取代的碳环基;其中每个r*独立地选自h、任选被卤素取代的c
1-6
烷基,任选被卤素或c
1-6
烷基取代的杂环基,和任选被卤素或c
1-6
烷基取代的碳环基;其中连接到相同氮原子的任何两个r*可以任选地连接,和
[0128]
其中任选取代的c
1-6
烷基和任选取代的c
1-6
亚烷基的任选取代基独立地选自-卤素、-cn、-no2、氧代、-c(o)r**、-coor**、-c(o)nr**r**、-nr**r**、-n(r**)-c(o)r**、-n(r**)-c(o)-or**、-n(r**)-c(o)-nr**r**、-n(r**)-s(o)2r**、-or**、-o-c(o)r**、-o-c(o)-nr**r**、-sr**、-s(o)r**、-s(o)2r**、-s(o)
2-nr**r**和-n(r**)-s(o)
2-nr**r**,其中r**独立地选自h、任选被卤素取代的c
1-6
烷基,任选被卤素或c
1-6
烷基取代的杂环基,和任选被卤素或c
1-6
烷基取代的碳环基;其中连接到相同氮原子上的任何两个r**可以任选地连接。
[0129]
实施方案13:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300抑制剂是式(a)的芳基咪唑基异噁唑
[0130]
其中
[0131]r°
和r相同或不同,各自为h或c
1-c6烷基,其未被取代或被oh、-oc(o)r’或or’取代,其中r’是未被取代的c
1-c6烷基;
[0132]
w为n或ch;
[0133]
r1是未被取代或被取代的基团,并且选自c-连接的4至6元杂环基;c
3-c6环烷基;未被取代或被c
6-c
10
芳基、5-12元含n杂芳基、c
3-c6环烷基、oh、-oc(o)r’或or’取代的c
1-c6烷基,其中r’如上定义;和下式的螺环基团:
[0134][0135]
y为-ch
2-、-ch2ch
2-或-ch2ch2ch
2-;
[0136]
n为0或1;
[0137]
r2是选自c
6-c
10
芳基、5至12元含n杂芳基、c
3-c6环烷基和c
5-c6环烯基的基团,其中该基团是未取代的或取代的,并且其中c
6-c
10
芳基任选稠合到5或6元杂环上;
[0138]
或其药学上可接受的盐,并且其中优选所述芳基咪唑基异噁唑具有式(aa*):
[0139]
(aa*;ccs1477[cas 2222941-37-7]).
[0140]
实施方案14:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300抑制剂是式(ba)的化合物
[0141]
其中
[0142]
r1为-o(c
1-c3烷基);
[0143]
r6是任选独立地被一个或多个rb取代的苯基,其中rb选自
–
o-c
1-6
烷基、-o-c
3-6
环烷基、-o-芳基或-o-杂芳基,其中每个烷基、环烷基、芳基或杂芳基任选独立地被一个或多个卤素取代;
[0144]
或者其中cbp/p300抑制剂是式(bc)的化合物
[0145]
其中
[0146]
r1为-or5;
[0147]
r5为
–c1-6
烷基、-c
3-8
环烷基、杂环基、芳基或杂芳基;
[0148]
r6为-oh、卤素、氧代、-no2、-cn、-nh2、-c
1-6
烷基、-c
3-8
环烷基、-c
4-8
环烯基、杂环基、芳基、螺环烷基、螺杂环基、杂芳基、-oc
3-6
环烷基、-o芳基、-o杂芳基、-(ch2)n-or8、-c(o)r8'、-c(o)or8或-c(o)nr8r9、-nhc
1-6
烷基、-n(c
1-6
烷基)2、-s(o)2nh(c
1-6
烷基)、-s(o)2n(c
1-6
烷基)2、-s(o)2c
1-6
烷基、-n(c
1-6
烷基)so2c
1-6
烷基、-s(o)(c
1-6
烷基)、-s(o)n(c
1-6
烷基)2或-n(c
1-6
烷基)s(o)(c
1-6
烷基),其中每个烷基、环烷基、环烯基、杂环基、螺环烷基、螺杂环基、杂芳基或芳基任选被一个或多个r
10
取代;
[0149]
r7每次出现时独立地为-h、卤素、-oh、-cn、-oc
1-6
烷基、-nh2、-nh(c
1-6
烷基)、-n(c
1-6
烷基)2、-s(o)2h(c
1-6
烷基)、-s(o)2n(c
1-6
烷基)2、-s(o)2(c
1-6
烷基、-s(o)2oh、-c(o)c
1-6
烷基、-c(o)nh2、-c(o)nh(c
1-6
烷基)、-c(o)n(c
1-6
烷基)2、-c(o)oh、-c(o)oc
1-6
烷基、-n(c
1-6
烷基)so2c
1-6
烷基、-s(o)(c
1-6
烷基)、-s(o)n(c
1-6
烷基)2、-s(o)2nh2、-n(c
1-6
烷基)s(o)(c
1-6
烷基)或四唑;
[0150]r10
每次出现时独立地为-c
1-6
烷基、-c
2-6
烯基、-c
2-6
炔基、-c
3-8
环烷基、-c
4-8
环烯基、杂环基、杂芳基、芳基、-oh、卤素、氧代、-no2、-cn、-nh2、-oc
1-6
烷基、-oc
3-6
环烷基、-o芳基、-o杂芳基、-nhc
1-6
烷基、-n(c
1-6
烷基)2、-s(o)2nh(c
1-6
烷基)、-s(o)2n(c
1-6
烷基)2、-s(o)2c
1-6
烷基、-c(o)c
1-6
烷基、-c(o)nh2、-c(o)nh(-c
1-6
烷基)、-nhc(o)c
1-6
烷基-c(o)n(c
1-6
烷基)2、-c(o)oc
1-6
烷基、-n(c
1-6
烷基)so2-c
1-6
烷基、-s(o)(c
1-6
烷基)、-s(o)n(c
1-6
烷基)2或-n(c
1-6
烷基)s(o)(c
1-6
烷基),其中每个烷基、烯基、炔基、环烷基环烯基、杂环基、杂芳基或芳基任选被一个或多个-r
12
取代;
[0151]r12
在每次出现时独立地为卤素;
[0152]
m是0至5的整数;
[0153]
r是从0到5的整数。
[0154]
实施方案15:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中使用recist 1.1反应标准对动物中的靶病变或非靶病变测量癌症的缓慢进展。
[0155]
实施方案16:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中所述癌症是非小细胞肺癌(nsclc)。
[0156]
实施方案17:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300溴结构域抑制剂是实施方案12的式(i)化合物,受体酪氨酸激酶抑制剂是egfr抑制剂,受体酪氨酸激酶是egfr,癌症是nsclc,更优选地,nsclc包含egfr t790m突变,更优选地,其中受体酪氨酸激酶抑制剂是奥希替尼.
[0157]
实施方案18:根据任一前述实施方案所述的用于所述用途或方法的cbp/p300溴结构域抑制剂或组合物,其中cbp/p300溴结构域抑制剂是实施方案13的式(a)化合物,优选ccs1477(cas 2222941-37-7),受体酪氨酸激酶抑制剂是egfr抑制剂,受体酪氨酸激酶是egfr,癌症是nsclc,更优选nsclc包含egfr t790m突变,更优选其中受体酪氨酸激酶抑制剂是奥希替尼。
[0158]
关于上述实施方案13,注意到式(a)的化合物已经在wo2016170324、wo2018073586和wo2019202332中描述,所有申请和它们的公开内容全部引入本文作为参考,特别是关于式(a)的化合物的合成。
[0159]
在另一个实施方案中,提供了一种治疗动物癌症的方法,包括给有需要的动物给药cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂,所述抑制剂选自egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂,或kras或braf抑制剂,其中所述癌症包含相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不会减缓癌症的
进展。
[0160]
在另一个实施方案中,提供了用组合物治疗癌症的方法,所述组合物包含cbp/p300溴结构域抑制剂或其药学上可接受的盐和选自egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂的受体酪氨酸激酶抑制剂或kras或braf抑制剂的协同组合,其中所述癌症包含相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不会减缓癌症的进展。
[0161]
在另一个实施方案中,提供了一种延长动物对受体酪氨酸激酶抑制剂或kras或braf抑制剂癌症疗法的反应持续时间的方法,包括给患有癌症的动物施用cbp/p300溴结构域抑制剂或其药学上可接受的盐,其中与没有给药cbp/p300抑制剂或其药学上可接受的盐时对癌症治疗的响应持续时间相比,给药cbp/p300抑制剂或其药学上可接受的盐时对癌症治疗的响应持续时间延长,并且其中所述受体酪氨酸激酶抑制剂选自egfr、alk、met、her2、ros1、ret、ntrk1和axl或者是kras或braf抑制剂。
[0162]
在另一个实施方案中,提供了抑制癌细胞生长的方法,该方法包括给癌细胞给药cbp/p300溴结构域抑制剂和选自egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂或kras或braf抑制剂的受体酪氨酸激酶抑制剂,其中癌细胞包含相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不抑制癌细胞的生长,并且其中以有效量给药cbp/p300溴结构域抑制剂以防止癌细胞对激酶抑制剂产生耐药性。
[0163]
在另一个实施方案中,提供了一种在癌细胞中诱导细胞死亡的方法,包括给癌细胞给药cbp/p300溴结构域抑制剂和选自egfr、alk、met、her2、ros1、ret、ntrk1和axl抑制剂的受体酪氨酸激酶抑制剂或kras或braf抑制剂,其中所述癌细胞包含相应受体酪氨酸激酶或kras或braf的改变,并且其中单独的cbp/p300溴结构域抑制剂不诱导癌细胞中的细胞死亡。
[0164]
在一个实施方案中,受体酪氨酸激酶的改变可以是致癌改变,其中在第4部分的该实施方案中,术语“致癌改变”可以指细胞原癌基因的遗传改变。这些遗传变化/改变的结果可能是赋予细胞生长优势。在一个实施方案中,突变、基因扩增、基因融合和/或染色体重排的遗传机制可以激活人类肿瘤中的致癌基因。
[0165]
在另一个实施方案中,致癌改变是选自以下的egfr基因突变:egfr-外显子19缺失、egfr-l858r、egfr-t790m、egfr-t854a、egfr-d761y、egfr-l747s、egfr-g796s/r、egfr-l792f/h、egfr-l718q、egfr-外显子20插入、egfr-g719x(其中x是任何其他氨基酸)、egfr-l861x、egfr-s768i或者egfr扩增。在一个优选实施方案中,该改变是egfr-t790m。在另一个实施方案中,癌症是nsclc,并且改变是包含egfr外显子19缺失、l858r或t790m的突变。
[0166]
在另一个实施方案中,致癌改变是选自以下的ret基因突变或重排:kif5b-ret、ccdc6-ret、ncoa4-ret、trim33-ret、ret-v804l、ret-l730、ret-e732、ret-v738、ret-g810a、ret-y806、ret-a807或ret-s904f。
[0167]
在另一个实施方案中,致癌改变是选自her2外显子20插入或突变和her2-c805s、her2 t798m、her2 l869r、her2 g309e、her2 s310f或her2扩增的her2基因突变。
[0168]
在另一个实施方案中,致癌改变是ros1基因融合或重排,其选自:cd74-ros1、gopc-ros1、ezr-ros1、cep85l-ros1、slc34a2-ros1、sdc4-ros1、fig-ros1、tpm3-ros1、lrig3-ros1、kdelr2-ros1、ccdc6-ros1、tmem106b-ros1、tpd52l1-ros1、cltc-ros1和
lima1-ros1,或包括ros1 g2032r、d2033n、s1986y/f、l2026m和/或l1951r的突变。
[0169]
在另一个实施方案中,致癌改变是met基因扩增、met基因突变如met y1230c、d1227n、d1228v、y1248h以及met外显子14跳跃,或者选自tpr-met、clip2-met、tfg-met融合、kif5b-met融合的基因融合或重排。
[0170]
在另一个实施方案中,致癌改变是选自g12c、g12v、g12d、g13d、q61h或l或r、k117n的kras基因突变。
[0171]
在另一个实施方案中,致癌改变是选自以下的alk基因突变或基因融合或重排:eml4-alk、tfg-alk、kif5b-alk、klc1-alk、nsclc中的strn-alk、eml4-alk、c2orf44-alk、eml4-alk、tpm-alk、vcl-alk、tpm3-alk、eml4-alk或vcl-alk。
[0172]
在另一个实施方案中,致癌改变是选自v600e或v600k的braf基因突变。
[0173]
在另一个实施方案中,致癌改变是选自以下的ntkr基因融合或重排:tpm3-ntrk1、etv6-ntrk3、tpm3-ntrk1、tpr-ntrk1、tfg-ntrk1、ppl-ntrk1、etv6-ntrk3、tpr-ntrk1、mprip-ntrk1、cd74-ntrk1、sqstm1-ntrk1、trim24-ntrk2、lmna-ntrk、etv6-ntrk3、bcan-ntrk1、etv6-ntrk3、aml、gist、nfasc-ntrk1、bcan-ntrk1、agbl4-ntrk2、vcl-ntrk2、etv6-ntrk3、btbd1-ntrk3、rfwd2-ntrk1、rabgap1l-ntrk1、tp53-ntrk1、afap1-ntrk2、nacc2-ntrk2、oki-ntrk2、pan3-ntrk2,或选自f589l、g595r、g667c/s、a608d的ntkr1基因突变,或选自g623r、g696a的ntrk3基因突变。
[0174]
在另一个实施方案中,受体酪氨酸激酶抑制剂是egfr抑制剂。在另一个实施方案中,所述egfr抑制剂选自西妥昔单抗、帕尼单抗、扎芦木单抗、尼妥珠单抗、马妥珠单抗、吉非替尼、厄洛替尼、拉帕替尼、来那替尼、凡德他尼、耐昔妥珠单抗、奥希替尼、阿法替尼、达可替尼、ap26113、波奇替尼、egfr抑制剂(cas no.879127-07-8)、egfr/erbb2/erbb-4抑制剂(cas no.881001-19-0)、egfr/erbb-2抑制剂(cas no.17924861-4)、egfr抑制剂ii(bibx 1382,cas no.196612-93-8)、egfr抑制剂iii(cas no.733009-42-2)、egfr/erbb-2/erbb-4抑制剂ii(cas no.944341-54-2)或pkcβii/egfr抑制剂(cas no.145915-60-2)。
[0175]
在另一个实施方案中,受体酪氨酸激酶的改变是egfr基因中的突变。
[0176]
在另一个实施方案中,受体酪氨酸激酶抑制剂是ret抑制剂。在另一个实施方案中,ret抑制剂选自卡博替尼、凡德他尼、乐伐替尼、阿雷替尼、阿帕替尼、帕纳替尼、loxo-292、blu-667、或rxdx-105。
[0177]
在另一个实施方案中,受体酪氨酸激酶抑制剂是her2抑制剂。在另一个实施方案中,her2抑制剂选自曲妥珠单抗、透明质酸酶/曲妥珠单抗fam-trastumzumab deruxtecan、恩美曲妥珠单抗(ado-trastuzumab emtansine)、拉帕替尼、来那替尼、帕妥珠单抗、图卡替尼、波奇替尼,或者达可替尼。
[0178]
在另一个实施方案中,受体酪氨酸激酶抑制剂是ros1抑制剂。在另一个实施方案中,ros1抑制剂选自克唑替尼、色瑞替尼、布加替尼、洛拉替尼、恩曲替尼(etrectinib)、卡博替尼、ds-6051b、tpx-0005。
[0179]
在另一个实施方案中,受体酪氨酸激酶抑制剂是met抑制剂。在另一个实施方案中,met抑制剂选自:克唑替尼、卡博替尼、mgcd265、amg208、奥曲替尼(altiratinib)、戈伐替尼(golvatinib)、格来替尼(glesantinib)、福瑞替尼、avumatinib、tivatinib、赛沃替尼、amg337、卡马替尼和特泊替尼、omo-1[jnj38877618]或抗met抗体奥妥珠单抗
(onartuzumab)和依玛妥珠单抗(emibetuzumab)[ly2875358]或抗hgf抗体非拉妥组单抗(ficlatuzumab)[av-299]和利妥木单抗(rilotumumab)[amg102]。
[0180]
在另一个实施方案中,抑制剂是kras抑制剂。在另一个实施方案中,kras抑制剂选自amg510、mrtx849、jnj-74699157/ars-3248、bi1701963、bay-293或“ras(on)”抑制剂。
[0181]
在另一个实施方案中,受体酪氨酸激酶抑制剂是alk抑制剂。在另一个实施方案中,alk抑制剂选自克唑替尼、色瑞替尼、阿来替尼、洛拉替尼或布加替尼。
[0182]
在另一个实施方案中,抑制剂是braf抑制剂。在另一个实施方案中,braf抑制剂选自维莫非尼、达拉菲尼、康奈非尼(encorafenib)或任何非特异性raf抑制剂。
[0183]
在另一个实施方案中,受体酪氨酸激酶抑制剂是ntrk抑制剂。在另一个实施方案中,ntrk抑制剂选自恩曲替尼、拉罗替尼(larotrectinib)(loxo-101)、loco-195、ds-6051b、卡博替尼、梅沙替尼(merestinib)、tsr-011、plx7486、mgcd516、克唑替尼、瑞格拉非尼(regorafenib)、多韦替尼(dovitinib)、来他替尼(lestaurtinib)、bms-754807、达鲁舍替(danusertib)、enmd-2076、米哚妥林(midostaurin)、pha-848125ac、bms-777607、altriratinib、azd7451、mk5108、pf-03814735、sns-314、福瑞替尼、尼达尼布(nintedanib)、普纳替尼(ponatinib)、ono-5390556或tpx-0005。
[0184]
在另一个实施方案中,与单独的cbp/p300抑制剂或单独的受体酪氨酸激酶或kras或braf抑制剂相比,cbp/p300溴结构域抑制剂或其药学上可接受的盐和受体酪氨酸激酶抑制剂或kras或braf抑制剂的组合物或组合在治疗癌症中具有协同作用。在第4节的实施方案中,术语“协同”是指两种或多种药物之间的相互作用,其导致药物的总作用大于每种药物的单独作用的总和。在一个优选的实施方案中,协同效应是动物对cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂的组合的响应率增加。在另一个实施方案中,响应率的增加被测量为癌症治疗中功效的增加。
[0185]
在另一个实施方案中,由cbp/p300溴结构域抑制剂或其药学上可接受的盐和受体酪氨酸激酶或kras或braf抑制剂的组合物或组合提供的抗癌作用大于用相同剂量的cbp/p300抑制剂或受体酪氨酸激酶抑制剂或kras或braf抑制剂的单一疗法提供的抗癌作用。如在第4节的实施方案的上下文中所使用的,术语“抗癌”是指恶性或癌性疾病的治疗。在另一个实施方案中,本发明提供了用于使用或方法的组合物,其中由cbp/p300溴结构域抑制剂或其药学上可接受的盐和受体酪氨酸激酶抑制剂或kras或braf抑制剂的组合物或组合提供的抗癌效果为单独的单一疗法的至少2倍、至少3倍、至少5倍或至少10倍。
[0186]
在另一个实施方案中,cbp/p300溴结构域抑制剂或其药学上可接受的盐和受体酪氨酸激酶抑制剂或kras或braf抑制剂的组合物或组合延迟或降低了癌症对受体酪氨酸激酶抑制剂或kras或braf抑制剂的耐药性风险。如在第4节的实施方案的上下文中所使用的,术语“癌症的耐药性”是指药物疗效的降低;更具体地说,该术语可以指癌细胞产生耐药性。在另一个实施方案中,癌症在至少3个月、6个月、9个月、12个月、24个月、48个月或60个月内不会对受体酪氨酸激酶抑制剂或kras或braf抑制剂产生耐药性。在另一个实施方案中,cbp/p300溴结构域抑制剂以有效量给药,以防止癌细胞对受体酪氨酸激酶抑制剂或kras或braf抑制剂产生耐药性。
[0187]
在另一个实施方案中,cbp/p300溴结构域抑制剂抑制cbp和/或p300的溴结构域。p300(也称为组蛋白乙酰转移酶p300、e1a结合蛋白p300、e1a相关蛋白p300)和cbp(也称为
creb结合蛋白或crebbp)是两种结构非常相似的转录共激活蛋白。
[0188]
如在第4节的实施方案的上下文中使用的,术语“cbp/p300溴结构域抑制剂”可以被认为是指与cbp溴结构域和/或p300溴结构域结合并抑制和/或降低cbp和/或p300的生物活性或功能的化合物。在一些实施方案中,cbp/p300溴结构域抑制剂可以主要(例如,单独)通过与cbp溴结构域和/或p300溴结构域的接触和/或相互作用来结合cbp和/或p300。在一些实施方案中,cbp/p300溴结构域抑制剂可以通过与cbp溴结构域和/或p300溴结构域以及额外的cbp和/或p300残基和/或结构域的接触和/或相互作用而结合至cbp和/或p300。在一些实施方案中,cbp/p300溴结构域抑制剂可以基本上或完全抑制cbp和/或p300的生物活性。在一些实施方案中,生物活性可以是cbp和/或p300的溴结构域与染色质(例如,与dna相关的组蛋白)和/或另一种乙酰化蛋白的结合。在第4节的实施方案的上下文中的某些实施方案中,抑制剂可具有小于约50μm、小于约1μm、小于约500nm、小于约100nm、小于约10nm或小于约1nm的ic
50
或结合常数。在一些实施方案中,cbp/p300溴结构域抑制剂可以结合并抑制cbp溴结构域。在一些实施方案中,cbp/p300溴结构域抑制剂可以结合并抑制p300溴结构域。在一些实施方案中,cbp/p300溴结构域抑制剂可能不抑制cbp/p300的组蛋白乙酰转移酶活性。
[0189]
在一个实施方案中,cbp/p300溴结构域抑制剂是式(i)的化合物。在一个实施方案中,cbp/p300溴结构域抑制剂是式(a)的化合物,优选ccs1477(cas 2222941-37-7)。在另一个实施方案中,cbp/p300溴结构域抑制剂是ft-7051。在另一个实施方案中,式(i)化合物、式(a)化合物、优选ccs1477或ft-7051是药物的日剂量,其浓度选自包括10mg、15mg、25mg、50mg、100mg、150mg或200mg的列表。在另一个实施方案中,ccs1477每周给药2、3、4、5、6或7天。在另一个实施方案中,ccs1477每天给药两次。在另一个实施方案中,对癌细胞的给药包括使癌细胞与cbp/p300抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂接触。
[0190]
在另一个实施方案中,剂量取决于多种因素,包括患者的年龄、体重和状况以及给药途径。每日剂量可以在很宽的范围内变化,并且将根据每个特定病例的个体需要进行调整。然而,典型地,当化合物单独给药于成年人时,每种给药途径所采用的剂量可以在0.0001至50mg/kg的范围内,最常见的是在0.001至10mg/kg体重的范围内,例如0.01至1mg/kg。这种剂量可以例如每天给药1至5次。对于静脉注射,合适的日剂量可以是0.0001至1mg/kg体重,优选0.0001至0.1mg/kg体重。日剂量可以单剂量给药或根据分剂量方案给药。
[0191]
在另一个实施方案中,可以使用recist 1.1反应标准对受试者/动物中靶病变或非靶病变来测量癌症的进展或对癌症治疗的反应持续时间。
[0192]
在另一个实施方案中,术语“不减缓癌症的进展”可以在第4节的实施方案中定义为没有实现任何recist 1.1临床反应的受试者。在另一个实施方案中,术语“不减缓癌症的进展”可以在第4节的实施方案中定义为没有实现部分recist 1.1临床反应的受试者/动物。在另一个实施方案中,根据recist 1.1,术语“不减缓癌症进展”被测量为无客观响应率和/或无进展生存期无增加。在另一个实施方案中,术语“不减缓癌症的发展”被测量为靶病变最长直径总和的减少小于30%,以靶病变最长直径的基线总和作为参考。
[0193]
在某些实施方案中,癌症选自听神经瘤、急性白血病、急性淋巴细胞白血病、急性髓细胞白血病、急性t细胞白血病、基底细胞癌、胆管癌、膀胱癌、脑癌、乳腺癌、支气管癌、宫颈癌、软骨肉瘤、脊索瘤、绒毛膜癌、慢性白血病、慢性淋巴细胞白血病、慢性髓细胞性白血
病、慢性髓细胞性白血病、结肠癌、结肠直肠癌、颅咽管瘤、囊腺癌、弥漫性大b细胞淋巴瘤、增殖障碍性病变、胚胎癌、子宫内膜癌、内皮肉瘤、室管膜瘤、上皮癌、红白血病、食道癌、雌激素受体阳性乳腺癌、原发性血小板增多症、尤文氏肉瘤、纤维肉瘤、滤泡性淋巴瘤、生殖细胞睾丸癌、神经胶质瘤、胶质母细胞瘤、胶质肉瘤、重链疾病、头颈癌、成血管细胞瘤、肝癌、肝细胞癌、激素不敏感的前列腺癌、平滑肌肉瘤、白血病、脂肪肉瘤、肺癌、淋巴管内皮肉瘤、淋巴管肉瘤、淋巴母细胞性白血病、淋巴瘤、t细胞或b细胞来源的淋巴恶性肿瘤、髓样癌、髓母细胞瘤、黑色素瘤、脑膜瘤、间皮瘤、多发性骨髓瘤、骨髓性白血病、骨髓瘤、粘液肉瘤、成神经细胞瘤、nut中线癌(nmc)、非小细胞肺癌(nsclc)、少突胶质细胞瘤、口腔癌、骨肉瘤、卵巢癌、胰腺癌、乳头状腺癌、乳头状癌、松果体瘤、真性红细胞增多症、前列腺癌、直肠癌、肾细胞癌、视网膜母细胞瘤、横纹肌肉瘤、肉瘤、皮脂腺癌、精原细胞瘤、皮肤癌、小细胞肺癌、实体瘤(癌和肉瘤)、小细胞肺癌、胃癌、鳞状细胞癌、滑膜瘤、汗腺癌、甲状腺癌、瓦尔登斯特伦巨球蛋白血症、睾丸肿瘤、子宫癌和肾母细胞瘤。在某些实施方案中,癌症是黑素瘤、nsclc、肾癌、卵巢癌、结肠癌、胰腺癌、肝细胞癌或乳腺癌。在任一方法的某些实施方案中,癌症是肺癌、乳腺癌、胰腺癌、结肠直肠癌和/或黑色素瘤。在某些实施方案中,癌症是肺癌。在某些实施方案中,肺癌是非小细胞肺癌nsclc。在某些实施方案中,癌症是乳腺癌。在某些实施方案中,癌症是黑素瘤。在某些实施方案中,癌症是结肠直肠癌。
[0194]
在另一个实施方案中,cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂作为单一组合物同时给药动物。在另一个实施方案中,cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂分别给药动物。在另一个实施方案中,cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂同时给药动物。在另一个实施方案中,在给药受体酪氨酸激酶抑制剂或kras或braf抑制剂之前,给药动物cbp/p300溴结构域抑制剂。在另一个实施方案中,动物是人。
[0195]
在一个实施方案中,术语药剂(例如药物制剂)的“有效量”可以指在必要的剂量和时间段内达到所需治疗或预防效果的有效量。在一些实施方案中,有效量是指cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂的量,其(i)治疗特定疾病、病症或障碍,(ii)减弱、改善或消除特定疾病、病症或障碍的一种或多种症状,或(iii)预防或延迟本文所述特定疾病、病症或障碍的一种或多种症状的发作。在一些实施方案中,有效量的cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂可以减少癌细胞的数量;可能缩小肿瘤大小;可以抑制(即,在一定程度上减缓并优选停止)癌细胞向外周器官的浸润;可以抑制(即,在一定程度上减缓并优选停止)肿瘤转移;可能在一定程度上抑制肿瘤生长;和/或可以在一定程度上缓解一种或多种与癌症相关的症状。对于癌症治疗,功效可以例如通过评估疾病进展时间(ttp)和/或确定响应率(rr)来测量。在一些实施方案中,有效量是本文所述的cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂实体足以显著降低药物耐受性或药物耐受性持续癌细胞的活性或数量的量。
[0196]
在一个实施方案中,本发明的化合物可以与用于治疗癌症的放射疗法或另一种化疗剂联合给药于人类或动物患者。在另一个实施方案中,可以提供联合治疗,其中cbp/p300抑制剂或rtk抑制剂或kras或braf抑制剂与放疗同时或相继给药;或者与另一种化疗剂或多种化疗剂同时顺序给药或作为联合制剂给药,用于治疗癌症。所述或每种其他化疗剂通常是常规用于所治疗的癌症类型的药剂。在一个实施方案中,用于组合的化疗剂的种类可
以是例如用于治疗前列腺癌的雄激素受体拮抗剂,例如恩杂鲁胺,和cyp17a1(17a-羟化酶/c 17,20裂解酶)的抑制剂,例如阿比特龙。在其他实施方案中,联合治疗中的其他化疗剂可以包括多西他赛。在一个实施方案中,第4节中的术语“组合”可以指同时、分别或顺序给药。当依次或分别给药时,第二种组分的延迟给药不应导致组合的有益效果丧失。
[0197]
在另一个实施方案中,对cbp/p300溴结构域抑制剂和受体酪氨酸激酶抑制剂或kras或braf抑制剂的反应是持续的反应。在一个实施方案中,“持续反应”可以指停止治疗后对减少肿瘤生长的持续作用。例如,与给药阶段开始时的大小相比,肿瘤大小可以保持相同或更小。
[0198]
在另一个实施方案中,术语“治疗”(以及诸如“治疗”或“处理”的变体)可以指试图改变被治疗的个体或细胞的自然进程的临床干预,并且可以为了预防或在临床病理学过程中进行。治疗的理想效果可能包括预防疾病的发生或复发、减轻症状、减少疾病的任何直接或间接病理后果、稳定(即不恶化)疾病状态、预防转移、降低疾病进展的速率、改善或减轻疾病状态、与未接受治疗时的预期生存相比延长生存或改善预后中的一种或多种。在某些实施方案中,cbp/p300溴结构域抑制剂和受体酪氨酸激酶或kras或braf抑制剂可用于延缓疾病或障碍的发展或减缓疾病或障碍的进展。在一个实施方案中,需要治疗的那些个体可以包括已经患有该病症或障碍的那些个体以及倾向于患有该病症或障碍的那些个体(例如,通过基因突变或基因或蛋白质的异常表达)或其中该病症或障碍将被预防的那些个体。
[0199]
在一个实施方案中,术语“延迟”可以指延缓、阻碍、减缓、迟延、稳定和/或推迟疾病(如癌症)的发展或疾病的耐药性。这种延迟时间长短不一,取决于疾病史和/或接受治疗的个体。对本领域技术人员来说显而易见的是,充分或显著的延迟实际上可以包括预防,因为个体不会发展成疾病。例如,晚期癌症,如转移的发展,可以被延迟。
5.实施例
[0200]
以下实施例仅仅是说明性的,并且将以进一步的方式描述本发明。这些实施例不应被解释为将本发明限制于此。
[0201]
化合物00003(化合物b)、00004(化合物a)、00030、00071和化合物c的制备描述如下。如果认为有帮助,给出了中间体化合物和/或接近上述化合物的化合物的合成路线。
[0202]
一般实验方法
[0203]
lcms方法:
[0204]
方法a:仪器:agilent 1260bin.泵:g1312b,脱气装置;自动取样器,colcom,dad:agilent g1315d,220-320nm,msd:agilent lc/msd g6130b esi,pos/neg 100-800,elsd alltech 3300气流1.5ml/min,气体温度:40℃;柱:waters xselect
tm c18,30x2.1mm,3.5μ,温度:35℃,流量:1ml/min,梯度:t0=5%a,t
1.6min
=98%a,t
3min
=98%a,后运行时间(posttime):1.3min,洗脱液a:0.1%甲酸于乙腈中,洗脱液b:0.1%甲酸于水中)。
[0205]
方法b:仪器:agilent 1260bin.泵:g1312b,脱气装置;自动取样器,colcom,dad:agilent g1315d,220-320nm,msd:agilent lc/msd g6130b esi,pos/neg 100-800,elsd alltech 3300气流1.5ml/min,气体温度:40℃;柱:waters xselect
tm c18,50x2.1mm,3.5μ,温度:35℃,流量:0.8ml/min,梯度:t0=5%a,t
3.5min
=98%a,t
6min
=98%a,后运行时间:2min;洗脱液a:0.1%甲酸于乙腈中,洗脱液b:0.1%甲酸于水中).
g4212b,220-320nm,柱:od-h 250x4.6mm,温度:25℃,流量:1ml/min,无梯度:90/10,time:30min,洗脱液a:庚烷,洗脱液b:乙醇)。
[0217]
制备型反相色谱:
[0218]
方法a:设备类型:reveleris
tm
prep mplc;柱:phenomenex luna c18(150x25mm,10μ);流量:40ml/min;柱温度:室温;洗脱液a:0.1%(v/v)甲酸于水中,洗脱液b:0.1%(v/v)甲酸于乙腈中;梯度:t=0min 5%b,t=1min 5%b,t=2min 30%b,t=17min 70%b,t=18min 100%b,t=23min 100%b;检测型uv:220/254nm。将适当的级分合并并冻干。
[0219]
方法b:设备类型:reveleris
tm
prep mplc;柱:waters xselect
tm csh c18(145x25mm,10μ);流量:40ml/min;柱温度:室温;洗脱液a:10mm碳酸氢铵的水溶液ph=9.0);洗脱液b:99%乙腈 1%10mm碳酸氢铵的水溶液;梯度:t=0min 5%b,t=1min 5%b,t=2min 30%b,t=17min 70%b,t=18min 100%b,t=23min 100%b;检测型uv:220/254nm。将适当的级分合并并冻干。
[0220]
手性(制备型)sfc
[0221]
方法a:(柱:sfc仪器模块:waters prep100q sfc系统,pda:waters 2998,馏分收集器:waters 2767;柱:phenomenex lux amylose-1(250x20mm,5μm),柱温度:35℃;流速:100ml/min;abpr:170bar;洗脱液a:co2,洗脱液b:20mm氨的甲醇溶液;无梯度10%b,时间:30min,检测:pda(210-320nm);基于pda的馏分收集)。
[0222]
方法b:(柱:sfc仪器模块:waters prep100q sfc系统,pda:waters2998,馏分收集器:waters 2767;柱:phenomenex lux celulose-1(250x20mm,5μm),柱温度:35℃;流速:100ml/min;abpr:170bar;洗脱液a:co2,洗脱液b:20mm氨的甲醇溶液;无梯度10%b,时间:30min,检测:pda(210-320nm);基于pda的级分收集)。
[0223]
方法c:(柱:sfc仪器模块:waters prep100q sfc system,pda:waters 2998;柱:chiralpak ic(100x4.6mm,5μm),柱温度:35℃;流速:2.5ml/min;abpr:170bar;洗脱液a:co2,洗脱液b:含20mm氨的甲醇;t=0min5%b,t=5min 50%b,t=6min 50%b,检测:pda(210-320nm);基于pda的馏分收集)。
[0224]
方法d:(柱:sfc仪器模块:waters prep 100sfc uv/ms定向系统;waters 2998 photodiode array(pda)检测器;waters acquity qda ms检测器;waters 2767样品管理器;柱:waters torus 2-pic 130a obd(250x19mm,5μm);柱温度:35℃;流量:70ml/min;abpr:120bar;洗脱液a:co2,洗脱液b:含20mm氨的甲醇;线性梯度:t=0min 10%b,t=4min 50%b,t=6min 50%b;检测:pda(210-400nm);基于pda tic的级分收集)。
[0225]
起始材料
[0226]
标准试剂和溶剂以最高商业纯度获得并照此使用,购买的具体试剂如下所述。
[0227]
[0228]
[0229][0230]
关键中间体的合成步骤
[0231]
中间体1:1-(5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮
[0232][0233]
在1l钢制高压釜中,向6-甲基烟酸甲酯(100g,662mmol)在乙酸(250ml)中的溶液中加入氧化铂(iv)(0.5g,2.202mmol),之后在10巴氢气气氛并于60℃搅拌反应混合物。观察到快速的氢气消耗,并且将高压釜再填充几次,直到氢气消耗停止并且还原完成。将混合物冷却至室温,用硅藻土过滤。浓缩滤液以提供作为非对映异构体混合物的6-甲基哌啶-3-羧酸甲酯乙酸盐(143.8g,100%),其原样用于下一步骤。gcms(方法a):tr 2.40(80%)和2.48min(20%),100%,ms(ei)157.1(m) ,142.1(m-me) .向6-甲基哌啶-3-羧酸甲酯乙酸盐(53克,244毫摩尔)在水(500ml)和二氯甲烷(500ml)混合物中的溶液中小心加入碳酸氢钠(82克,976mmol)(泡腾!!)之后缓慢加入乙酸酐(29.9克,293mmol)。将反应混合物在室温下搅拌2小时。分离有机层,用硫酸钠干燥,过滤并真空浓缩,得到1-乙酰基-6-甲基哌啶-3-羧酸甲酯(49g,100%),为黄色油状物。将1-乙酰基-6-甲基哌啶-3-羧酸甲酯(49g,246mmol)的氨的甲醇(7n,500ml,3.5mol)溶液在压力容器中于120℃搅拌40小时。将混合物
冷却至室温并浓缩,得到浅黄色固体。将该固体溶解在二氯甲烷中,并通过硅胶塞过滤。浓缩滤液,得到1-乙酰基-6-甲基哌啶-3-甲酰胺,为灰白色固体,其原样用于下一步。将上一步得到的1-乙酰基-6-甲基哌啶-3-甲酰胺(266mmol)在三氯氧磷(500ml,5.37mol)中的溶液在室温搅拌16小时。真空蒸发反应混合物,得到稠油。该油与甲苯共蒸发两次,并小心在冷饱和碳酸钠(泡腾!)和乙酸乙酯之间区分。将有机层与碱性水层分离,用硫酸钠干燥,过滤并真空浓缩,得到稠油状产物,静置后固化。将粗产物溶于二氯甲烷中,用硅胶塞过滤(用10%甲醇二氯甲烷溶液洗脱)。这提供了1-乙酰基-6-甲基哌啶-3-甲腈(28g,63%),为静置时固化的油状物。gcms(方法a):tr 3.78(63%)和3.89min(378%),100%,ms(ei)166.1(m) .向1-乙酰基-6-甲基哌啶-3-甲腈(23g,138mmol)在乙醇(300ml)中的溶液中加入羟胺溶液(50%溶于水,25.4ml,415mmol),然后将反应混合物在回流下搅拌16小时。将反应混合物浓缩并与乙酸乙酯共蒸发三次至干,得到粘性固体形式的1-乙酰基-n-羟基-6-甲基哌啶-3-甲脒(carboximidamide)。lcms(方法a):tr 0.13min,100%,ms(esi)200.2(m h) .假设定量产率,则在下一步中按原样使用产物。向来自前一步骤的1-乙酰基-n-羟基-6-甲基哌啶-3-甲脒(23g,138mmol)在乙醇(500ml)中的溶液中加入乙酸(23.79ml,416mmol)和50%雷尼镍在水(5ml)中的浆液,然后将反应混合物在氢气气氛下于50℃搅拌2天。将混合物用硅藻土过滤,用一些乙醇洗涤并浓缩,得到70g稠油。将其与乙酸乙酯共蒸发两次,并在真空中充分干燥,得到1-乙酰基-6-甲基哌啶-3-甲脒乙酸盐(33g,98%),为绿黄色油状物,用于下一步。lcms(方法a):tr 0.14min,90%,ms(esi)184.1(m h) .在氮气气氛(60ml)下,向钠(18.14g,789mmol)在无水甲醇中的溶液中加入1-乙酰基-6-甲基哌啶-3-甲脒乙酸盐(32g,132mmol)和丙二酸二甲酯(26.1g,197mmol),然后将反应混合物在50℃搅拌16小时。浓缩反应混合物,溶于水(300ml),用6n盐酸酸化至ph 4,并使其沉淀。滤出沉淀,得到1-(5-(4,6-二羟基嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮为黄色固体(10.4克,31%),在下一步骤中原样使用。1-(5-(4,6-二羟基嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(10.4g,41.4mmol)的三氯氧磷(200ml,2146mmol)悬浮液在50℃搅拌。约3小时后固体缓慢溶解。5小时后,真空浓缩反应混合物,并用甲苯共蒸发两次。剩余的油用冰小心淬灭,用饱和碳酸氢钠水溶液中和,并用乙酸乙酯(2x 100ml)萃取。合并的有机层用硫酸钠干燥并真空浓缩,得到1-(5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(中间体1,6.8g,57%),为静置时固化的黄色油状物。lcms(方法a):tr 1.88min,100%,ms(esi)288.1(m h) .
[0234]
中间体2:1-((2s,5r)-5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮
[0235][0236]
向n-乙酰基-d-亮氨酸(1kg,5.77mol)在乙醇(1.5l)中的溶液中加入6-甲基哌啶-3-羧酸甲酯(934g,2.38mol,根据中间体1制备)的乙酸乙酯(3l)溶液,并将混合物加热至40
℃。使所得溶液在16小时内达到室温,在此期间发生沉淀。滤出沉淀,用乙醚(500ml)洗涤并风干,得到白色固体形式的粗(3r,6s)-6-甲基哌啶-3-羧酸甲酯乙酰-d-亮氨酸盐(287g,34%)。将粗(3r,6s)-6-甲基哌啶-3-羧酸甲酯乙酰-d-亮氨酸盐(287g,869mmol)从乙醇和乙酸乙酯1:2(1l)的热混合物中结晶出来。滤出沉淀,将滤饼在乙醚和正戊烷1:1(500ml)的混合物中研磨。滤出沉淀并风干,得到白色固体形式的(3r,6s)-6-甲基哌啶-3-羧酸甲酯乙酰-d-亮氨酸盐(128g,44%)。向(3r,6s)-6-甲基哌啶-3-羧酸甲酯乙酰-d-亮氨酸盐(128g,387mmol)的二氯甲烷(1l)溶液中加入饱和碳酸钠溶液(1l)。将两相系统剧烈搅拌10分钟,分离各层。有机层用硫酸钠干燥并过滤,得到透明溶液。然后,加入三乙胺(65ml,465mmol)和乙酸酐(44ml,465mmol),混合物在室温搅拌1小时。用饱和碳酸氢钠溶液洗涤混合物,用硫酸钠干燥并浓缩,得到淡黄色固体形式的(3r,6s)-1-乙酰基-6-甲基哌啶-3-羧酸甲酯(93g)。向高压釜中加入溶于甲醇(600ml,4200mmol)的7n氨水中的(3r,6s)-1-乙酰基-6-甲基哌啶-3-羧酸甲酯(93g,387mmol),并加热至60℃持续3天。将混合物浓缩,得到淡黄色油状的(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲酰胺(102g)。假设定量产率,则在下一步中按原样使用产物。手性lc(方法a)tr=12.35min,》98%ee。向(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲酰胺(50g,271mmol)的二氯甲烷(500ml)溶液中逐份加入三乙基氧鎓四氟硼酸盐(77g,407mmol),并将混合物在室温下搅拌4小时。缓慢加入甲醇中的7n氨水(200ml,9.15mol),并将混合物在室温下搅拌16小时。将混合物浓缩,得到粉红色固体形式的(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲脒(50g),用于下一步。向5.4m甲醇钠的甲醇(99ml,535mmol)溶液于甲醇(200ml)中加入(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲脒(49g,267mmol)的甲醇(400ml)和丙二酸二甲酯(61.4ml,535mmol)溶液。将混合物加热至50℃,并搅拌24小时。用浓盐酸将混合物酸化(ph~3),并浓缩至较小体积。残留物用二氧化硅(20%甲醇二氯甲烷溶液)过滤并浓缩,得到橙色油状物。粗产物用硅胶柱色谱法(0%至20%甲醇二氯甲烷溶液)纯化,得到1-((2s,5r)-5-(4,6-二羟基嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(12g,17%)为无色胶状物。lcms(方法c):tr 0.17min,100%,ms(esi)252.1(m h) .1-((2s,5r)-5-(4,6-二羟基嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(12g,47.8mmol)的三氯氧磷(80ml,858mmol)溶液在60℃搅拌24小时。浓缩反应混合物,并用甲苯共蒸发两次,得到黄色油状物。将油溶于乙酸乙酯中,并用饱和碳酸氢钠溶液洗涤。水层用乙酸乙酯萃取两次。用盐水洗涤合并的有机层,用硫酸钠干燥并浓缩,得到黄色油状物。用硅胶柱色谱法(0%至20%四氢呋喃于甲苯中)纯化油,得到1-((2s,5r)-5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(中间体2,1.5g,11%),为无色胶状物。lcms(方法b):tr 3.34min,100%,ms(esi)288.0(m h) ;手性uplc(方法:a)tr 2.54min,》95%ee和de.
[0237]
中间体3:1-((2s,5r)-5-(4-氯-6-(吡嗪-2-基)嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮的合成
3-羧酸甲酯(93g)。1h-nmr(400mhz,cdcl3,旋转异构体混合物)δ5.02
–
4.87(m,0.5h),4.84
–
4.68(m,0.5h),4.18
–
4.05(m,0.5h),3.89
–
3.77(m,0.5h),3.71(d,j=11.6hz,3h),3.31
–
3.18(m,0.5h),2.79
–
2.67(m,0.5h),2.51
–
2.31(m,1h),2.11(d,j=6.7hz,3h),2.01
–
1.90(m,1h),1.88
–
1.55(m,3h),1.33
–
1.21(m,1.5h),1.20
–
1.06(m,1.5h).向高压釜中加入(3r,6s)-1-乙酰基-6-甲基哌啶-3-羧酸甲酯(93g,387mmol)的7n氨的(600ml,4200mmol)甲醇溶液,并加热至60℃持续3天。将混合物浓缩,得到淡黄色油状的(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲酰胺(102g)。假设定量产率,则在下一步中按原样使用产物。1h-nmr(400mhz,dmso-d6,旋转异构体混合物)δ7.38(s,1h),6.89(d,j=24.7hz,1h),4.76
–
4.59(m,0.5h),4.39
–
4.24(m,0.5h),4.16
–
4.01(m,0.5h),3.72
–
3.51(m,0.5h),3.14
–
2.99(m,0.5h),2.68
–
2.51(m,0.5h),2.30
–
2.12(m,0.5h),2.11
–
1.92(m,3.5h),1.78
–
1.38(m,4h),1.23
–
1.11(m,1.5h),1.09
–
0.94(m,1.5h);chiral lc(方法a)tr=12.35min,》98%ee.向(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲酰胺(50g,271mmol)的二氯甲烷(500ml)溶液中逐份加入三乙基氧鎓四氟硼酸盐(77g,407mmol),并将混合物在室温下搅拌4小时。缓慢加入7n氨的甲醇(200ml,9.15mol)溶液,并将混合物在室温下搅拌16小时。将混合物浓缩,得到粉红色固体形式的(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲脒(50g),用于下一步。向5.4m甲醇钠的甲醇(99ml,535mmol)溶液于甲醇(200ml)中加入(3r,6s)-1-乙酰基-6-甲基哌啶-3-甲脒(49g,267mmol)的甲醇(400ml)和丙二酸二甲酯(61.4ml,535mmol)溶液。将混合物加热至50℃,并搅拌24小时。用浓盐酸将混合物酸化(ph~3),并浓缩至较小体积。残留物用二氧化硅(20%甲醇的二氯甲烷溶液)过滤并浓缩,得到橙色油状物。粗产物用硅胶柱色谱法(0%至20%甲醇的二氯甲烷溶液)纯化,得到1-((2s,5r)-5-(4,6-二羟基嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(12克,17%)为无色胶状物。lcms(方法c):t
r 0.17min,100%,ms(esi)252.1(m h)
.1-((2s,5r)-5-(4,6-二羟基嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(12g,47.8mmol)的三氯氧磷(80ml,858mmol)溶液在60℃搅拌24小时。浓缩反应混合物,并用甲苯共蒸发两次,得到黄色油状物。将油溶于乙酸乙酯中,并用饱和碳酸氢钠溶液洗涤。水层用乙酸乙酯萃取两次。用盐水洗涤合并的有机层,用硫酸钠干燥并浓缩,得到黄色油状物。用硅胶柱色谱法(甲苯中0%至20%四氢呋喃)纯化油,得到1-((2s,5r)-5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(1.5g,11%)为无色胶状物。1h-nmr(400mhz,dmso-d6,旋转异构体混合物)δ7.95(d,j=7.3hz,1h),4.85
–
4.72(m,1h),4.69
–
4.62(m,1h),4.23
–
4.13(m,1h),4.07
–
3.98(m,1h),3.97
–
3.88(m,1h),3.00
–
2.89(m,1h),2.81
–
2.67(m,1h),2.09
–
1.72(m,7h),1.71
–
1.58(m,2h),1.25
–
1.14(m,3h),1.12
–
1.05(m,2h);lcms(方法b):tr3.34min,ms(esi)288.0(m h)
;手性uplc(方法:a)t
r 2.54min,》95%ee和de.在氩气下,2-三丁基甲锡烷基吡嗪(607mg,1.65mmol),1-((2s,5r)-5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(500mg,1.74mmol)和双(三苯基膦)氯化钯(ii)(244mg,0.34mmol)的1,4-二噁烷(20ml)溶液加热至100℃并搅拌32小时。用含1%三乙胺的二氯甲烷稀释混合物,并涂在二氧化硅上。用硅胶柱色谱法(含1%三乙胺的0%至40%乙腈的二氯甲烷)纯化,得到1-((2s,5r)-5-(4-氯-6-(吡嗪-2-基)嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(中间体3,134mg,18%)为橙色胶状物。1h-nmr(400mhz,dmso-d6,旋转异构体混合物)δ9.46
–
9.41(m,1h),8.80
–
8.76(m,1h),8.65
–
8.59(m,1h),8.33
–
8.29(m,1h),7.66
–
7.59(m,1h),4.86
–
4.70(m,0.5h),4.27
–
4.17(m,0.5h),4.09
–
3.97(m,0.5h),3.55
–
3.41(m,0.5h),3.06
–
2.98(m,0.5h),2.88
–
2.82
(m,0.5h),2.10
–
1.90(m,6h),1.89
–
1.76(m,0.5h),1.75
–
1.61(m,1.5h),1.29
–
1.20(m,1.5h),1.17
–
1.10(m,1.5h);lcms(方法c):t
r 1.81min,ms(esi)331.1(m h)
.
[0240]
最终产品的合成程序
[0241]
实施例1:合成1-((2s,5r)-2-甲基-5-(4-((5-甲基吡啶-3-基)氨基)-6-(吡嗪-2-基)嘧啶-2-基)哌啶-1-基)乙-1-酮(00001)和1-((2r,5s)-2-甲基-5-(4-((5-甲基吡啶-3-基)氨基)-6-(吡嗪-2-基)嘧啶-2-基)哌啶-1-基)乙-1-酮(00002)
[0242][0243]
向3-氨基-5-甲基吡啶(0.751g,6.94mmol)在四氢呋喃(20ml)中的溶液中加入1m双(三甲基甲硅烷基)氨基锂在四氢呋喃(6.94ml,6.94mmol)中的溶液,并将混合物在室温下搅拌10分钟。接下来,添加1-(5-(4,6-二氯嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(中间体1.1g,3.47mmol)于四氢呋喃(20ml)中的溶液,混合物在室温下搅拌2小时。将混合物倒入饱和氯化铵溶液中,用乙酸乙酯萃取两次。合并的有机层用盐水洗涤一次,用硫酸钠干燥并浓缩,得到黄色固体。用硅胶柱色谱法(0%至5%甲醇的二氯甲烷溶液)纯化固体,得到1-(5-(4-氯-6-((5-甲基吡啶-3-基)氨基)嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(788mg,60%)为黄色泡沫。lcms(方法b):tr 1.81min,100%,ms(esi)360.1(m h) .在氮气下,2-(三丁基甲锡烷基)吡嗪(103mg,0.28mmol),1-(5-(4-氯-6-((5-甲基吡啶-3-基)氨基)嘧啶-2-基)-2-甲基哌啶-1-基)乙-1-酮(50mg,0.14mmol)和双(三苯基膦)二氯化钯(ii)(9.75mg,0.01mmol)溶于n,n-二甲基甲酰胺(3ml)中。将混合物加热至80℃并保持24小时,然后冷却至室温。使用乙腈通过c18塞洗脱混合物,用反相色谱法(方法b)纯化滤液并冻干,得到1-(2-甲基-5-(4-((5-甲基吡啶-3-基)氨基)-6-(吡嗪-2-基)嘧啶-2-基)哌啶-1-基)乙-1-酮(22mg,37%)为白色固体。将获得的顺式对映异构体混合物提交给手性制备sfc(方法a)并冻干,得到两种立体异构体。1-((2s,5r)-2-甲基-5-(4-((5-甲基吡啶-3-基)氨基)-6-(吡嗪-2-基)嘧啶-2-基)哌啶-1-基)乙-1-酮(5mg,22%)lcms(方法d):tr 3.17min,100%,ms(esi)404.1(m h) ;chiral uplc(方法:a):tr 3.17min,》95%ee和de.1-((2r,5s)-2-甲基-5-(4-((5-甲基吡啶-3-基)氨基)-6-(吡嗪-2-基)嘧啶-2-基)哌啶-1-基)乙-1-酮(6mg,27%)lcms(方法d):tr 3.17min,100%,ms(esi)404.2(m h) ;chiral uplc(方法a):tr 4.60min,》95%ee和de。
[0244]
化合物00003(其在本文中也称为化合物b)和00004(其在本文中也称为化合物a)
((3-(1-甲基1h-1,2,3-三唑-4-基)苯基)氨基)-6-(吡嗪-2-基)嘧啶-2-基)哌啶-1-基)乙-1-酮(化合物c,102mg,60%)为白色固体。1h-nmr(400mhz,dmso-d6,旋转异构体混合物)δ10.01(d,j=5.6hz,1h),9.56(dd,j=11.0,1.1hz,1h),8.80(d,j=1.5hz,2h),8.54
–
8.42(m,2h),7.72
–
7.54(m,2h),7.53
–
7.39(m,2h),4.86
–
4.76(m,1h),4.27
–
4.16(m,0.5h),4.15
–
4.03(m,3.5h),3.58
–
3.42(m,0.5h),3.00
–
2.86(m,1h),2.86
–
2.68(m,0.5h),2.17
–
1.96(m,5h),1.93
–
1.77(m,0.5h),1.76
–
1.64(m,1.5h),1.27(d,j=6.8hz,1.5h),1.13(d,j=7.0hz,1.5h);lcms(方法d):t
r 3.31min,ms(esi)470.2(m h)
.
[0257]
实施例4:与化合物00004复合的人crebbp溴结构域的晶体结构以及化合物a、化合物c和ccs1477的bromoscan
tm
的结果
[0258]
结晶化
[0259]
实验设置:用于结晶的构建体包含残基1081至1197。使用悬滴蒸汽扩散装置获得与化合物00004复合的crebbp晶体。将浓度为20.3mg/ml的crebbp(10mm hepes,500mm nacl,5%甘油,0.5mm tcep,ph 7.4)用4.3mm(3.0倍摩尔过量)的00004(150mm在dmso中)预孵育1小时。然后将1μl蛋白质溶液与1μl储库溶液(0.1m mgcl2、0.1m mes/naoh ph 6.3、18%(w/v)peg 6000和10%(v/v)乙二醇)混合,并在4℃下用0.4ml储库溶液平衡。良好的衍射晶体出现,并在4天内生长至全尺寸。
[0260]
数据收集
[0261]
安装前,通过向结晶液滴中加入10%甘油(最终浓度)对晶体进行低温保护。用钻石光源(diamond light source)(英国didcot,光束线i03)收集了crebbp/00004晶体的完整数据集,数据由xds、pointless和aimless在autoproc管道内整合、分析和缩放(表1)。
[0262]
表1:数据收集统计
[0263][0264]
结构确定和细化
[0265]
使用先前确定的crebbp结构作为起始模型进行分子置换。使用refmac5进行几轮交替的手动重建和改进,产生了最终模型(表2)。原子位移因子用每个原子一个各向同性b因子建模。
[0266]
表2:精细统计
[0267][0268]
结果:我们制备了crebbp/00004晶体,衍射至分辨率,并测定了蛋白质-配体复合物的三维结构。在初始模型的fo-fc省略图(omit map)中,crebbp每条链中化合物结合位点的清晰电子密度揭示了整个化合物的结合(图7)并允许其明确放置。此外,该结构还证实了化合物00004(哌啶部分上的2s,5r)的绝对立体化学。
[0269]
bromokdmax-测定
[0270]
在discoverx进行了bromokdmax。该测定可用于确定化合物是否以特定kd(如100nm或更低)与p300的溴结构域和/或cbp的溴结构域结合。
[0271]
测定原理如下:bromoscan
tm
是用于识别小分子溴结构域抑制剂的新型行业领先的平台。基于成熟的kinomescan
tm
技术,bromoscan
tm
采用专有的配体结合定点竞争分析来定量测量测试化合物和溴结构域之间的相互作用。该稳健可靠的检测面板适用于高通量筛选,可提供定量配体结合数据,便于鉴定和优化强效和选择性小分子溴结构域抑制剂。bromoscan
tm
测定包括痕量溴结构域浓度(《0.1nm),从而在广泛的亲和力范围内报告真实的热力学抑制剂kd值(《0.1nm至》10um)。
[0272]
该测定如下进行:对于溴结构域分析,展示溴结构域的t7噬菌体菌株在来自bl21菌株的大肠杆菌宿主中平行生长在24孔块中。大肠杆菌生长至对数生长期,用来自冷冻原液的t7噬菌体感染(感染多样性=0.4),并在32℃振荡温育直至裂解(90-150分钟)。将裂解物离心(5000x g)并过滤(0.2μm),以去除细胞碎片。室温下,用生物素化小分子或乙酰化肽配体处理链霉亲和素包被的磁珠30分钟,以生成用于溴结构域测定的亲和树脂。用过量的生物素封闭配体化的(liganded)珠子,并用封闭缓冲液(seablock(pierce),1%bsa,0.05%吐温20,1mm dtt)洗涤,以除去未结合的配体并减少非特异性噬菌体结合。结合反应是通过在1x结合缓冲液(17% seablock,0.33x pbs,0.04% tween 20,0.02% bsa,0.004%叠氮化钠,7.4mm dtt)中结合溴结构域、配体化亲和珠和试验化合物(即化合物a、化合物c或ccs1477)组装的。试验化合物在100% dmso中以1000x贮备物(stocks)制备。使用11点3倍化合物稀释系列和一个dmso对照点测定kd。用于kd测量的所有化合物均通过声传递(非接触分配)法在100% dmso中进行分配。然后将化合物直接稀释到测定中,使dmso的最终浓度为0.09%。所有反应均在聚丙烯384孔板中进行。每份的最终体积为0.02ml。在室温下振荡培养测定板1小时,并用洗涤缓冲液洗涤亲和珠(1x pbs,0.05%吐温20)。然后将珠子重新悬浮在洗脱缓冲液(1x pbs,0.05%吐温20,2μm非生物素化亲和配体)中,并在室温下振荡温育30分钟。通过qpcr测量洗脱液中的溴结构域浓度。
[0273]
结果如下:
[0274]
[0275]
[0276][0277]
相应的数据是公开的i)对于sgc-cbp30,例如在wu等人,nature communications(2019)10:1915https://doi.org/10.1038/s41467-09672-2的补充资料中;ii)对于gne-781例如在romero等人,j.med.chem.2017,60,9162-9183中;和iii)对于ft-6876例如在aacr年会2020,虚拟会议ii,2020年6月22日至24日(标题为“ft-6876,a potent and selective inhibitor of cbp/p300 with antitumor activity in ar-positive breast cancer”)的海报#3079中。
[0278]
实施例5
[0279]
材料和方法
[0280]
基因表达分析:
[0281]
在含有10%fcs和2mm l-谷氨酰胺的rpmi培养基中进行药物治疗的前一天将250000hcc827(atcc;crl 2868;具有egfr外显子19缺失e746-a750)细胞/孔或200000nci-h1975(atcc;crl 5908;具有egfr l858r和t790m)将细胞/孔接种在6孔培养皿中(greiner bio-one,7657160)。然后用dmso、egfr抑制剂(对于hcc827吉非替尼[最终100nm,lc laboratories g-4408];对于nci-h1975奥希替尼[最终20nm,lc laboratories;o-7200])有或没有1μm化合物a (化合物a在本文中也称为化合物00004),ccs1477(chemgood;c1505),a-485(lucerna-chem;hy-107455)或icg-001(selleckchem,s2662)处理24小时。随后用2ml pbs洗涤细胞3次,并在300μl裂解缓冲液(ra1 1% tcep[sigma 646547])中裂解。根据用于rna提取的macherey-nagel nucleospin 8rna kit protocol for vacuum(740698.5)提取rna,在h2o中洗脱rna。使用thermo scientific高容量cdna逆转录试剂盒(4368813)逆转录0.5
ꢀ‑
2μg rna。然后在roche light cycler 480中使用384孔形式的kapa sybr快速试剂盒(kk4611)对等量的cdna进行qpcr分析。通过从感兴趣基因的ct中减去看守基因ct值(b-actin),并通过从感兴趣样本中减去dmso对照值计算δδct来评估基因的基因表达,最终计算处理样本与对照处理样本的倍数变化差异。
[0282]
引物:
[0283]
actb(b-actin):fwd 5`gcc ccagctcaccatggat 3`(seq id no:1),rev 5`tgggcctcgtcgcccacata 3`(seq id no:2);
[0284]
alpp:fwd 5`agaaagcagggaagtcagtgg 3`(seq id no:3),rev5`cgagtaccagttgcggttca 3`(seq id no:4);
[0285]
hopx:fwd 5`gaccatgtcggcggagacc 3`(seq id no:5),rev 5`
gcgctgcttaaaccatttctggg 3`(seq id no:6)。
[0286]
溴结构域和hat域结合的cbp/p300抑制剂,但不是阻止cbp和β-连环蛋白相互作用的抑制剂,钝化了egfr抑制剂诱导的egfr突变非小细胞肺癌细胞(nsclc)的基因表达。
[0287]
与具有不同作用模式(即,与cbp/p300的不同蛋白质结构域结合)的其他cbp/p300抑制剂(cbp-i)平行地评估化合物a的基因表达。化合物a和ccs1477结合到cbp/p300的溴结构域(brd-i),a-485靶向cbp/p300的催化组蛋白乙酰转移酶(hat)活性(hat-i),icg-001破坏cbp与β-连环蛋白(cbp/β-cat-i)的相互作用。
[0288]
图1显示了结果,显示的受调节基因是alpp(碱性磷酸酶,胎盘型;图1a和图1c)和hopx(仅同源结构域蛋白;图1b和图1d)。暴露于20nm吉非替尼(egfr抑制剂)或在20nm吉非替尼存在下暴露于1μm不同cbp抑制剂24小时的hcc827中的基因表达(图1a-b)。nci-h1975(携带导致吉非替尼耐药性的egfr t790m突变)中的基因表达暴露于20nm奥希替尼(第三代egfr抑制剂)或在20nm奥希替尼存在下于1μm不同的cbp/p300抑制剂中24小时。数据来自两次qpcrs独立实验(平均值
±
sem)(图1c-d)。
[0289]
结果:两种egfr突变的nsclc细胞系对用两种不同的egfr抑制剂(第一代吉非替尼和第三代奥希替尼)治疗的实例基因alpp和hopx的上调作出响应。化合物a和ccs1477(均为cbp/p300溴结构域结合剂)和a-485(cbp/p300的催化抑制作用)可逆转egfr抑制剂诱导的基因表达。阻止cbp与β-连环蛋白相互作用的不同作用模式化合物(icg001)不会使egfr诱导的基因表达变钝。
[0290]
实施例6
[0291]
材料和方法
[0292]
细胞计数:
[0293]
在含有10% fcs和2mm l-谷氨酰胺的rpmi培养基中进行药物处理前一天,将2000hcc 827(atcc;crl 2868)细胞/孔接种到96孔板(greiner bioone 655090)中。接种几个平板,对于细胞计数的每个时间点,固定一个平板并染色用于分析。第二天,用指定的化合物和浓度与dmso或吉非替尼(图2中20nm或图3中300nm)组合处理细胞。[图2显示了化合物a和b;图3显示了化合物a,ccs 1477(chemgood;c1505)、sgc-cbp 30(selleckchem;s7256 cas no.:1613695-14-9)、a-485(lucerna-chem;hy-107455)、化合物00071和化合物00030]。细胞培养持续到每个平板的固定和分析时间。对于延长的时间点,培养基和药物每周补充两次。
[0294]
固定和成像:
[0295]
在给定时间,用pbs洗涤平板3次,用80μl的4% pfa固定细胞10min,rt。用pbs洗涤3次后,用10μg/ml hoechst 33342(thermo scientific h 12492)在100μl pbs中对细胞染色2h,rt,黑暗。在3次pbs洗涤步骤后,使用具有电动x/y平台和5倍物镜的zeiss apotome以自动成像模式采集hoechst33342信号。使用imagej进行hoechst33342斑点(细胞核)的图像分析和测定。使用graphpad prism将细胞核数目作为时间的函数作图。
[0296]
细胞增殖的无标记测定:
[0297]
在含10% fcs和2mm l-谷氨酰胺的rpmi培养基中进行药物治疗前一天,2000hcc 827(atcc;crl 2868)细胞/孔或2000nci-h1975(atcc;crl 5908)接种到96孔板(greiner bioone 655090)中。第二天,在celigo成像血细胞计数器上使用明场(brightfield)成像对
孔进行无标记成像,以确定初始细胞数。随后,用dmso、单一药物或药物组合处理细胞,并使用明场模式(celigo成像血细胞计数器)定期成像数周,以随时间追踪每个孔中的细胞增殖。生长培养基和处理每周补充两次。hcc827的药物和浓度:300nm吉非替尼、1μm化合物a和300nm吉非替尼 1μm化合物a。药物和浓度:nci-h1975:50nm奥希替尼(lc laboratories;o-7200)、0.125、0.5或2μm ccs 1477(chemgood;c1505)和0.125、0.5或2μm化合物a或图中所示的组合。使用celigo软件的内置“直接细胞计数”分析工具在明场模式下测定细胞数。
[0298]
只有结合cbp/p300溴结构域的对映异构体(化合物a)而非不结合cbp/p300溴结构域的对映异构体(化合物b)以浓度依赖性方式增强egfr抑制剂介导的nsclc细胞增殖的抑制作用。
[0299]
随着时间的推移,监测egfr突变的hcc827细胞的细胞数量。图2a显示了指定的化合物浓度下用以下处理的细胞:单独用dmso(实心圆圈)、单独用20nm egfr抑制剂(吉非替尼;第一代egfr抑制剂,空心圆圈),或与cbp/p300 brd抑制剂化合物a的活性对映异构体(上图)或其不与cbp/p300的溴结构域结合的对映异构体化合物b(化合物b在本文中也称为化合物00003)(下图)联合使用。图2b hcc827细胞在没有egfr抑制剂的情况下暴露于化合物a&b。给出的图来自一个实验,每个时间点和条件三次重复(平均值
±
sd)。
[0300]
结果:在20nm吉非替尼治疗下,hcc827细胞数量最初减少,但在吉非替尼连续暴露下开始重新生长。在研究期间,使用brd-i化合物a通过抑制cbp/p300抑制再生长,而使用相应的非溴结构域结合对映异构体化合物b则没有抑制。有趣的是,尽管brd-i作为单一药物不起作用,但其在联合治疗中防止再生长的作用存在。
[0301]
化合物a和基准cbp/p300抑制剂与egfr抑制剂组合介导的hcc827细胞增殖抑制。
[0302]
图3(a)、(b)、(c)、(d)和(e)显示了使用核荧光染色在96孔板中测量的egfr突变的nsclc细胞系hcc827的细胞数随药物处理时间[天数]的函数(图表图例中的符号)。图3(a)和(b)和(c)左图:用化合物a(图3a)、ccs1477(图3b)、sgc-cbp30(图3c)或a485对细胞进行的单剂处理。图3(a)和(b)和(c)右图和图3(c)和(d):在300nm吉非替尼存在下化合物a(图3a)和ccs1477(cbp/p300 brd-i)(图3b)和化合物sgc-cbp30(图3c)和化合物00071(图3d)以及化合物00030(图3e)和a485(cbp/p300 hat-i)的抗增殖活性。呈现的曲线来自一式三份的一个实验(平均值
±
sd),在类似的实验中重复获得类似的结果。
[0303]
图4a显示了对hcc827细胞数量随时间[h]变化的评估。在不存在egfr抑制剂的情况下,化合物a不影响egfr突变的nsclc细胞的细胞增殖,但当与egfr抑制剂联合用药时,可防止产生耐药性(显示1个示例板,吉非替尼n=24个孔,吉非替尼 化合物a处理n=24个孔,dmso:n=6,化合物a:n=6,平均值
±
sd)。图4b描绘了用吉非替尼或吉非替尼 化合物a处理0或22天(来自如a中的2个实验板,每个条件下n=48个孔)的每个孔的细胞数,如点图所示。图4c显示了图4a中分析的使用300nm吉非替尼或300nm吉非替尼 1μm化合物a处理的2块板(n=48个孔/每种条件)的孔的瀑布图。在药物治疗前(第0天),以距每孔初始细胞数的对数倍数变化计算每孔第22天细胞数的增加。
[0304]
结果:
[0305]
图3a、3b、3c、3d和3e:在300nm吉非替尼处理后,hcc827细胞数量最初减少,但在吉非替尼连续暴露下开始重新生长。在研究期间,通过使用五种独立的brd-i或hat-i抑制cbp/p300来抑制再生长。有趣的是,尽管brd-i作为单一药物不起作用,但其在联合治疗中
预防再生长的作用存在。
[0306]
图4a 4b和4c:化合物a本身对细胞数量有微弱/无影响,而300nm吉非替尼最初可完全阻断细胞增殖。然而,在长期培养中,如果仅用吉非替尼处理,细胞会重新生长,而在研究时间内用化合物a共同处理会显著延迟或完全阻止重新生长(》22日)。
[0307]
化合物a和基准cbp/p300抑制剂联合egfr抑制剂介导的nsclc细胞增殖抑制-nci-h1975。
[0308]
图5(a)显示了在存在dmso、50nm奥希替尼、2μm化合物a或50nm奥希替尼与2.0、0.5或0.125μm化合物a的组合的情况下,nci-h1975细胞数作为时间[h]的函数的评估。图5(b)显示了在dmso、50nm奥希替尼、2μm ccs1477或50nm奥希替尼与2.0、0.5或0.125μm ccs1477的组合存在下,nci-h1975细胞生长的评估。(示例图显示了每个数据和时间点的副本,并在graphpad prism中计算了逻辑增长曲线拟合)。
[0309]
结果:图5:对于进一步egfr突变的nsclc细胞系和不同的egfr-i化合物,cbp/p300 brd-i和egfr-i的联合效应成立。化合物a和ccs1477本身对细胞数量没有影响/具有弱影响,而50nm奥希替尼最初阻断细胞增殖。然而,长期来看,即使在50nm奥希替尼的存在下,细胞仍以缓慢的速率继续生长,其通过与化合物a或ccs1477联合治疗而延迟,呈剂量依赖性。
[0310]
实施例7
[0311]
异种移植物:200万个nci-h1975细胞(atcc;crl 5908)注射到nmri裸小鼠(janvier)的两侧。当较大肿瘤达到200mm3左右时,将小鼠分为治疗组。小鼠每天用以下口服处理:媒介物(30% peg300/h2o;202371sigma-aldrich),20mg/kg ccs 1477(chemietek;ct-ccs1477),2mg/kg奥希替尼(o-7200lc laboratories)或2mg/kg奥希替尼与20mg/kg ccs1477的组合(预混合dmso储备液)。每周用手动测径器测量肿瘤体积两到三次。使用以下公式计算肿瘤体积:较大肿瘤直径
×
较小肿瘤直径的平方除以2。基于平均肿瘤体积的线性拟合,通过回归分析(双尾)比较曲线的斜率。奥希替尼组与奥希替尼/ccs1477组之间检测到显著差异。未观察到媒介物和ccs1477单剂治疗之间的显著差异。
[0312]
通过将时间t时的肿瘤体积变化与其基线进行比较来确定响应:%肿瘤体积变化=δvolt=100%
×
((vt
–
v初始)/v初始)。最佳响应是t≥10d时δvolt的最小值。对于每个时间t,还计算了t=0至t的δvolt的平均值。最佳平均响应定义为t≥10d时该平均值的最小值。该度量(metric)将响应的速度、强度和持久性的组合捕获为单一值。响应标准(mrecist)改编自recist标准21,定义如下(按此顺序应用):mcr,最佳响应《-95%和最佳平均响应《-40%;mpr,最佳响应《-50%和最佳平均响应《-20%;msd,最佳响应《35%和最佳平均响应《30%;mpd,没有另外分类。在完成14天试验之前由于不良事件而被处死的小鼠被从数据集中移除。
[0313]
egfri与cbp/p300i的组合的体内疗效
[0314]
图6(a)显示了随时间绘制的egfr突变nci-h1975异种移植物的平均肿瘤体积( sem)。描述了四个不同的治疗组:媒介物(30% peg300/h2o;交叉圆;n=4)、20mg/kg ccs1477(空心圆圈;n=4),2mg/kg奥希替尼(实心圆;n=9)或2mg/kg奥希替尼与20mg/kg ccs1477的组合(半实心圆圈;n=10)。图6(b)显示了瀑布图所示的所有4个治疗组的最佳平均反应(媒介物为灰色,20mg/kg ccs1477为白色,2mg/kg奥希替尼为黑色和2mg/kg奥希替尼与20mg/kg ccs1477的组合为方格。虚线表示初始肿瘤体积减少30%。
[0315]
结果:图6:当在没有egfr抑制剂的情况下使用cbp/p300的溴结构域抑制剂时,它们对egfr突变的非小细胞肺癌异种移植瘤生长没有影响。然而,当它们与egfr抑制剂联用时,在整个治疗过程中,对治疗的响应增加,肿瘤大小得到更好的控制。令人惊讶的是,当溴结构域结合抑制剂(ccs1477)与egfr抑制剂(奥希替尼)联用时,尽管对单独的溴结构域结合抑制剂(ccs1477)无应答,但对治疗的应答率有所提高。
[0316]
实施例8
[0317]
材料和方法:
[0318]
cbp溴结构域结合试验(tr-fret):
[0319]
将10mm的化合物dmso溶液在dsmo预稀释成25x的dmso储备溶液。然后在检测缓冲液中稀释至4x。在检测缓冲液中进行了一系列稀释,以保持dmso浓度稳定。将测定缓冲液中的5μl化合物移入测定板(由测定试剂盒提供)且tr-fret测定(cayman chemicals;600850)是按照制造商的说明进行的。在室温下在黑暗中孵育1小时后,使用tr-fret模式在tecan m1000读板仪中读取分析板(顶部读数;激发340nm带宽20nm;发射620nm带宽7nm;为第一孔确定的最佳增益,闪光次数:5;闪光频率100hz;积分时间:500μs,滞后时间:100μs,室温)。将670nm发射除以620nm发射计算tr-fret比值。根据归一化值(dmso=1)和阳性对照(0)计算ec50。对值进行对数转换,并使用具有可变斜率的非线性回归(4个参数)将数值拟合至剂量-反应曲线,以评估ec50值(见下表3)。
[0320]
表3:
[0321]
说明ec50:a*《0.2μm《a《1μm《b《10μm《c
[0322][0323]
tr-fret数据表明,ec50》10μm的化合物00003不符合本文定义的cbp/p300溴结构域抑制剂。
[0324]
实施例9
[0325]
材料和方法
[0326]
细胞增殖的无标记测定:
[0327]
在含有10% fcs和2mm l-谷氨酰胺的rpmi培养基中药物处理的前一天,将2000hcc4006(atcc;crl2871;具有egfr外显子19缺失l747至e749)接种到96孔板(greiner bioone 655090)中。第二天,使用celigo imagecytometer上的明场成像对孔进行无标记成像,以测定初始细胞数。随后,使用dmso、单一药物或药物组合对细胞进行处理,并使用明场模式(celigo成像血细胞计数器)在数周内定期成像,以跟踪每个孔中细胞随时间的增殖情况。每周补充两次生长培养基和处理物。hcc4006的药物和浓度:300nm吉非替尼(lc laboratories;g-4408)、1μm化合物a和300nm吉非替尼 1μm化合物a。使用celigo软件内置的“直接细胞计数”分析工具在明场模式下测定细胞数量。
[0328]
图8a显示了对hcc4006细胞数量随时间[以天为单位]变化的评估。在不存在egfr
4006(atcc;crl2871)接种到96孔板(greiner bioone655090)中。第二天,使用celigo图像流式细胞仪上的明场成像对孔进行无标记成像,以测定初始细胞数。随后,使用dmso、单一药物(奥希替尼和每种cbp/p300溴结构域抑制剂)或100nm奥希替尼和一种cbp/p300溴结构域抑制剂的药物组合(药物浓度如下所示)对细胞进行处理。使用明场模式(celigo成像血细胞计数器)对平板进行数周的定期成像,以跟踪每个孔中细胞随时间的增殖。生长培养基和处理物每周补充两次。hcc827的药物和浓度:100nm奥希替尼(egfr-抑制剂,lc laboratories;o-7200)和对于cbp/p300溴结构域抑制剂:1μm化合物a,0.2μm化合物c,0.2μm ccs1477(chemitek;ct-ccs1477),1μm ft-6876(“cbp/p300-in-8”,medchemexpress;hy-136920)和0.2μm gne-781(medchemexpress;hy-108696)。使用celigo软件内置的“直接细胞计数”分析工具在明场模式下测定细胞数量。
[0340]
图10a-e显示了对21天内hcc4006细胞数量的评估。在没有egfr抑制剂的情况下,cbp/p300溴结构域抑制剂[(a)化合物a,(b)化合物c,(c)ccs1477,(d)ft-6876和(e)gne-781)]不影响egfr突变的nsclc细胞增殖,但当与奥希替尼联合用药时,可防止对100nm奥希替尼产生耐药性。请注意,在图(a-e)中,100nm奥希替尼处理的dmso曲线和时间过程是相同的,因为所有条件都是并行运行的(dmso:18孔,cbp/p300溴结构域抑制剂:每种6孔,奥希替尼:12孔,和奥希替尼 cbp/p300溴结构域抑制剂的所有组合:12孔,平均
±
sd)。
[0341]
结果:图10a-e:cbp/p300溴结构域抑制剂本身对hcc4006细胞数量没有/至多有微弱的影响,而100nm奥希替尼最初会阻止细胞增殖。在长期培养中,如果仅用奥希替尼处理,hcc4006细胞重新生长,而与不同的cbp/p300溴结构域抑制剂共同处理在21天的研究时间内显著延迟或完全阻止重新生长。
[0342]
实施例12
[0343]
该异种移植实施例按照上述实施例7的方法进行。因此,200万个nci-h1975细胞(atcc;crl 5908)注射到nmri裸小鼠(janvier)的两侧。当较大肿瘤达到200mm3左右体积时,将小鼠分为治疗组。每天用媒介物(0.8%(vol)dmso(cas[67-68-5];5%(vol)nmp(cas[872-50-4]),4.2%(vol)dma(cas[127-19-5]),90%(vol)的40%(wt/vol)captisol(cas[182410-00-0])于ph4醋酸盐缓冲液0.1m中),90mg/kg化合物c,2mg/kg奥希替尼(o-7200lc laboratories)或2mg/kg奥希替尼与90mg/kg化合物c(预先混合的)的组合口服治疗小鼠。每周使用测径器测量肿瘤体积两至三次。肿瘤体积的计算公式为:较大肿瘤直径x较小肿瘤直径的平方除以2。基于平均肿瘤体积的线性拟合,通过回归分析(双尾)比较曲线的斜率。检测到奥希替尼和奥希替尼/化合物c组之间存在显著差异。未观察到媒介物与化合物c单一药剂治疗之间的显著差异。
[0344]
通过将时间t时的肿瘤体积变化与其基线进行比较来确定响应:%肿瘤体积变化=δvolt=100%
×
((vt
–
v初始)/v初始)。最佳响应是t≥6d时δvolt的最小值。对于每个时间t,还计算了t=0至t的δvolt的平均值。最佳平均响应定义为t≥10d时该平均值的最小值。该度量将响应的速度、强度和持久性的组合捕获为一个值。响应标准(mrecist)改编自recist标准21,定义如下(按此顺序应用):mcr,最佳响应《-95%和最佳平均响应《-40%;mpr,最佳响应《-50%和最佳平均响应《-20%;msd,最佳响应《35%和最佳平均响应《30%;mpd,没有另外分类。在完成14天试验之前由于不良事件而被处死的小鼠被从数据集中移除。
[0345]
图11(a)显示了随时间绘制的egfr突变nci-h1975异种移植物的平均肿瘤体积( sem)。描述了四个不同的治疗组:媒介物;交叉圆圈;n=8,90mg/kg化合物c(空心圆圈;n=6),2mg/kg奥希替尼(实心圆圈;n=9)或2mg/kg奥希替尼联合90mg/kg化合物c(半实心圆;n=12)。对平均肿瘤体积随治疗时间进行线性回归拟合,并比较奥希替尼和奥希替尼与化合物c联合用药的斜率。可以检测到两组之间斜率的显著差异,分别在奥希替尼组中为正( 6.4),在组合组中为负(-7.4)。图11(b)显示了瀑布图中所有4个治疗组的最佳平均响应(媒介物为灰色,90mg/kg化合物c为白色,2mg/kg奥希替尼为黑色,2mg/kg奥希替尼与90mg/kg化合物c的组合为方格)。虚线表示初始肿瘤体积减少了30%。
[0346]
结果:结果证实了上述实施例7中获得的结果,这次是化合物c代替ccs1477作为cbp/p300溴结构域抑制剂,其中两种抑制剂在结构上不相关但具有相同的功能。因此,当在没有egfr抑制剂的情况下使用cbp/p300溴结构域抑制剂时,对egfr突变的nsclc异种移植肿瘤生长没有影响。然而,当cbp/p300溴结构域抑制剂(实施例7中的ccs1477,本实施例中的化合物c)与egfr抑制剂(此处为奥希替尼)组合时,对治疗的响应显著增加,并且在治疗过程中肿瘤大小得到更好的控制。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
