草乌甲素多晶型及其制备方法和应用
1.本案是以申请号为2020106364524,申请日为2020年7月3日,名称为《草乌甲素多晶型及其制备方法和应用》的专利申请的分案申请。
技术领域
2.本发明涉及草乌甲素制备技术领域,尤其涉及一种草乌甲素晶型vi及其制备方法和应用。
背景技术:
3.草乌甲素(bulleyaconitine a,cas-rn为79592-91-9)是一种现代植物药,由毛莨科乌头属植物滇西嘟拉中分离出来,属于二萜双酯型生物碱,是不同于乌头碱、次乌头碱的一种生物碱。该药物无耐受性,无成瘾性,可作为强效镇痛药和抗炎药,具有中枢镇痛和外用镇痛的双重药理作用。目前,国家食品药品监督管理总局(china food and drug administration,cfda)已批准草乌甲素注射剂和片剂用于慢性疼痛及类风湿性关节炎的治疗。临床上,草乌甲素也已被广泛用于治疗类风湿性关节炎、骨关节炎、肌纤维炎、颈椎病、癌性疼痛及各种原因导致的慢性疼痛。
4.草乌甲素对炎症和疼痛均有明显的抑制作用。前列腺素pge2是在致炎因子作用下产生,局部组织产生和释放的具有致炎、致痛作用的炎症介质,使局部血管通透性增加,进一步加重炎症反应,并由此产生一系列的变化,例如:滑膜细胞和成纤维细胞增殖、血管新生、产生胶原酶等,使得骨和软骨细胞被破坏。降低血清pge2是草乌甲素的抗炎作用的机理之一,pge2能激活外周痛觉感受器,传递痛觉信号。β-内啡肽是一种具有较强镇痛作用的神经肽,草乌甲素的镇痛作用可能与其拮抗脑内5-羟色胺(5-ht),抑制pge2释放,从而解除对β-内啡肽的抑制有关。
5.同时,据文献报道,草乌甲素可以直接诱导脊髓小胶质细胞强啡肽a的表达,从而表现出镇痛作用,并且在脊髓后角的c-纤维突触面显示了对神经性疼痛的抑制作用,同时也证明了草乌甲素可以增强吗啡的镇痛作用,并抑制吗啡的镇痛耐受性,此外,草乌甲素还可抑制炎性趋化因子。
6.目前,关于草乌甲素的研究及应用还仅仅局限于抗炎镇痛方面,例如:cn101468000公开了草乌甲素作为制备治疗原发性红斑肢痛症药物的应用;cn107245054a则公开了一种无定形草乌甲素化合物在制备治疗由风湿或类风湿性关节炎引起的疼痛疾病的药物中的用途;cn106943402a中的草乌甲素则用于制备防治骨质疏松或骨溶解的药物。
7.本发明人的前期研究(cn110478350a)发现草乌甲素具有明显抑制因毒品引发的自发活动症状的作用,表明草乌甲素及其衍生物在抑制药物成瘾性方面具有巨大的应用潜力。
8.然而,现有技术中关于草乌甲素的多晶型研究还相对较少,文献报道中多有涉及通过诸如结晶,重结晶得到白色、无色透明的草乌甲素。但未见关于草乌甲素多晶型的xrpd
报道。cn107245054a公开了一种无定形草乌甲素化合物,其溶解性和镇痛效果略优于对照品。进一步开发其他多晶型及稳定性更好的无定形草乌甲素具有重要意义。
技术实现要素:
9.本发明的一个方面涉及一种草乌甲素的稳定的多晶型形式。
10.本发明的另一个方面涉及草乌甲素的多晶型,其与已知的无定形形式相比,具有更好的稳定性或药物动力学特性。
11.具体而言,本发明的第一个方面涉及一种草乌甲素的无定形形式,其xrpd图基本如附图1所示;优选地,其tga和dsc图基本如图2所示。
12.根据本发明的另一个方面涉及草乌甲素的晶型i,其xrpd图基本如附图3所示。
13.优选地,所述晶型i的tga和dsc图基本如图4所示。
14.根据本发明的另一个方面涉及草乌甲素的晶型vi,其xrpd图基本如附图5所示。
15.优选地,所述晶型vi的tga和dsc图基本如图6所示。
16.根据本发明的另一个方面还涉及上述草乌甲素多晶型的制备方法。
17.具体而言,无定形草乌甲素的制备方法为:草乌甲素溶于低级醇(优选甲醇、乙醇),将所得溶液快速滴加至水中,抽滤分离,将固体干燥,即得。
18.草乌甲素的晶型i的制备方法,其特征在于,将草乌甲素溶于常用有机溶剂,常温挥发。优选地,所述常用有机溶剂包括乙醚、四氢呋喃、甲醇、乙醇、正丙醇、异丙醇、丙酮、乙酸乙酯、甲酸乙酯、乙腈、甲苯、二氯甲烷、氯仿、四氢呋喃或二甲基亚砜中的至少一种;优选乙醚、四氢呋喃、乙醇、丙酮、乙酸乙酯、乙腈、二氯甲烷、氯仿、四氢呋喃;最优选乙醚、四氢呋喃、乙醇、丙酮、乙酸乙酯或二氯甲烷中的至少一种。
19.草乌甲素的晶型vi的制备方法,以乙腈和水的混合溶剂结晶获得;优选地,所述乙腈与水的比例为1:2-4;更优选地,乙腈与水的比例为1:3。
20.根据本发明的另一个方面还涉及一种药物组合物,其包括根据本发明的草乌甲素的无定形形式、草乌甲素晶型i或vi;以及药学可接受的辅料。
21.优选地,所述药用辅料选自崩解剂、稀释剂、润滑剂、粘合剂、湿润剂、矫味剂、助悬剂、表面活性剂或防腐剂中的至少一种;更优选地,所述崩解剂选自玉米淀粉、马铃薯淀粉、交联聚乙烯吡咯烷酮、羧甲基淀粉钠、低取代羟丙基纤维素、交联羧甲纤维素钠、羧甲基纤维素、羧甲基纤维素钙或藻酸中的至少一种;更优选地,所述稀释剂选自乳糖、蔗糖、甘露醇、玉米淀粉、马铃薯淀粉、磷酸钙、柠檬酸钙或结晶纤维素中的至少一种;更优选地,所述润滑剂选自微粉硅胶、硬脂酸镁、硬脂酸钙、硬脂酸、滑石粉或无水硅胶中的至少一种;更优选地,所述粘合剂选自阿拉伯胶、明胶、糊精、羟丙基纤维素、甲基纤维素或聚乙烯吡咯烷酮中的至少一种;更优选地,所述湿润剂选自十二烷基硫酸钠;更优选地,所述矫味剂选自阿斯巴甜、甜菊甙、蔗糖、麦芽糖醇或柠檬酸中的至少一种;更优选地,所述助悬剂选自阿拉伯胶、明胶、甲基纤维素、羧甲基纤维素钠、羟甲基纤维素或硬脂酸铝凝胶中的至少一种;更优选地,所述表面活性剂选自卵磷脂、山梨糖醇酐单油酸酯或单硬脂酸甘油酯中的至少一种;更优选地,所述防腐剂选自对羟苯甲酸甲酯或对羟苯甲酸丙酯中的至少一种。
22.根据本发明的另一个方面,所述药物组合物为固体剂型,优选口服剂。
23.根据本发明的另一个方面还涉及根据本发明的草乌甲素多晶型的应用。
24.本领域技术人员可以理解,根据本发明的草乌甲素多晶型可以应用于任意已知的草乌甲素的已知用途中。
25.具体而言,根据本发明的草乌甲素无定形或草乌甲素晶型i或vi可以用于制备抗炎、镇痛药物,也可以用于制备抑制药物成瘾性的药物。
26.本发明的有益效果是:
27.1)根据本发明的草乌甲素的多晶型具有良好的稳定性形式。
28.2)草乌甲素的多晶型,其与已知的无定形形式相比,药物动力学特性更佳。
29.3)与草乌甲素原料相比,根据本发明的草乌甲素的多晶型具有更低的毒性。
30.4)与草乌甲素原料相比,根据本发明的草乌甲素的多晶型具有更好的镇痛效果。
附图说明
31.图1:草乌甲素无定形的xrpd图。
32.图2:草乌甲素无定形的tga和dsc图。
33.图3:草乌甲素晶型i的xrpd图。
34.图4:草乌甲素晶型i的tga和dsc图。
35.图5:草乌甲素晶型vi的xrpd图。
36.图6:草乌甲素晶型vi的tga和dsc图。
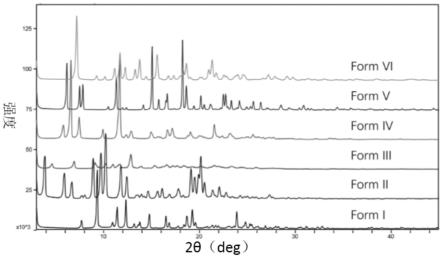
37.图7:草乌甲素晶型i-vi的xrpd比较图。
38.图8:空白血浆质谱图。
39.图9:标曲样品中草乌甲素晶型ⅰ(a)、ⅵ(b)实测样品图谱。
40.图10:大鼠给药后血浆样品中草乌甲素晶型ⅰ(a)、ⅵ(b)实测样品图谱。
41.图11:lc-ms/ms法测定大鼠血浆中草乌甲素晶型ⅰ(1)、vi(2)标准曲线图。
42.图12:单次灌胃给予草乌甲素晶型ⅰ后血药浓度-时间曲线。
43.图13:单次灌胃给予草乌甲素晶型ⅵ后血药浓度-时间曲线。
44.图14:大鼠单次灌胃给予天然草乌甲素后血药浓度-时间曲线。
45.图15:大鼠单次灌胃给予无定形草乌甲素后血药浓度-时间曲线
46.图16:晶体i的死亡率-对数剂量曲线(a)和机率单位-对数剂量曲线(b)。
47.图17:晶体vi的死亡率-对数剂量曲线(a)和机率单位-对数剂量曲线(b)。
具体实施方式
48.原料化合物:草乌甲素,分子式c35h49no10,分子量643.77,其结构如下所示:
[0049][0050]
纯度≥98%,购于上海源叶生物科技有限公司。
[0051]
批号:hs0903xa13
[0052]
保存条件:4℃保存
[0053]
分析方法
[0054]
1.1核磁分析(nuclear magnetic resonance spectroscopy,1h nmr)
[0055]
将若干毫克固体样品溶解于二甲基亚砜-d6溶剂中,在bruker avance-iii(bruker,ger)上进行核磁分析。
[0056]
1.2x射线粉末衍射(x-ray powder diffractometer,xrpd)
[0057]
实验所得固体样品用x射线粉末衍射仪bruker d8 advance(bruker,ger)进行分析。2θ扫描角度从3
°
到45
°
,扫描步长为0.02
°
,曝光时间为0.2秒。测试样品时光管电压和电流分别为40kv和40ma,样品盘为零背景样品盘。
[0058]
1.3热重分析(thermogravimetric analysis,tga)
[0059]
热重分析仪的型号为ta discovery 55(ta,us)。将2-5mg样品置于已平衡的开口铝制样品盘中,在tga加热炉内自动称量。样品以10℃/min的速率加热至最终温度,样品处氮气吹扫速度为60ml/min,天平处氮气吹扫速度为40ml/min。
[0060]
1.4差式扫描量热分析(differential scanning calorimetry,dsc)
[0061]
差示扫描量热分析仪的型号为ta discovery 2500(ta,us)。1-2mg样品经精确称重后置于扎孔的dsc tzero样品盘中,以10℃/min的速率加热至最终温度,炉内氮气吹扫速度为50ml/min。
[0062]
1.5动态水分吸脱附分析(dynamic moisture desorption analysis,dvs)
[0063]
动态水分吸脱附分析采用dvs intrinsic(sms,uk)进行测定。测试采用梯度模式,湿度变化为50%—95%—0%—50%,在0%至90%范围内每个梯度的湿度变化量为10%,梯度终点采用dm/d
t
方式进行判断,以dm/d
t
小于0.002%并维持10分钟为梯度终点。测试完成后的样品进行xrpd分析。
[0064]
实施例1
[0065]
将80mg草乌甲素溶于1.6ml甲醇,将所得澄清溶液快速滴加至80ml水中,将溶液抽滤分离,将固体真空干燥,得到白色絮状固体。
[0066]
xrpd结果(图1)显示所得样品为无定形。tga(图2)结果显示样品有少量吸附水;dsc结果显示无定形样品的玻璃化转变信号不明显,在120℃左右发生重结晶,然后重结晶后的固体在160℃左右发生熔化。
[0067]
实施例2
[0068]
1)将97.3mg原料溶于6ml乙醚,过滤,常温挥发;或者,
[0069]
2)将200.4mg原料溶于0.3ml四氢呋喃,过滤,常温挥发;
[0070]
得晶型i(formi)。对其进行xrpd(图3)、dsc和tga(图4)表征。tga表征中form i加热至150℃无明显失重,在dsc表征中在162.8℃有熔融吸热峰,说明该晶型为无水物。
[0071]
form i的nmr结果与无水草乌甲素一致。根据dvs结果,form i在0-95%湿度范围增重失重不超过0.13%,表明form i随着湿度变化没有明显失水或吸水,且测试前后xrpd图无实质改变,表明dvs测试后的样品并未发生晶型改变。综上,form i是不易吸湿的无水物。
[0072]
实施例3
[0073]
二元溶剂降温法
[0074]
分别采用多种良溶剂与3种不良溶剂组合,进行二元溶剂的冷却结晶实验,每个实验使用样品20mg,结果如表1所示。在甲酸丁酯/正己烷体系中得到form v,二氧六环/环己烷体系中得到form ii,二氯甲烷/环己烷体系中得到form iii,氯仿/环己烷体系中得到form iv,其他有晶体析出的实验均得到form i。
[0075]
form ii在tga表征加热至150℃的过程中失重10.6%,且呈现明显失重台阶,说明该晶型可能为水合物或溶剂合物。dsc表征中,在81.7℃有吸热峰,对应tga的脱溶剂失重过程;在99.7℃出现对应重结晶的放热峰,重结晶所得晶体在158.9℃呈现熔融吸热峰(该峰位置与form i的熔融吸热峰接近)。将form ii样品加热至120℃后进行xrpd测试,结果显示form ii在此条件下转晶为form i。对在室温保存若干天后的form ii进行xrpd测试,结果样品为form ii和form i的混晶,说明form ii不稳定,室温下自发转晶为form i。formii的nmr图中,1.4ppm出现环己烷的特征峰。综合以上信息,form ii应为环己烷溶剂合物。
[0076]
form iii在tga表征加热至130℃的过程中失重5.1%,且呈现明显失重台阶,说明该晶型可能为水合物或溶剂合物。dsc表征中,在94.6℃有吸热峰,对应tga的脱溶剂失重过程;在109.0℃出现对应重结晶的放热峰,重结晶所得晶体在159.2℃呈现熔融吸热峰(该峰位置与formi的熔融吸热峰接近);在170.8℃亦呈现熔融吸热峰(该峰位置与form vi的熔融吸热峰接近)。将form iii样品加热至150℃后进行xrpd测试,结果form iii在此条件下转晶为form i。对在室温保存若干天后的form iii进行xrpd测试,结果样品完全转化成form i,说明form iii不稳定,在室温下自发转晶为form i。form iii的nmr表征图中,1.4ppm出现环己烷的特征峰。综合以上信息,form iii应为环己烷溶剂合物。
[0077]
form iv在tga表征加热至120℃的过程中失重5.0%,且呈现明显失重台阶,说明该晶型可能为水合物或溶剂合物。dsc表征中,在84.6℃有吸热峰,对应tga的脱溶剂失重过程;在92.9℃出现对应重结晶的放热峰,重结晶所得晶体在158.1℃呈现熔融吸热峰(该峰位置与formi的熔融吸热峰接近);在171.3℃亦呈现微弱的熔融吸热峰(该峰位置与formvi的熔融吸热峰接近)。对在0℃保存三天后的form iv进行xrpd测试,样品呈现出form i的特征峰,说明form iv不稳定,即使在0℃也会自发转晶为form i。form iv的nmr表征图中,1.4ppm的峰是环己烷的特征峰。综合以上信息,form iv应为环己烷溶剂合物。
[0078]
form v在tga表征加热至120℃的过程中失重5.8%,且呈现明显失重台阶,说明该晶型可能为水合物或溶剂合物。dsc表征中,在112.0℃有吸热峰,对应tga的脱溶剂失重过程;随后从126℃开始呈现较宽的吸热信号。将form v样品加热至115℃后进行xrpd测试,结果显示,form v在此条件下转晶为form i。
[0079]
表1
[0080]
[0081][0082]
实施例4
[0083]
二元溶剂悬浮法
[0084]
1)称取20mg样品在0.8ml的选定二元混合溶剂(乙腈/水(1:3))中于室温悬浮5天,得到新晶型form vi,或者;
[0085]
2)在1.2ml乙腈/水(1:3)混合溶剂中加入249.3mg原料和007-23-15form vi晶种,室温下悬浮5天,得到新晶型form vi。
[0086]
form vi的xrpd结果见图5。tga及dsc测试结果如图6所示,form vi在tga表征中加热至150℃无失重,在dsc表征中仅174.6℃有熔融吸热峰。form vi的nmr结果与无水草乌甲素结构一致。综上,form vi应为无水晶型。
[0087]
晶型form i-vi的xrpd对比图如图7所示,不同晶型的性质汇总如下表2:
[0088]
表2
[0089][0090][0091]
晶型研究结果表明,form ii、form iii、form iv和form v不稳定,受热会转晶为
form i,即使在室温也会自发脱溶剂转晶为form i。
[0092]
实施例5药代动力学实验
[0093]
1材料与方法
[0094]
1.1仪器
[0095]
waters超高效液相色谱-质谱联用系统(含在线脱气机,超高压梯度泵,柱温箱,自动进样器,waters i-class uplc液相系统,xevo tq-s质谱,unifi工作站);eppendorf 5810r全自动高速冷冻离心机;eppendorf mixmate涡旋仪;mettler toledo xp105dr电子天平。
[0096]
1.1.1试剂
[0097]
甲醇为色谱纯(merck公司,德国);乙腈为色谱纯(merck公司,德国);水为市售屈臣氏蒸馏水。
[0098]
1.2试验动物
[0099]
品系:sd大鼠
[0100]
性别:雄性
[0101]
体重:约250g
[0102]
来源:上海西普尔-必凯实验动物有限责任公司
[0103]
实验动物生产许可证号:scxk(沪)2018-0006
[0104]
饲养:每笼4只,饲养于空调恒温室内,室温20-24℃,湿度40-70%,光照12h。自由进食与饮水。1.3给药方法及样品采集
[0105]
1.3.1给药剂量及给药途径
[0106]
给药方式:灌胃给药;
[0107]
灌胃给药给药剂量:2.7mg/kg(草乌甲素晶型ⅰ、ⅵ、草乌甲素原料、无定形草乌甲素);0.5mg/kg(无定形草乌甲素)。
[0108]
将草乌甲素晶型ⅰ、ⅵ粉末加总溶液体积1%的吐温-80研磨使透明,再加羧甲基纤维素钠补足体积,边加边研磨,配制成0.27mg/ml的药液用于灌胃给药。
[0109]
1.3.2给药及取血
[0110]
sd大鼠12只,分为三组。一组给予草乌甲素晶型ⅰ,一组给予草乌甲素晶型ⅵ,一组给予无定形草乌甲素。给予草乌甲素晶型ⅰ、ⅵ、无定形草乌甲素后0.083,0.25,0.5,1,1.5,2,2.5,3,6,9,12,24h每个时间点样品取全血约0.2ml后8000r/min,离心10min,分离血浆,一份50μl,剩余血浆备份。均冻于-80℃保存。
[0111]
1.4血浆样品处理方法
[0112]
血浆样品50μl加入5μl is(500ng/ml甲苯磺丁脲(jbhdn))混匀后加入450μl甲醇沉淀蛋白后,涡旋30s,13000r/min离心5min,取上清过滤,后经lc/ms/ms分析。
[0113]
1.5建立uplc-ms/ms法测定sd大鼠血浆中草乌甲素晶型ⅰ、ⅵ、草乌甲素原料和无定形草乌甲素含量
[0114]
1.5.1溶液配制
[0115]
1.5.1.1草乌甲素晶型ⅰ、ⅵ标曲工作液配制
[0116]
精密称取草乌甲素晶型ⅰ、ⅵ标准品适量,用甲醇溶解后,再用80%甲醇水稀释为浓度为1mg/ml的草乌甲素晶型ⅰ、ⅵ储备液。取草乌甲素晶型ⅰ、ⅵ储备液以80%甲醇水稀
释,获得草乌甲素晶型ⅰ、ⅵ浓度分别为10,50,100,500,1000,2500,5000ng/ml标曲工作液,并保存至-20℃冰箱。
[0117]
草乌甲素原料分析以草乌甲素晶型ⅰ工作液作为标准进行分析。
[0118]
1.5.1.2草乌甲素晶型ⅰ、ⅵ质控工作液配制
[0119]
精密称取草乌甲素晶型ⅰ、ⅵ标准品适量,用甲醇溶解后,再用80%甲醇水稀释为浓度为1mg/ml的质控储备液。取质控储备液以80%甲醇水稀释,获得草乌甲素晶型ⅰ、ⅵ浓度分别为30,400,4000ng/ml质控工作液,并保存至-20℃冰箱。
[0120]
1.6草乌甲素晶型ⅰ、ⅵ标曲样品及质控样品制备
[0121]
将“1.5.1.1”项下草乌甲素晶型ⅰ、ⅵ标曲系列溶液按照1:10的比例配制于sd大鼠空白血浆中获得浓度分别为1,5,10,50,100,250,500ng/ml的标曲系列样品。按同样的方法稀释“1.5.1.2”项下质控工作液,得到浓度分别为3,40,400ng/ml的血浆质控样品。
[0122]
草乌甲素原料分析以草乌甲素晶型ⅰ工作液作为标准进行分析。
[0123]
1.7样品测定条件
[0124]
1.7.1色谱条件
[0125]
分析柱为acquity uplc beh c18(1.7μm 2.1
×
100mm);流动相为水(0.1%fa):乙腈;梯度洗脱:流速为0.3ml/min;进样量为1μl;柱温为40℃;洗脱梯度见下表3:
[0126]
表3草乌甲素晶型ⅰ、ⅵ流动相梯度表
[0127][0128]
1.7.2质谱条件
[0129]
离子源为电喷雾离子化源(electrospray ionization,esi);源温度为150℃;雾化气体温度为350℃;毛细管电压为3kv;检测方式为正离子检测;扫描方式为多反应监测(multiple reaction monitoring,mrm);待测物及内标的质谱检测参数见下表4:
[0130]
表4待测物的质谱检测参数表
[0131][0132]
1.8数据处理方法
[0133]
采用das2.0软件的非房室模型计算sd大鼠给药后的药代动力学参数。包括达峰浓度c
max
:采用实测值;达峰时间t
max
:采用实测值;药时曲线下面积auc
(0-t)
值:采用梯形法计算;auc
(0-∞)
=auc
(0-t)
c
t
/ke,c
t
为最后一个可测得时间点的血药浓度,ke为消除速率常数;消除半衰期t
1/2
=0.693/ke;平均滞留时间mrt=aumc/auc;分布容积vz=cl/ke。
[0134]
采用excel软件计算各样品血药浓度的平均值、标准差、精密度及准确度。
[0135]
2结果与分析
[0136]
2.1样品分析方法
[0137]
2.1.1测定方法专属性
[0138]
本试验在空白大鼠血浆中添加草乌甲素晶型ⅰ、ⅵ标曲工作液,按“1.4”项下方法处理,分别采用“1.7”项下色谱质谱条件进行lc-ms/ms分析,测试过程中参数设置如下:通道名称:积分式,平滑次数2次,三重四级杆mrm模式,电喷雾电压为30ev,电喷雾正离子模式,母离子分子量为644.29,子离子分子量为583.99。实验结果可见,基质中不存在对草乌甲素晶型ⅰ、ⅵ测定干扰的成分。草乌甲素晶型ⅰ、ⅵ的保留时间为:1.92min。图8为空白血浆质谱图;图9为标曲样品中实测样品质谱图;图10为大鼠给药后血浆中草乌甲素晶型ⅰ、ⅵ实测样品质谱图。
[0139]
2.1.2随行标准曲线及质控
[0140]
以草乌甲素晶型ⅰ、ⅵ的峰面积(as,y)对相应的浓度(c,x)进行加权(1/x2)线性回归,得到草乌甲素晶型ⅰ、ⅵ在大鼠血浆中的标准曲线,见图11,其中,图11中(1)的标准曲线拟合方程为y=2.15e-002 3.19e-002*x,rsd为6.372%,r2为0.997827;图11中(2)标准曲线拟合方程为y=2.01e-002 3.67e-002*x,rsd为5.112%,r2为0.998776。各分析批质控准确度结果见表5:
[0141]
表5样品分析批qc质控样品的计算浓度
[0142][0143]
接受标准:每个分析批应含有至少2套3个水平的质控样品,质控样品总数不低于待测样品数的5%。每个分析批至少67%的质控样品应符合准确度在
±
15%范围内,精密度不超过15%的标准,且每一浓度的质控样品中至少50%符合上述标准。
[0144]
2.2sd大鼠草乌甲素晶型ⅰ、ⅵ和无定形草乌甲素给药后血药浓度及药代动力学参数
[0145]
2.2.1sd大鼠草乌甲素晶型ⅰ、ⅵ和无定形草乌甲素单次灌胃给药血药浓度
[0146]
sd大鼠单次灌胃给予草乌甲素晶型ⅰ后血药浓度见表6-1,血药浓度-时间曲线见图12;单次灌胃给予草乌甲素晶型ⅵ后血药浓度见表6-2,血药浓度-时间曲线见图13;大鼠单次灌胃给予天然草乌甲素后血药浓度见表6-3,血药浓度-时间曲线见图14;大鼠单次灌胃给予无定形草乌甲素后血药浓度见表6-4、6-5,血药浓度-时间曲线见图15;
[0147]
表6-1大鼠灌胃给予受试药单次灌胃给予草乌甲素晶型ⅰ后不同时间点血浆药物浓度(2.7mg/kg)
[0148][0149][0150]
表6-2大鼠灌胃给予受试药草乌甲素晶型ⅵ后不同时间点血浆药物浓度(2.7mg/kg)
[0151][0152]
表6-3大鼠灌胃给予受试药草乌甲素原料后不同时间点血浆药物浓度(2.7mg/kg)
[0153][0154]
表6-4大鼠0.5mg/kg剂量经口灌胃给予无定形草乌甲素后血药浓度
[0155][0156][0157]
备注:*代表低于检测限
[0158]
表6-5大鼠2.7mg/kg剂量经口灌胃给予无定形草乌甲素后血药浓度
[0159][0160]
备注:/代表大鼠死亡
[0161]
2.2.2sd大鼠给药后药代动力学参数
[0162]
sd大鼠单次给予草乌甲素晶型ⅰ、ⅵ、草乌甲素原料及无定形草乌甲素后相应药代动力学参数见表7-1,7-2,7-3及7-4:
[0163]
表7-1大鼠灌胃给予受试药草乌甲素晶型ⅰ后药代动力学参数(2.7mg/kg)
[0164][0165]
表7-2大鼠灌胃给予受试药草乌甲素晶型ⅵ后药代动力学参数(2.7mg/kg)
[0166][0167][0168]
表7-3大鼠灌胃给予受试药天然草乌甲素后药代动力学参数(2.7mg/kg)
[0169][0170]
表7-4大鼠灌胃给予受试药无定型草乌甲素后药代动力学参数(0.5mg/kg)
[0171][0172]
上述试验结果表明大鼠单次灌胃给予草乌甲素晶型ⅰ、ⅵ、草乌甲素原料及无定形草乌甲素后体内草乌甲素血药浓度平均达峰时间分别为(0.38
±
0.18)h、(1.063
±
0.591)h、(0.875
±
0.8)h和(0.5
±
0.354)h,平均达峰浓度分别为(10.38
±
1.85)ng/ml、(20.575
±
8.32)ng/ml、(21.098
±
4.40)ng/ml和(11.809
±
4.153)ng/ml,平均药时曲线下面积auc
0-t
分别为(73.2
±
18.6)ng/ml*h、(115.544
±
29.598)ng/ml*h、(124.25
±
32.85)ng/ml*h和(34.363
±
6.582)ng/ml*h;大鼠灌胃给药后体内消除半衰期平均分别为(21.48
±
2.12)h、(12.547
±
14.311)h、(4.700
±
2.489)h和(2.085
±
0.539)h。
[0173]
实施例6镇痛比较试验
[0174]
1.动物:spf级小鼠(上海西普尔-必凯实验动物有限公司,动物合格证号:scxk(沪)2018-0006,体重约18~22g)
[0175]
2.方法:取雄性小鼠110只,随机分为11组:溶媒对照组;阳性对照(阿斯匹林200mg/kg)组;草乌甲素原料组,0.2、0.4、0.8mg/kg;草乌甲素晶型ⅰ组0.1、0.2、0.4mg/kg;无定形草乌甲素组0.04、0.08、0.16mg/kg组;每组10只。各组动物分别按剂量灌胃给药一次,溶媒对照组按20ml/kg给予1%cmc-na给药后30min,腹腔注射0.6%冰醋酸溶液0.1ml/10g,观察并记录15min内各鼠疼痛扭体次数,比较各组对醋酸致疼痛扭体的抑制率结果如下表8所示:
[0176]
抑制率(%)=(溶媒组平均扭体次数-药物组平均扭体次数)/溶媒组平均扭体次数*100%
[0177]
表8草乌甲素晶型对醋酸致小鼠疼痛的影响
[0178][0179]
从上表8可以看出,草乌甲素晶型i和草乌甲素无定形的效果均优于草乌甲素原料,尤其是草乌甲素无定形显著优于草乌甲素原料。
[0180]
实施例7草乌甲素对km鼠急毒实验的研究
[0181]
1、实验动物
[0182]
种属:km小鼠
[0183]
等级:spf级
[0184]
购入动物数量和性别:30只,雄性
[0185]
来源:上海西普尔-必凯实验动物有限公司
[0186]
生产许可证号:scxk(沪)2018-0006
[0187]
实验动物使用许可证:syxk(沪)2014-0018
[0188]
2、实验方法
[0189]
2.1分组
[0190]
小鼠适应性饲养1-2天,草乌甲素晶型ⅰ和晶型ⅵ各15只小鼠,草乌甲素晶型ⅰ随机分为5组,每组3只:空白组、晶型ⅰ2.8mg/kg、晶型ⅰ3.79mg/kg、晶型ⅰ5.06mg/kg、晶型ⅰ6.75mg/kg、晶型ⅰ9.0mg/kg;晶型ⅵ同晶型ⅰ分组一致。
[0191]
2.2实验方法
[0192]
给药前,禁食并观察动物生理状态。除空白组外,其余组采用小鼠灌胃给予各剂量草乌甲素晶型i或晶型vi,各组剂量分别为2.8、3.79、5.06、6.75、9.0mg/kg(两种晶型一
致)。观察动物中毒症状,以及记录死亡数量。
[0193]
2.3计算方法
[0194]
采用bliss法软件计算小鼠半数致死量。得出数据并绘制死亡率-对数剂量曲线以及机率单位-对数剂量曲线。
[0195]
3、实验结果
[0196]
晶型i和晶型vi的ld
50
的计算报告分别如表9和10所示,结果如表晶型i和晶型vi死亡率-对数剂量曲线以及机率单位-对数剂量曲线分别如图16和17所示。从表9~10及图16和17可以看出,给予草乌甲素后数据显示,晶型ⅰ的ld
50
为4.4mg/kg,95%可信限为3.6~5.3mg/kg;晶型ⅵ的ld
50
为5.9mg/kg,95%可信限为4.8~7.3mg/kg
[0197]
表9晶型i的ld
50
计算报告
[0198][0199]
表10晶型ⅵ的ld
50
计算报告
[0200][0201]
4、结论
[0202]
草乌甲素晶型ⅰ和ⅵ均有一定毒性。
[0203]
晶型ⅰ的ld
50
为4.4mg/kg,95%可信限为3.6~5.3mg/kg,feiller校正后ld
50
为4.3mg/kg;晶型ⅵ的ld
50
为5.9mg/kg,95%可信限为4.8~7.3mg/kg;feiller校正后ld
50
为6.1mg/kg。
[0204]
根据不规范的草乌甲素的两个晶型ⅰ和ⅵ在相应的溶剂溶解后用吐温80调整至口服溶液测得的急性毒性。
[0205]
本发明人对剂量摸索后,对晶型药物的药代动力学进行测试。证明了晶型ⅰ和晶型ⅵ较无定形草乌甲素c
max
明显升高,表明晶型i和晶型vi的毒性均明显低于无定形草乌甲素。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。