1.本发明涉及有机合成技术领域,特别是涉及一种多官能团化环状硝酮衍生物的合成方法。
背景技术:
2.γ,δ-不饱和肟的环化反应是一种常用的构建硝酮化合物的合成方法,且硝酮化合物是有机合成中重要的结构单元,也是农药、医药、染料以及功能材料的重要合成中间体。在以往的研究中,基于硝酮杂环化合物的这些优良的性质,很多化学家踊跃参与到硝酮及其衍生物的合成方法开发中,从而发展并丰富了此类化合物的合成方法。尽管现在已经具有很多此类化合物的合成方法,但是合成化学家依旧不断开拓发展更加温和、简洁和实用的合成路线来实现一步高效构建硝酮杂环化合物。
3.目前,已报道的通过分子内烯烃双官能团化构建硝酮化合物的方法均存在一些缺点。例如,使用较高的反应温度和当量的引发剂(han b.angew.chem.int.ed.2012,51,8816-8820;wang q.chemical science,2015,6,4279-4283.),或者使用过量的氧化剂(han b.acs catalysis,2016,6,6525-6530.)因而亟需寻找一种温和且无需额外氧化剂参与,价格低廉的合成环状硝酮化合物的方法。
技术实现要素:
4.本发明提供一种多官能团化环状硝酮衍生物的合成方法,本发明致力于发展一种反应条件温和,价格低廉,简单高效的非贵金属催化γ,δ-不饱和酮肟和苯甲酰羟胺的自由基串级环化反应构建邻位二胺化的硝酮化合物的方法,该方法在无需任何氧化剂的参与下,以温和的条件,高效地合成环状衍生物,并获得较好的收率。
5.为实现上述目的,本发明提供了如下方案:
6.本发明提供一种多官能团化环状硝酮衍生物的合成方法,以γ,δ-不饱和酮肟和苯甲酰羟胺为原料,以二异丙基乙基胺(dipea)为有机碱添加剂,二价铜为催化剂,50℃加热反应,得到多官能团化环状硝酮衍生物,所述多官能团化环状硝酮衍生物为分子内烯烃二胺化的环状硝酮化合物。
7.本发明以γ,δ-不饱和酮肟为底物,以廉价的二价铜盐作为催化剂,在dipea碱性条件下,室温搅拌,串级环化反应一步构建两个新的碳-氮键,得到一系列官能团化的硝酮杂环化合物,并且所合成的二胺化硝酮化合物可以进一步衍生化成氮氧lewis配体,为有机中间体和新型配体的开发提供思路。
8.优选地,所述多官能团化环状硝酮衍生物的合成方法,合成路线如下:
[0009][0010]
其中r1=r4=ch3、cl、br或cf3,r2、r3、r5、r6=烷基(alkyl),r7=芳基(ar);
[0011]
具体包括以下步骤:
[0012]
1)多官能团化环状硝酮衍生物的合成
[0013]
氩气条件下,将γ,δ-不饱和酮肟、苯甲酰羟胺、二异丙基乙基胺(dipea)和二价铜加入到反应器中反应,控制温度在50℃,加入乙腈(ch3cn,2.0ml),磁力搅拌反应24-36h;
[0014]
2)反应结束后,旋蒸除去乙腈,加水淬灭,乙酸乙酯萃取,合并有机相,减压蒸馏去除大部分溶剂,柱层析分离提纯,得到多官能团化环状硝酮衍生物。
[0015]
本发明采用γ,δ-不饱和酮肟为底物与苯甲酰羟胺进行亲电胺化环化反应,以dipea为有机碱,在铜催化的条件下,室温反应合成官能团化的环状硝酮杂环化合物。与已报道的制备方法相比,本发明有着反应条件温和、底物普适性强、官能团兼容性好、反应收率高、无需过氧化物添加剂以及使用的铜催化剂价格低廉、后处理简单易操作等诸多优点,具有很高的应用价值。同时获得的多官能团化环状硝酮衍生物是有机合成,医药,催化和配体中重要的结构片段。
[0016]
优选地,所述二价铜为cucl2。
[0017]
优选地,γ,δ-不饱和酮肟和苯甲酰羟胺的摩尔比为1:2。
[0018]
优选地,二价铜的加入量为20%摩尔量。
[0019]
优选地γ,δ-不饱和酮肟的结构式如式1所示:苯甲酰羟胺的结构式如式2所示:多官能团化环状硝酮衍生物的结构式如式3所示:
[0020]
其中r1=r4=ch3、cl、br或cf3,r2、r3、r5、r6=烷基,r7=芳基。
[0021]
r6=柱层析分离提纯中的洗脱剂为石油醚与乙酸乙酯。
[0022]
r6=石油醚与乙酸乙酯的体积比为5:1~10:1。
[0023]
本发明公开了以下技术效果:
[0024]
1)本发明使用苯甲酰羟胺作为亲电氮源,无需外加氧化剂的参与,利用廉价的金属铜盐作为催化剂一定程度上降低了经济成本,同时也成功实现了烯丙基肟分子内的二胺化反应,而二胺化产物是有机合成中间体,医药,催化和配体的重要结构片段,收益可观。
[0025]
2)本发明含有吗啉取代基的环状硝酮骨架,可作为有机合成中间体,该方法有着良好的官能团耐受性和宽广的底物范围等优势,可对药物,农药和功能材料的新型合成提
供有效片段。
[0026]
3)本发明采用铜催化自由基串级环化的方式构建氮杂环化合物,并且无需添加当量的氧化剂,符合绿色环保的理念;以便宜的铜盐为催化剂,在温和的条件下促使两个新的碳-氮键形成,实现环化反应,实用性强。
附图说明
[0027]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0028]
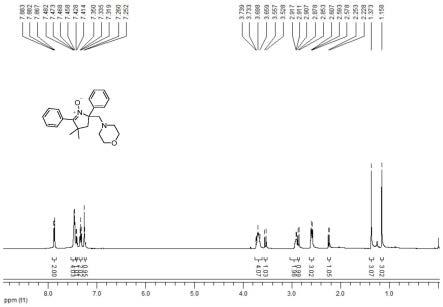
图1为实施例1制备的4,4-二甲基-2-(吗啉甲基)-2,5-二苯基-3,4-二氢吡咯-1-氮氧化物的1h-nmr核磁共振谱;
[0029]
图2为实施例2制备的4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-甲基苯基)-3,4-二氢吡咯-1-氮氧化物的1h-nmr核磁共振谱;
[0030]
图3为实施例3制备的4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-氯苯基)-3,4-二氢吡咯-1-氮氧化物的1h-nmr核磁共振谱;
[0031]
图4为实施例4制备的4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-三氟甲基苯基)-3,4-二氢吡咯-1-氮氧化物的1h-nmr核磁共振谱;
[0032]
图5为实施例5制备的4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-甲氧基苯基)-3,4-二氢吡咯-1-氮氧化物的1h-nmr核磁共振谱;
[0033]
图6为实施例6制备的5-苯基-5-(2,2,2-三氟乙基)-3-(4-甲硫基苯基)-4,5-二氢异噁唑的1h-nmr核磁共振谱。
具体实施方式
[0034]
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
[0035]
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值,以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
[0036]
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
[0037]
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见得的。本发明说明书和实施例仅是示例性的。
[0038]
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
[0039]
本发明利用γ,δ-不饱和酮肟,苯甲酰羟胺为起始原料,在氩气保护下,加入1.2摩尔当量的dipea(二异丙基乙基胺)和20%摩尔催化量的cucl2,并控制温度在50℃,于乙腈(2.0ml)中,加热反应36h,其中γ,δ-不饱和酮肟与三氟甲基亚磺酸钠的摩尔比为1:2,实现了不饱和酮肟分子内烯烃的双胺化反应,高效合成了亲电胺化的环状硝酮衍生物。其中dipea为二异丙基乙基胺。
[0040]
实施例1:4,4-二甲基-2-(吗啉甲基)-2,5-二苯基-3,4-二氢吡咯-1-氮氧化物的合成
[0041][0042]
氩气氛围下,向25ml的史莱克反应管中加入2,2-二甲基-1,4-二苯基-丁-4-烯-1-酮肟(0.2mmol,53.2mg)、吗啉苯甲酰羟胺(0.4mmol,41.4mg),dipea(二异丙基乙基胺)(0.24mmol,31.0mg),cucl2(0.04mmol,5.4mg)乙腈3ml,50℃磁力搅拌反应24小时;反应完毕后,减压蒸馏除去乙腈,加水淬灭,乙酸乙酯萃取三次,合并有机相,减压浓缩得到粗品,粗品用石油醚:乙酸乙酯(体积比10:1)作为淋洗剂洗脱,经柱层析分离提纯后得到目标产物为淡黄色固体,收率74%。
[0043]
其核磁数据如下:
[0044]1h nmr(500mhz,cdcl3):δ7.89-7.86(m,2h),7.49-7.41(m,4h),7.43-7.41(m,1h),7.35-7.31(m,2h),7.26-7.25(m,1h),3.74-3.65(m,4h),3.54(d,j=14.5hz,1h),2.92-2.90(m,2h),2.86(d,j=12.5hz,1h),2.61-2.57(m,3h),2.24(d,j=12.5hz,1h),1.37(s,3h),1.16(s,3h).
[0045]
实施例2:4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-甲基苯基)-3,4-二氢吡咯-1-氮氧化物的合成
[0046][0047]
氩气氛围下,向25ml的史莱克反应管中加入2,2-二甲基-1-(4-甲基苯基)-4-苯基-丁-4-烯-1-酮肟(0.2mmol,58.6mg)、吗啉苯甲酰羟胺(0.4mmol,41.4mg),dipea(二异丙基乙基胺)(0.24mmol,31.0mg),cucl2(0.04mmol,5.4mg)乙腈3ml,50℃磁力搅拌反应24小时;反应完毕后,减压蒸馏除去乙腈,加水淬灭,乙酸乙酯萃取三次,合并有机相,减压浓缩
得到粗品,粗品用石油醚:乙酸乙酯(体积比5:1)作为淋洗剂洗脱,经柱层析分离提纯后得到目标产物为淡黄色泡状物,收率57%。
[0048]
其核磁数据如下:
[0049]1h nmr(500mhz,cdcl3):δ7.85(d,j=8.0hz,2h),7.47-7.44(m,2h),7.42-7.41(m,1h),7.34-7.30(m,2h),7.28-7.24(m,2h),3.73-3.63(m,4h),3.53(d,j=14.5hz,1h),2.93-2.84(m,2h),2.85(d,j=12.5hz,1h),2.61-2.56(m,3h),2.39(s,3h),2.21(d,j=12.0hz,1h),1.38(s,3h),1.16(s,3h).
[0050]
实施例3:4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-氯苯基)-3,4-二氢吡咯-1-氮氧化物的合成
[0051][0052]
氩气氛围下,向25ml的史莱克反应管中加入2,2-二甲基-1-(4-氯苯基)-4-苯基-丁-4-烯-1-酮肟(0.2mmol,62.6mg)、吗啉苯甲酰羟胺(0.4mmol,41.4mg),dipea(二异丙基乙基胺)(0.24mmol,31.0mg),cucl2(0.04mmol,5.4mg)乙腈3ml,50℃磁力搅拌反应36小时;反应完毕后,减压蒸馏除去乙腈,加水淬灭,乙酸乙酯萃取三次,合并有机相,减压浓缩得到粗品,粗品用石油醚:乙酸乙酯(体积比5:1)作为淋洗剂洗脱,经柱层析分离提纯后得到目标产物为棕色絮状物,收率51%。
[0053]
其核磁数据如下:
[0054]1h nmr(500mhz,cdcl3):δ7.94(d,j=9.0hz,2h),7.45-7.42(m,4h),7.35-7.31(m,2h),7.26-7.24(m,1h),3.72-3.63(m,4h),3.52(d,j=14.5hz,1h),2.90-2.84(m,3h),2.24(d,j=12.0hz,1h),1.38(s,3h),1.16(s,3h).
[0055]
实施例4:4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-三氟甲基苯基)-3,4-二氢吡咯-1-氮氧化物的合成
[0056][0057]
氩气氛围下,向25ml的史莱克反应管中加入2,2-二甲基-1-(4-三氟甲基苯基)-4-苯基-丁-4-烯-1-酮肟(0.2mmol,66.8mg)、吗啉苯甲酰羟胺(0.4mmol,41.4mg),dipea(二异丙基乙基胺)(0.24mmol,31.0mg),cucl2(0.04mmol,5.4mg)乙腈3ml,50℃磁力搅拌反应36小时;反应完毕后,减压蒸馏除去乙腈,加水淬灭,乙酸乙酯萃取三次,合并有机相,减压浓缩得到粗品,粗品用石油醚:乙酸乙酯(体积比5:1)作为淋洗剂洗脱,经柱层析分离提纯后
得到目标产物为白色固体,收率45%。
[0058]
其核磁数据如下:
[0059]1h nmr(500mhz,cdcl3):δ8.05(d,j=8.5hz,2h),7.72(d,j=8.5hz,2h),7.44(d,j=7.5hz,2h),7.28-7.26(m,1h),3.74-3.64(m,4h),3.54(d,j=15.0hz,1h),2.92-2.86(m,3h),2.63-2.59(m,3h),2.29(d,j=12.5hz,1h),1.39(s,3h),1.18(s,3h).
[0060]
实施例5:4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-甲氧基苯基)-3,4-二氢吡咯-1-氮氧化物的合成
[0061][0062]
氩气氛围下,向25ml的史莱克反应管中加入2,2-二甲基-1-(4-甲氧基苯基)-4-苯基-丁-4-烯-1-酮肟(0.2mmol,62.0mg)、吗啉苯甲酰羟胺(0.4mmol,41.4mg),dipea(二异丙基乙基胺)(0.24mmol,31.0mg),cucl2(0.04mmol,5.4mg)乙腈3ml,50℃磁力搅拌反应24小时;反应完毕后,减压蒸馏除去乙腈,加水淬灭,乙酸乙酯萃取三次,合并有机相,减压浓缩得到粗品,粗品用石油醚:乙酸乙酯(体积比5:1)作为淋洗剂洗脱,经柱层析分离提纯后得到目标产物为黄色固体,收率74%。
[0063]
其核磁数据如下:
[0064]1h nmr(500mhz,cdcl3):δ8.05(d,j=8.5hz,2h),7.72(d,j=8.5hz,2h),7.44(d,j=7.5hz,2h),7.28-7.26(m,1h),3.74-3.64(m,4h),3.54(d,j=15.0hz,1h),2.92-2.86(m,3h),2.63-2.59(m,3h),2.29(d,j=12.5hz,1h),1.39(s,3h),1.18(s,3h).
[0065]
实施例6:4,4-二甲基-2-(吗啉甲基)-2-苯基-5-(4-甲硫基苯基)-3,4-二氢吡咯-1-氮氧化物的合成
[0066][0067]
氩气氛围下,向25ml的史莱克反应管中加入2,2-二甲基-1-(4-甲硫基苯基)-4-苯基-丁-4-烯-1-酮肟(0.2mmol,65.1mg)、吗啉苯甲酰羟胺(0.4mmol,41.4mg),dipea(二异丙基乙基胺)(0.24mmol,31.0mg),cucl2(0.04mmol,5.4mg)乙腈3ml,50℃磁力搅拌反应24小时;反应完毕后,减压蒸馏除去乙腈,加水淬灭,乙酸乙酯萃取三次,合并有机相,减压浓缩得到粗品,粗品用石油醚:乙酸乙酯(体积比5:1)作为淋洗剂洗脱,经柱层析分离提纯后得到目标产物为白色固体,收率73%。
[0068]
其核磁数据如下:
[0069]1h nmr(500mhz,cdcl3):δ7.99-7.96(m,2h),7.45-7.43(m,2h),7.34-7.30(m,2h),7.25-7.22(m,1h),3.72-3.62(m,4h),3.53(d,j=14.5hz,1h),2.92-2.88(m,2h),2.85(d,j=12.5hz,1h),2.61-2.58(m,3h),2.50(s,3h),2.22(d,j=12.5hz,1h),1.40(s,3h),1.17(s,3h).
[0070]
本发明合成的多官能团化环状硝酮衍生物在医药,农药,功能材料,有机合成等诸多领域有着广泛的应用,另外,本发明操作简单,成本低廉,反应条件温和,后处理简便,底物范围广,拥有广阔的应用前景。
[0071]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。